

ABSTRACT
During infections with the protozoan parasite Toxoplasma gondii, gamma-aminobutyric acid (GABA) is utilized as a carbon source for parasite metabolism and also to facilitate parasite dissemination by stimulating dendritic-cell motility. The best-recognized function for GABA, however, is its role in the nervous system as an inhibitory neurotransmitter that regulates the flow and timing of excitatory neurotransmission. When this pathway is altered, seizures develop. Human toxoplasmosis patients suffer from seizures, suggesting that Toxoplasma interferes with GABA signaling in the brain. Here, we show that while excitatory glutamatergic presynaptic proteins appeared normal, infection with type II ME49 Toxoplasma tissue cysts led to global changes in the distribution of glutamic acid decarboxylase 67 (GAD67), a key enzyme that catalyzes GABA synthesis in the brain. Alterations in GAD67 staining were not due to decreased expression but rather to a change from GAD67 clustering at presynaptic termini to a more diffuse localization throughout the neuropil. Consistent with a loss of GAD67 from the synaptic terminals, Toxoplasma-infected mice develop spontaneous seizures and are more susceptible to drugs that induce seizures by antagonizing GABA receptors. Interestingly, GABAergic protein mislocalization and the response to seizure-inducing drugs were observed in mice infected with type II ME49 but not type III CEP strain parasites, indicating a role for a polymorphic parasite factor(s) in regulating GABAergic synapses. Taken together, these data support a model in which seizures and other neurological complications seen in Toxoplasma-infected individuals are due, at least in part, to changes in GABAergic signaling.
IMPORTANCE
Infections of the central nervous system can cause seizures. While inflammation in the brain has been proposed to initiate the onset of the seizures, relatively little is known about how inflammation impacts the structure and function of the neurons. Here we used a parasite called Toxoplasma gondii that infects the brain and showed that seizures arise due to a defect in signaling of GABA, which is the neurotransmitter primarily responsible for preventing the onset of seizures.
INTRODUCTION
Neural activity within the brain is a balance of excitatory and inhibitory neurotransmission, and when excitatory activity becomes uncontrolled, seizures develop. A number of pathological conditions, including trauma, cancer, and inflammation, trigger seizures (1–3). Bacterial, viral, fungal, and parasitic infections of the brain also cause seizures, and several mechanisms by which seizures develop in response to infection have been proposed. These include breakdown of the blood-brain barrier, which allows an influx of either potassium, which increases neuronal firing, or autoantibodies, which target a number of proteins involved in neurotransmission (4). In addition, proinflammatory proteins, such as interleukin-1 beta (IL-1β) and cyclooxygenase 2, can cause immune system-mediated damage to neurons and glia that leads to the onset of seizures (4, 5). Although there is a general understanding of the initial infection-induced events that trigger seizures, significantly less is understood about how they actually impact the structure and function of neurons.
Toxoplasma gondii is a protozoan parasite that infects ~30% of the world’s population (6, 7). People and animals become infected following digestion of oocysts found in feline fecal material or tissue cysts in undercooked meat. Once in the gut, the parasite infects intestinal epithelial cells and converts to the rapidly growing tachyzoite form. Inflammatory cells are recruited to combat the infection and in turn become infected and hijacked by the parasite to disseminate from the gut to target tissues, including the brain, retina, and muscle (8–11). Within these tissues, gamma interferon (IFN-γ) is key for killing most of the tachyzoites, but some evade killing and transform into the quiescent bradyzoite form (7, 12). Bradyzoites then form tissue cysts by encasing themselves within a highly glycosylated and impermeable cyst wall that renders the bradyzoites resistant to killing by either the immune response or antiparasitic drugs (13). Tissue cysts can spontaneously rupture, and the released bradyzoites develop back into tachyzoites, which continue to replicate until an immune response is mounted (7). The parasite has a clonal population structure, and the strain types display differences in virulence and clinical presentation. North America and Europe are dominated by four major strain types (types I, II, III, and 12 [or X]) (14, 15). In mice, type I strains are very virulent, with a 100% lethal dose (LD100) of 1 parasite and death occurring within 10 days postinfection, which is a time before a chronic infection is established. Type II and III strains, with LD50s of 103 and 105, respectively, are less virulent in mice (16). Regardless, all strain types can establish long-lived chronic infections, although the numbers and sizes of cysts can differ, even between parasites belonging to the same strain type (17).
Three factors contribute to the onset and severity of toxoplasmosis. First, a host’s ability to control parasite replication is key and those who are immunocompromised or were infected in utero are highly susceptible to developing disease (18, 19). Second, improperly regulated immune responses can cause immune system-mediated tissue damage (20). Finally, the disease type and severity are dictated by the genotypes of both the parasite and host (21–23). Symptomatic brain infection with Toxoplasma is known as toxoplasmic encephalitis (TE) and can clinically present with dizziness, headaches, and seizures. Despite the fact that TE has been clinically recognized for over 60 years (24), the effect that Toxoplasma and many other intracerebral pathogens have on neuronal structure and function is unknown. Therefore, the goal of this study was to use a murine TE model to identify the changes to neurons during TE that may underlie these neurological symptoms. Here, we report that mice with TE develop seizures due, in part, to specific changes in the synaptic localization of glutamic acid decarboxylase 67 (GAD67). Since GAD67 is the key enzyme for the neuronal biosynthesis of gamma-aminobutyric acid (GABA), which is the major inhibitory neurotransmitter in the brain, these data indicate that Toxoplasma directly interferes with inhibitory neurotransmission and provides a molecular basis for the development of neurological complications in TE patients.
RESULTS
Toxoplasma specifically alters GABAergic synapses.
Since Toxoplasma modulates GABAergic signaling in monocytes and uses GABA as a carbon source, and since GABA is the major inhibitory neurotransmitter in the brain (25, 26), we examined the effect of type II ME49 Toxoplasma infection on GABAergic neurons. Thus, mice were mock infected or infected with Toxoplasma tissue cysts of the type II ME49 strain and, 30 days later, their brains were harvested and stained to detect GAD67, which catalyzes the conversion of glutamate to GABA and is normally clustered at presynaptic GABAergic terminals in the mature brain (27, 28). Low-magnification images of whole-hemisphere brain sections revealed an apparent loss of GAD67 immunoreactivity in the caudal areas of hippocampi from type II ME49-infected mice as well as in the cortex and thalamus (Fig. 1A and B). The effect on GAD67 immunoreactivity appeared to be more widespread than the parasites were in the brain (according to the results of an assay using an anti-Toxoplasma antisera that detects both tachyzoite and bradyzoite antigens) (Fig. 1C and D). The effect of type II ME49 Toxoplasma on inhibitory GABAergic synapses appeared to be specific, as no apparent differences were seen in staining of the presynaptic glutamatergic markers vesicular glutamate transporter 1 (VGluT1) in the hippocampus, VGluT1 or VGluT2 in the cortex, and VGluT1 in the ventral horn of the spinal cord (Fig. 2; see also Fig. S1 in the supplemental material).
FIG 1 .
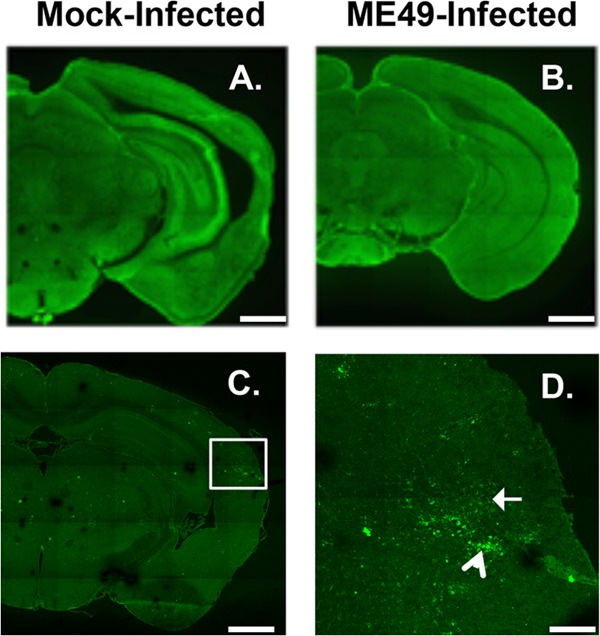
Decreased GABAergic synaptic protein staining in type II ME49 Toxoplasma-infected brains. (A and B) GAD67 immunoreactivity in mock-infected (A) or type II ME49 Toxoplasma-infected (B) brains. (C and D) Detection of type II ME49 Toxoplasma tachyzoites (white arrow in panel D) and bradyzoites (arrowhead in panel D). The image in panel D is a high-magnification image of the boxed in area in panel C. Scale bars, 1 mm (A, B, and C) and 0.25 mm (D). Lines appear in the images due to lack of overlap during image acquisition of each brain.
FIG 2 .
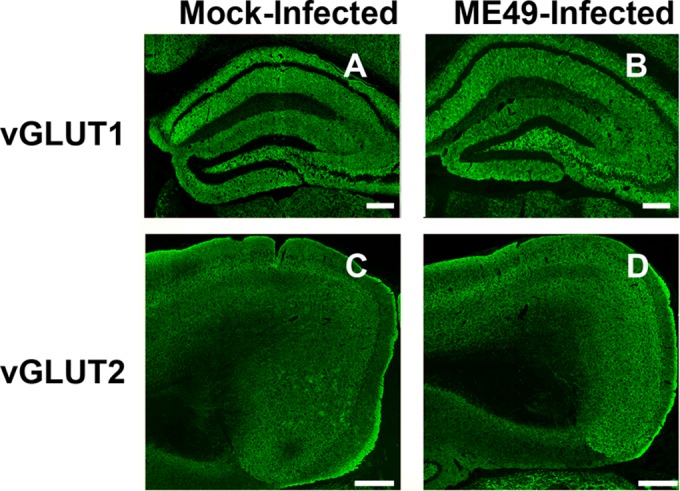
Unaltered glutamatergic synaptic protein staining in type II ME49 Toxoplasma-infected brains. (A and B) VGluT1 immunoreactivity in mock-infected or type II ME49 Toxoplasma-infected hippocampi. Scale bars, 0.2 mm. (C and D) VGluT2 immunoreactivity in mock-infected or type II ME49 Toxoplasma-infected cortexes. Scale bars, 0.4 mm.
Toxoplasma disrupts GAD67 presynaptic clustering.
Decreased GAD67 immunoreactivity could be due to either changes in GAD67 expression or its relocalization from distinct puncta at synaptic terminals to a more diffuse staining within the neuron. In fact, redistribution of presynaptic machinery is known to occur during development, when transsynaptic signals induce the clustering of diffusely localized presynaptic molecules into discrete clusters specifically at synaptic sites (29, 30). To discriminate between these two possibilities, we compared brains of mock-infected and type II ME49-infected mice for mRNA levels of gad1 (the gene that encodes GAD67) and gphn, which encodes gephyrin, a scaffolding protein localized to the postsynaptic specializations of GABAergic synapses. No significant differences were observed in gad1 or gphn transcript levels (Fig. 3A). Similarly, no statistically significant differences in the abundances of the proteins were noted in parasite-infected brains (P > 0.7 [two-tailed, unpaired t test]) (Fig. 3B).
FIG 3 .

Type II ME49 Toxoplasma infections induce mislocalization of GAD67. (A) RT-PCR quantification of relative gad1 and gphn transcript levels in mock-infected and type II ME49 parasite-infected brains. (B) Western blot detection of GAD67, gephyrin, and β-actin in cortical tissues harvested from mock-infected and type II ME49 parasite-infected brains. (C) High-magnification images of GAD67 staining in hippocampal CA3 regions in mock-infected and type II ME49 parasite-infected mice. Note the change from punctate staining in the mock-infected brains to a more diffuse staining in the parasite-infected brains. (D) Quantification of percent GAD67 and VGlut1 staining following binarizing of 5 images/brain (n = 3 mice/group). *, P < 0.02 (unpaired, two-tailed t test).
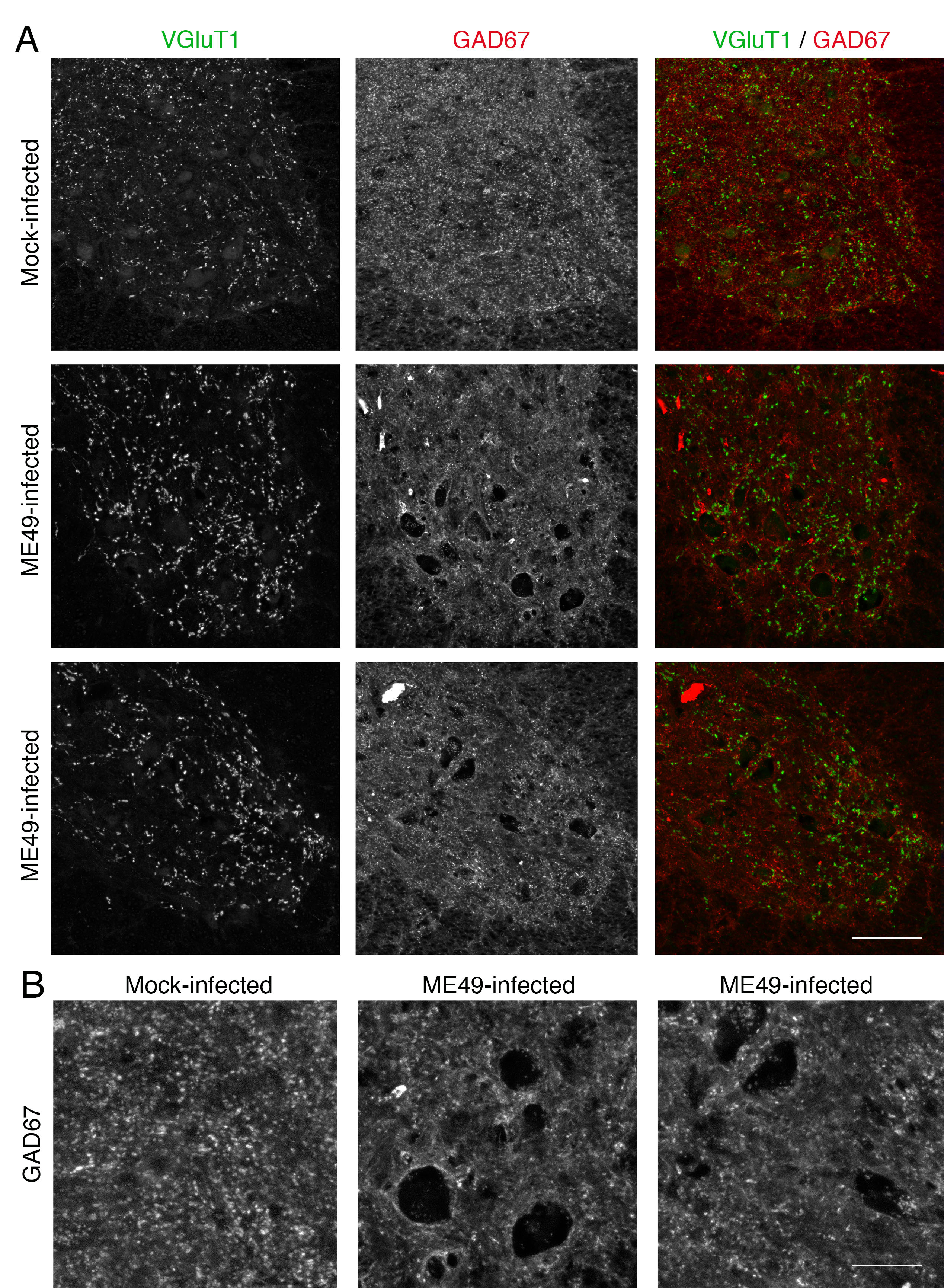
Similarly to previous work (28), high-magnification analysis revealed that GAD67 localized to discrete synaptic puncta in the stratum pyramidale and other regions in the hippocampi of mock-infected mice (Fig. 3C). In contrast, GAD67 staining in these regions from the infected animals no longer appeared enriched at synaptic release sites. To quantify these differences, GAD67-stained images were binarized and the percent area of binarized immunoreactivity was calculated. Despite the minimal changes in overall GABA levels (Fig. 3A and B), these analyses revealed a statistically significant increase in the area containing binarized GAD67 immunoreactivity in the parasite-infected brains (Fig. 3D). Similarly to VGluT1 staining in Fig. 2, binary image analysis revealed no significant differences in the levels of VGluT1 coverage between mock-infected and parasite-infected brains. The impact of type II ME49 Toxoplasma on GAD67 localization was not restricted to the hippocampus. GAD67 localization was similarly affected in the lateral geniculate nucleus (LGN) of the thalamus and in the ventral horn of the spinal cord (see Fig. S1 and S2 in the supplemental material). Moreover, GAD67 remained restricted to neurons since it did not colocalize with either Iba1 (a macrophage marker)-expressing or glial fibrillary acidic protein (GFAP) (an astrocyte marker)-expressing cells (see Fig. S3). These data indicate that type II ME49 Toxoplasma globally redistributes GAD67 away from GABAergic nerve terminals.
Toxoplasma-infected mice develop spontaneous seizures.
GAD67 clusters within presynaptic terminals due to its tethering to GABAergic synaptic vesicles, which is a process thought to support efficient and rapid neurotransmitter loading into the synaptic vesicles (28, 31). Thus, alterations in GAD67 localization significantly impact GABAergic neurotransmission. We therefore assessed the neural and muscular activity of mock-infected or type II ME49 parasite-infected mice with electroencephalography (EEG) and electromyography (EMG) over a period of 5 days. EEGs were recorded from skull electrodes implanted over motor and sensory cortices of the mice, and EMGs were recorded from the nuchal musculature. EEG and EMG analyses revealed significant increases in seizure activity in the infected mice, as defined as greater than 30 s of sustained high-amplitude, high-frequency neural and muscular activity (Fig. 4A and B). Within the 5 days of recording, infected mice experienced an average of 11.3 ± 5.3 (standard error of the mean [SEM]) of these seizure events (n = 8) (Fig. 4D). In contrast, mock-infected mice experienced an average of 0.5 ± 0.3 (SEM) of these events during the same recording period (n = 6), a difference that was statistically significant (P < 0.05 by Tukey-Kramer honestly significant difference [HSD] test) (Fig. 4D). EMG recordings further revealed examples in type II ME49 parasite-infected mice of generalized motor seizures that persisted for >15 min (Fig. 4C). These data indicate that, similarly to human toxoplasmosis patients, type II ME49 Toxoplasma-infected mice develop seizures.
FIG 4 .

Type II ME49 Toxoplasma infection leads to spontaneous seizures in mice. (A and B) Examples of EEG and EMG recordings from control (left column) and type II ME49-infected (right column) mice. Data represent 2 EEG traces and 1 EMG trace that were obtained simultaneously from each animal and are shown in a time-locked state. The recordings represented in panel A show 60 s of neural and muscular activity in control and type II ME49-infected mice. The recordings represented in panel B show 120 s of neural and muscular activity in control and type II ME49-infected mice. (C) EMG recordings from control (left column) and type II ME49-infected (right column) mice. Note that motor seizures persisted for as long as 15 min in this type II ME49-infected mouse. (D) Quantification of the number of seizure events that exceeded 30 s in the 5-day recording period. Bar graph data represent means ± SEM. Circles represent individual data points. P < 0.05 (Tukey-Kramer HSD test). (E) Individual and sparse spikes of neural and muscular activity were observed at similar frequencies in mock-infected and type II ME49-infected mice. (F) Quantification of the number of spiking events in the 5-day recording period. Bar graph data represent means ± SEM. Circles represent individual data points. The numbers of spikes seen with mock-infected mice were not statistically significantly different from those seen with type II ME49-infected mice (P > 0.1 [Tukey-Kramer HSD test]).
It has been widely speculated that interictal spikes of neural activity (rapid [<250 ms] single spikes of neural activity in EEG traces) may be predictive of seizure activity (32). For this reason, we also quantified the number of “spiking” events (defined as high-amplitude brain activity with <1 spike/s) in mock-infected and type II ME49 parasite-infected mice (33). In contrast to the significant impact of infection on seizure activity, no significant differences were seen in the numbers of “spiking” events between infected and mock-infected brains: an average of 18.5 ± 9.4 (SEM) spiking events were observed in type II ME49-infected brains during 5 days of recording versus 9.5 ± 4.6 spiking events in mock-infected controls (P > 0.1 [Tukey-Kramer HSD test]) (Fig. 4E and F).
Toxoplasma-infected mice develop more-severe seizures in response to GABA antagonists.
To assess the impact of altered GAD67 localization on epileptogenic activity in type II ME49 Toxoplasma-infected mice, we compared the levels of duration and intensity of seizures in mock-infected and parasite-infected mice treated with pentylenetetrazol (PTZ)—a GABA receptor antagonist that induces seizures (34, 35). Thirty days after mice were either mock infected or infected with type II ME49 parasite, a single injection of PTZ was administered, EMG traces were recorded, and visual seizure scores were noted every minute for 15 min. Results demonstrated that Toxoplasma infection increased seizure scores and also decreased the time needed for the mouse data to reach plateau scores (Fig. 5). Thus, the onset and magnitude of PTZ-induced seizures were increased in parasite-infected mice.
FIG 5 .
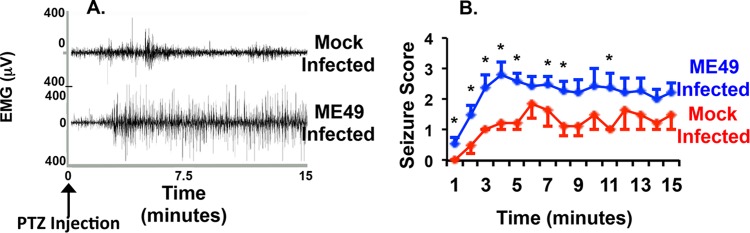
Increased susceptibility of type II ME49 Toxoplasma-infected mice to drug-induced seizures. (A) Five days after mice were analyzed for spontaneous seizures, they were injected with PTZ (40 mg/kg), and EEG/EMG data were recorded for 15 min. The data shown represent typical EMG traces from a mock-infected mouse (top) and a type II ME49 parasite-infected mouse (bottom). Notice the increased strength and intensity of muscular activity in the type II ME49 Toxoplasma-infected mouse. (B) Averages (± SEM) of seizure score data recorded every minute for 15 min after injection of PTZ. n = 5 mice for wild-type mice and 9 for type II ME49 mice. *, P < 0.05 (unpaired, two-tailed t test).
GAD67 relocalization between Toxoplasma strains is a polymorphic phenotype.
Four Toxoplasma haplotypes (I to III and X [or 12]) predominate in North America and Europe (15, 36–38). Of these, both type II and III strains are able to disseminate to the brain, establish chronic infections, and form tissue cysts (17, 36, 39). But genetic differences between these strains impact virulence, drug resistance, and specific host-pathogen interactions (40–45). Thus, we tested whether GAD67 localization was similarly affected by type II and III strains by infecting mice with either type II ME49 or CEP type III CEP strain parasites. We found that GAD67 staining in the hippocampi of type III CEP-infected mice closely resembled the punctate staining in uninfected animals (Fig. 6A to C). Binarized images of GAD67 immunoreactivity from mock-infected and type II ME49-infected or type III CEP-infected brains were quantified. We found that GAD67 immunoreactivity significantly increased (P < 0.0001 by analysis of variance [ANOVA] with Tukey’s post hoc test]) in the type II ME49-infected but not type III CEP-infected brains (Fig. 6D).
FIG 6 .

GAD67 mislocalization and drug-induced seizure are polymorphic responses in Toxoplasma-infected brains. (A to C) Representative images of coronal sections from mock-infected (A), type II ME49-infected (B), and type III CEP-infected (C) mice 30 days postinfection. Scale bars, 500 µm. Expanded images show high-magnification views of hippocampus CA3 regions. Scale bars, 50 µm. (D) Quantification of percent GAD67 staining following binarizing of images. *, P < 0.001 (one-way ANOVA with Tukey’s post hoc test; n ≥ 3 mice for each group). (E) Averages (± SEM) of seizure scores of mock-infected, type II ME49-infected, and type III CEP-infected mice (30 days postinfection) recorded every minute for 15 min after injection of PTZ. Each group represents score data for a minimum of 6 mice. *, statistically significant difference between type II ME49-infected and mock-infected mice only (P < 0.05 [one-way ANOVA with Tukey’s post hoc test]). #, statistically significant difference between type II ME49 and both mock-infected and type III CEP-infected mice (P < 0.05 [one-way ANOVA with Tukey’s post hoc test]).
To further test our hypothesis that GAD67 mislocalization is associated with the development of seizures in Toxoplasma-infected mice, we compared the responses of mock-infected, type II ME49-infected, and type III CEP-infected mice to PTZ. We found that, in contrast to the severe PTZ-induced seizures in the type II ME49-infected mice, the seizures in the type III CEP-infected mice were comparable to those in the mock-infected mice (Fig. 6E). These data therefore indicate that GAD67 mislocalization and seizure susceptibility represent a polymorphic phenotype following Toxoplasma infections and are not a general feature of the presence of Toxoplasma in the brain.
DISCUSSION
A key function for GABA in the nervous system is to regulate the timing, magnitude, and flow of excitatory neurotransmission, and defects in GABAergic signaling cause seizures (46). Decreasing synaptic release from inhibitory nerve terminals can reduce GABA signaling, and GAD localization to synaptic vesicles in presynaptic terminals leads to decreased GABA release since this ensures efficient GABA synthesis at synaptic termini (28, 31). Here, we show for the first time that a microbial infection of the CNS leads to mislocalization of GAD67. These data are significant because GAD67 is the GAD isoform responsible for the majority of GABA synthesis in the mammalian brain, whereas other GAD isoforms appear to have a more auxiliary role in GABA production and inhibitory neurotransmission (47–49). The anticipated outcome of such alterations would be increased spontaneous and drug-induced epileptogenic activity (50), and we showed that both occur in type II ME49 Toxoplasma-infected mice. Thus, these data provide an underlying mechanistic insight into the basis for seizures in toxoplasmic encephalitis patients (51, 52). These data also add to the number of mechanisms (including blood-brain-barrier breakdown, infiltration of immune cells and antibodies, and neuronal cell death) implicated in seizure formation in individuals suffering from intracerebral infections and inflammation (4).
GAD67 localizes to synaptic vesicles through 2 distinct mechanisms. First, it dimerizes via domains within its C-terminal half with GAD65, which is a second GAD that is tethered to synaptic vesicles (31). Second, and independently of GAD65 dimerization, GAD67 interacts with an unknown synaptic-vesicle-associated factor (28). Currently, we do not know which of these two mechanisms is affected by Toxoplasma. Type II ME49 Toxoplasma infection of cultured hippocampal neurons does not have a reproducible effect on GAD67 localization (J. M. Brooks and I. J. Blader, unpublished data), suggesting that GAD67 mislocalization is not a direct consequence of Toxoplasma-GABAergic neuron interactions. This conclusion is consistent with the finding that altered GAD67 mislocalization did not occur only in regions where either tachyzoites or bradyzoites are found. Toxoplasma can inject parasite-encoded effector proteins into neurons and other cells in the brain independently of infection (53). However, GAD67 mislocalization appears to be a more widespread phenomenon than parasite effector injection. Therefore, we hypothesize that alterations in GAD67 distribution and onset of seizures are likely controlled by a host-derived factor(s) whose expression is regulated by a polymorphic Toxoplasma gene product. Thus, unraveling the mechanism by which GAD67 is mislocalized will require identification of this polymorphic factor through either forward genetic quantitative trait locus (QTL) screening or reverse genetic candidate gene approaches.
Over the past decade, studies have revealed a connection between T. gondii and the onset of various neurological disorders (54). For example, a recent meta-analysis study demonstrated a link between chronic Toxoplasma infections and epilepsy (52), which is defined as an ongoing ability to spontaneously develop seizures. Other reports have also indicated that mice and humans chronically infected with Toxoplasma have altered behaviors such as reduced fear and inhibitions (55–57). It is tempting to speculate that the changes in GAD67 localization and GABAergic signaling reported here may underlie these long-term effects on the host’s nervous system. However, note that our studies were performed in mice infected for 30 days, and although these mice had large numbers of cysts in their brains, they also contained tachyzoites and activated microglia and astrocytes (Fig. 1D; see also Fig. S3 in the supplemental material), indicating that these mice had active toxoplasmic encephalitis. Thus, longer-term studies will be needed to assess how these early effects on GABAergic signaling alter a host’s behavior and neurological health.
MATERIALS AND METHODS
Animal infections.
All analyses conformed to National Institutes of Health guidelines and protocols approved by the University of Buffalo (no. MIC12093Y and MIC23035Y) and Virginia Polytechnic Institute and State University (no. 12-136 and 14-090) Institutional Animal Care and Use Committees. The Toxoplasma type II ME49 and type III CEP strains were routinely cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum as previously described (58). Tissue cysts were passed between C57BL/6 mice by intraperitoneal injection of 10 cysts. Experimental infections were performed by injecting 10 brain cysts into 0.2 ml of DMEM. All mice were purchased from Jackson Laboratories and used at between 6 and 12 weeks of age.
Immunohistochemistry.
Mice were exsanguinated and then perfused with 4% paraformaldehyde–phosphate-buffered saline (PBS). Brains were harvested, cryosectioned (16 µM), and stained as described previously (59, 60). Briefly, tissue slides were air dried for 15 min before being incubated with blocking buffer (5% normal goat serum–2.5% bovine serum albumin–0.1% Triton X-100–PBS) for 30 to 60 min. Primary antibodies were diluted in fresh blocking buffer and incubated on coronal brain sections overnight at 4°C. On the following day, slides were washed in PBS and secondary antibodies (diluted 1:1,000 in blocking buffer) were applied to slides for 1 h at room temperature. After being washed in PBS, tissue slides were coverslipped with Vectashield (Vector Laboratories, Burlingame, CA) and images were acquired on either a Zeiss Examiner Z1 LSM 510 or Zeiss LSM 700 confocal microscope. For comparisons of different experimental conditions, images were acquired with and processed using identical parameters. Whole-brain images were captured with a 10× objective using an automated motorized stage that imaged individual tiles without overlap. Composite images of each brain were generated by stitching together the individual images using Zeiss imaging software. A minimum of three animals (per experimental condition) were compared in all imaging experiments.
The following antibodies were used: rabbit anti-vGluT1 (1:500 dilution) and rabbit anti-vGluT2 (1:500 dilution) were from Synaptic Systems, Göttingen, Germany, rabbit anti-GFAP (1:1,000 dilution) was from Dako, Carpenteria, CA, rabbit anti-Iba1 (1:1,000 dilution) was from WAKO Chemicals USA, Inc., Richmond, VA, and mouse anti-GAD67 (1:500 dilution) was from Millipore, Temecula, CA. Neurotrace 640 deep red fluorescent Nissl stain was also used according to the protocol of the manufacturer (Life Technologies, Eugene, OR). Anti-Toxoplasma sera (1:250 dilution) that recognize both tachyzoites and bradyzoites were kindly provided by Tajie Harris. All conjugated secondary antibodies were purchased from Invitrogen (Carlsbad, CA) or Jackson ImmunoResearch (West Grove, PA).
Binary quantification.
The area within an image containing immunoreactivity was determined as previously described (61). Briefly, single channels of each image were converted to binary images using ImageJ software (see Fig. S1 in the supplemental material as an example). The percent area containing positive immunoreactivity in each image was then determined in ImageJ. A Student’s t test was used to test for statistical significance, and at least 9 images were collected from a minimum of three mock-infected and type II ME49-infected animals.
Western blotting.
Brains were harvested from decapitated mock-infected or parasite-infected mice, and coronal cortical sections (500 µm thick) were cut in ice-cold diethylpyrocarbonate (DEPC)-PBS and then flash frozen in liquid nitrogen. The tissue was then homogenized in radioimmunoprecipitation assay (RIPA) buffer supplemented with Protease Inhibitor Cocktail (Roche, Basel, Switzerland) as previously described (58). Lysates (30 µg/sample) were separated by SDS-PAGE and subjected to Western blotting with antibodies against GAD67 and β-actin (Cell Signaling, Inc., Danvers, MA) and anti-gephyrin antibodies (Synaptic Systems) diluted in Odyssey blocking buffer (Li-COR, Lincoln, NE) containing 0.1% Tween 20. Secondary antibodies were purchased from Li-COR. Images were captured using a Li-COR Odyssey Imaging system and processed using the manufacturer’s software.
RT-PCR.
RNA was prepared from coronal cortical sections using a Stratagene Absolutely RNA purification kit (La Jolla, CA) and converted to cDNA with Superscript III reverse transcriptase (RT; Invitrogen). Target gene expression levels were determined by RT-PCR using the threshold cycle (2−CTΔΔ) method as previously described (62). The following primer sets were used: for the β-actin gene, 5′-GACGGCCAGGTCATCACTATTG-3′ and 5′-CCACAGGATTCCATACCCAAGA-3′; for the gephyrin gene, 5′-ACCCTCGCCCAGAATACCAC-3′ and 5′-GCTCATCAGACGGCEPCTCA-3′; and for the GAD1 gene, 5′-CTTCCAGCCAGACAAGCAGTATGA-3′ and 5′-TGGGTTGGAGATGACCATCCGGAA-3′.
Spontaneous-seizure recordings.
Unless otherwise noted, all EEG/EMG hardware and software (including a 3-channel, tethered EEG/EMG system) were from Pinnacle Technologies (Lawrence, KS). Mock-infected or parasite-infected mice (30 days postinfection) were anesthetized with ketamine, the skull was exposed, and EEG/EMG head mounts were affixed to the skull with four recording electrode screws, ensuring that all 4 screws were implanted into the skull overlying the motor or sensory cortex in each cerebral hemisphere. EMG leads were implanted into nuchal muscles of the dorsal neck. The skull was then encased in a thick layer of dental acrylic (United States Dental Depot Inc., Ft. Lauderdale, FL) to prevent detachment of the head mount and EMG leads. Mice were allowed to recover for 3 to 5 days and were then tethered to the EEG/EMG recording chamber by connecting a 100× preamplifier (containing a 1.0-Hz high-pass EEG filter and a 10-Hz high-pass EMG filter) into the ports of the head mount. Mice remained tethered for 2 to 4 days before recording commenced, and EEG/EMG data were recorded for 5 days under normal light/dark conditions. Mice had free access to water, food pellets, and nestlets. EEG and EMG activity was recorded using Sirenia Acquisition software, and data acquisition was performed at a sampling rate of 10,000 Hz. Spontaneous seizures and spikes in EEG/EMG activity were identified and analyzed with Sirenia Seizure software. Generalized motor seizures were identified as consisting of high-amplitude, high-frequency neural and muscular activity that persisted for more than 30 s. A total of 8 type II ME49-infected mice and 6 mock-infected C57/B6 mice were analyzed in these studies.
PTZ-induced seizures.
Mice were injected with PTZ (40 mg/kg of body weight)–0.2 ml PBS and then visually monitored for 15 min while blind to their infection status. Seizures were scored manually every minute thereafter with the following scoring criteria: 0, normal activity; 1, reduced motility and prostate position; 2, partial clonus; 3, generalized clonus; 4, tonic-clonic seizure; 5, death. The PTZ dose was chosen since it reproducibly triggers high rates of mild seizures in C57/B6 mice (90% of wild-type mice develop mild seizures) but with a low (~10%) fatality rate (63).
SUPPLEMENTAL MATERIAL
Type II ME49 Toxoplasma alters GAD67 distribution in the dorsal lateral geniculate nucleus (dLGN). Retinal and inhibitory terminals were labeled in mock-infected and type II ME49-infected dLGN (A and B, respectively) with antibodies against VGluT2 and GAD67, respectively. As in the hippocampus, GAD67 appeared diffusely localized in type II ME49-infected dLGN, in contrast to its normal punctate appearance in controls. To quantify this phenotype, single-channel fluorescent images were subjected to binarizing in ImageJ (shown in black and white in panels A and B). (C) The area of binary signal was measured in ImageJ and revealed significant increases in the GAD67 immunoreactivity distribution in type II ME49-infected dLGN (P < 0.0001 by Student’s t test; n = 4 mice). No differences were observed in the area occupied by VGluT2 immunoreactivity. Scale bars in panels A and B, 20 µm. Download
{kind=link}
Type II ME49 Toxoplasma alters GAD67 distribution in the ventral horn of the thoracic spinal cord. (A) Excitatory and inhibitory terminals were labeled in mock-infected and type II ME49-infected spinal cord with antibodies against VGluT2 and GAD67, respectively. Examples of spinal cords from two type II ME49-infected mice are shown. As in the hippocampus and dLGN, GAD67 appeared diffusely localized in ME49-infected spinal cord, in contrast to its normal punctate appearance in the controls. (B) High-magnification images of the changes in GAD67 immunoreactivity in the ventral horn of mock-infected and 2 type II ME49-infected mice. Download
{kind=link}
GAD67 is not expressed in macrophages or astrocytes in Toxoplasma-infected brains. Hippocampal CA3 regions from mock-infected, type II ME49-infected, and type III CEP-infected mice 30 days postinfection were stained with anti-GAD67, anti-Iba1 (macrophages), or anti-GFAP (astrocytes). Note the lack of GAD67 expression in Iba1-positive (Iba1+) or GFAP+ cells. Download
ACKNOWLEDGMENTS
We thank Tajie Harris for providing the anti-Toxoplasma sera and Emma Wilson for helpful discussions and sharing of unpublished data.
Footnotes
Citation Brooks JM, Carrillo GL, Su J, Lindsay DS, Fox MA, Blader IJ. 2015. Toxoplasma gondii infections alter GABAergic synapses and signaling in the central nervous system. mBio 6(6):e01428-15. doi:10.1128/mBio.01428-15.
REFERENCES
- 1.Xu D, Miller SD, Koh S. 2013. Immune mechanisms in epileptogenesis. Front Cell Neurosci 7:195. doi: 10.3389/fncel.2013.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Annegers JF, Hauser WA, Coan SP, Rocca WA. 1998. A population-based study of seizures after traumatic brain injuries. N Engl J Med 338:20–24. doi: 10.1056/NEJM199801013380104. [DOI] [PubMed] [Google Scholar]
- 3.Avila EK, Graber J. 2010. Seizures and epilepsy in cancer patients. Curr Neurol Neurosci Rep 10:60–67. doi: 10.1007/s11910-009-0080-z. [DOI] [PubMed] [Google Scholar]
- 4.Marchi N, Angelov L, Masaryk T, Fazio V, Granata T, Hernandez N, Hallene K, Diglaw T, Franic L, Najm I, Janigro D. 2007. Seizure-promoting effect of blood-brain barrier disruption. Epilepsia 48:732–742. doi: 10.1111/j.1528-1167.2007.00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vezzani A, Maroso M, Balosso S, Sanchez M, Bartfai T. 2011. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun 25:1281–1289. doi: 10.1016/j.bbi.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 6.Pappas G, Roussos N, Falagas ME. 2009. Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. Int J Parasitol 39:1385–1394. doi: 10.1016/j.ijpara.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Weiss LM, Kim K. 2000. The development and biology of bradyzoites of Toxoplasma gondii. Front Biosci 5:D391–D405. doi: 10.2741/Weiss. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bierly AL, Shufesky WJ, Sukhumavasi W, Morelli AE, Denkers EY. 2008. Dendritic cells expressing plasmacytoid marker PDCA-1 are Trojan horses during Toxoplasma gondii infection. J Immunol 181:8485–8491. doi: 10.4049/jimmunol.181.12.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lambert H, Hitziger N, Dellacasa I, Svensson M, Barragan A. 2006. Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination. Cell Microbiol 8:1611–1623. doi: 10.1111/j.1462-5822.2006.00735.x. [DOI] [PubMed] [Google Scholar]
- 10.Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gatel D, Tardieux I. 2006. CD11c− and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood 107:309–316. doi: 10.1182/blood-2005-02-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coombes JL, Charsar BA, Han S-, Halkias J, Chan SW, Koshy AA, Striepen B, Robey EA. 2013. Motile invaded neutrophils in the small intestine of Toxoplasma gondii-infected mice reveal a potential mechanism for parasite spread. Proc Natl Acad Sci U S A 110:E1913–E1922. doi: 10.1073/pnas.1220272110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sturge CR, Yarovinsky F. 2014. Complex immune cell interplay in the gamma interferon response during Toxoplasma gondii infection. Infect Immun 82:3090–3097. doi: 10.1128/IAI.01722-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomita T, Bzik DJ, Ma YF, Fox BA, Markillie LM, Taylor RC, Kim K, Weiss LM. 2013. The Toxoplasma gondii cyst wall protein CST1 is critical for cyst wall integrity and promotes bradyzoite persistence. PLoS Pathog 9:e1003823. doi: 10.1371/journal.ppat.1003823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sibley LD, Ajioka JW. 2008. Population structure of Toxoplasma gondii: clonal expansion driven by infrequent recombination and selective sweeps. Annu Rev Microbiol 62:329–351. doi: 10.1146/annurev.micro.62.081307.162925. [DOI] [PubMed] [Google Scholar]
- 15.Dubey JP, Velmurugan GV, Rajendran C, Yabsley MJ, Thomas NJ, Beckmen KB, Sinnett D, Ruid D, Hart J, Fair PA, McFee WE, Shearn-Bochsler V, Kwok OCH, Ferreira LR, Choudhary S, Faria EB, Zhou H, Felix TA, Su C. 2011. Genetic characterisation of Toxoplasma gondii in wildlife from North America revealed widespread and high prevalence of the fourth clonal type. Int J Parasitol 41:1139–1147. doi: 10.1016/j.ijpara.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 16.Howe DK, Summers BC, Sibley LD. 1996. Acute virulence in mice is associated with markers on chromosome VIII in Toxoplasma gondii. Infect Immun 64:5193–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fux B, Nawas J, Khan A, Gill DB, Su C, Sibley LD. 2007. Toxoplasma gondii strains defective in oral transmission are also defective in developmental stage differentiation. Infect Immun 75:2580–2590. doi: 10.1128/IAI.00085-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Zhao M, Xu X, Liu X, Zhang H, Jiang Y, Zhang L, Hu X. 2014. Adoptive transfer of Treg cells counters adverse effects of Toxoplasma gondii infection on pregnancy. J Infect Dis 210:1435–1443. doi: 10.1093/infdis/jiu265. [DOI] [PubMed] [Google Scholar]
- 19.Luft BJ, Brooks RG, Conley FK, McCabe RE, Remington JS. 1984. Toxoplasmic encephalitis in patients with acquired immune deficiency syndrome. JAMA 252:913–917. [PubMed] [Google Scholar]
- 20.Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kuhn R, Muller W, Trinchieri G, Sher A. 1996. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J Immunol 157:798–805. [PubMed] [Google Scholar]
- 21.Grigg ME, Bonnefoy S, Hehl AB, Suzuki Y, Boothroyd JC. 2001. Success and virulence in Toxoplasma as the result of sexual recombination between two distinct ancestries. Science 294:161–165. doi: 10.1126/science.1061888. [DOI] [PubMed] [Google Scholar]
- 22.Witola WH, Mui E, Hargrave A, Liu S, Hypolite M, Montpetit A, Cavailles P, Bisanz C, Cesbron-Delauw M-, Fournie GJ, McLeod R. 2011. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect Immun 79:756–766. doi: 10.1128/IAI.00898-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan TG, Mui E, Cong H, Witola WH, Montpetit A, Muench SP, Sidney J, Alexander J, Sette A, Grigg ME, Maewal A, McLeod R. 2010. Identification of T. gondii epitopes, adjuvants, and host genetic factors that influence protection of mice and humans. Vaccine 28:3977–3989. doi: 10.1016/j.vaccine.2010.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabin AB, Feldman HA. 1949. Chorioretinopathy associated with other evidence of cerebral damage in childhood; a syndrome of unknown etiology separable from congenital toxoplasmosis. J Pediatr 35:296–309. doi: 10.1016/S0022-3476(49)80002-3. [DOI] [PubMed] [Google Scholar]
- 25.MacRae J, Sheiner L, Nahid A, Tonkin C, Striepen B, McConville M. 2012. Mitochondrial metabolism of glucose and glutamine is required for intracellular growth of Toxoplasma gondii. Cell Host Microbe 12:682–692. doi: 10.1016/j.chom.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuks JM, Arrighi RBG, Weidner JM, Kumar Mendu S, Jin Z, Wallin RPA, Rethi B, Birnir B, Barragan A. 2012. GABAergic signaling is linked to a hypermigratory phenotype in dendritic cells infected by Toxoplasma gondii. PLoS Pathog 8:e1003051. doi: 10.1371/journal.ppat.1003051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erlander MG, Tillakaratne NJK, Feldblum S, Patel N, Tobin AJ. 1991. Two genes encode distinct glutamate decarboxylases. Neuron 7:91–100. doi: 10.1016/0896-6273(91)90077-D. [DOI] [PubMed] [Google Scholar]
- 28.Kanaani J, Kolibachuk J, Martinez H, Baekkeskov S. 2010. Two distinct mechanisms target GAD67 to vesicular pathways and presynaptic clusters. J Cell Biol 190:911–925. doi: 10.1083/jcb.200912101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fox MA, Umemori H. 2006. Seeking long-term relationship: axon and target communicate to organize synaptic differentiation. J Neurochem 97:1215–1231. doi: 10.1111/j.1471-4159.2006.03834.x. [DOI] [PubMed] [Google Scholar]
- 30.Jin Y, Garner CC. 2008. Molecular mechanisms of presynaptic differentiation. Annu Rev Cell Dev Biol 24:237–262. doi: 10.1146/annurev.cellbio.23.090506.123417. [DOI] [PubMed] [Google Scholar]
- 31.Kanaani J, Diacovo MJ, El-Husseini Ael D, Bredt DS, Baekkeskov S. 2004. Palmitoylation controls trafficking of GAD65 from Golgi membranes to axon-specific endosomes and a Rab5a-dependent pathway to presynaptic clusters. J Cell Sci 117:2001–2013. doi: 10.1242/jcs.01030. [DOI] [PubMed] [Google Scholar]
- 32.Staley KJ, Dudek FE. 2006. Interictal spikes and epileptogenesis. Epilepsy Curr 6:199–202. doi: 10.1111/j.1535-7511.2006.00145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergstrom RA, Choi JH, Manduca A, Shin H, Worrell GA, Howe CL. 2013. Automated identification of multiple seizure-related and interictal epileptiform event types in the EEG of mice. Sci Rep 3:1483. doi: 10.1038/srep01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramanjaneyulu R, Ticku MK. 1984. Interactions of pentamethylenetetrazole and tetrazole analogues with the picrotoxinin site of the benzodiazepine-GABA receptor-ionophore complex. Eur J Pharmacol 98:337–345. doi: 10.1016/0014-2999(84)90282-6. [DOI] [PubMed] [Google Scholar]
- 35.Huang RQ, Bell-Horner CL, Dibas MI, Covey DF, Drewe JA, Dillon GH. 2001. Pentylenetetrazole-induced inhibition of recombinant gamma-aminobutyric acid type A (GABA(A)) receptors: mechanism and site of action. J Pharmacol Exp Ther 298:986–995. [PubMed] [Google Scholar]
- 36.Howe DK, Sibley LD. 1995. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J Infect Dis 172:1561–1566. doi: 10.1093/infdis/172.6.1561. [DOI] [PubMed] [Google Scholar]
- 37.Khan A, Dubey JP, Su C, Ajioka JW, Rosenthal BM, Sibley LD. 2011. Genetic analyses of atypical Toxoplasma gondii strains reveal a fourth clonal lineage in North America. Int J Parasitol 41:645–655. doi: 10.1016/j.ijpara.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller M, Miller W, Conrad P, James E, Melli A, Leutenegger C, Dabritz H, Packham A, Paradies D, Harris M, Ames J, Jessup DA, Worcester K, Grigg ME. 2008. Type X Toxoplasma gondii in a wild mussel and terrestrial carnivores from coastal California: new linkages between terrestrial mammals, runoff and toxoplasmosis of sea otters. Int J Parasitol 38:1319–1328. doi: 10.1016/j.ijpara.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 39.Sibley LD, Boothroyd JC. 1992. Virulent strains of Toxoplasma gondii comprise a single clonal lineage. Nature 359:82–85. doi: 10.1038/359082a0. [DOI] [PubMed] [Google Scholar]
- 40.Saeij JPJ, Boyle JP, Coller S, Taylor S, Sibley LD, Brooke-Powell ET, Ajioka JW, Boothroyd JC. 2006. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314:1780–1783. doi: 10.1126/science.1133690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saeij JPJ, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. 2007. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445:324–327. doi: 10.1038/nature05395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Behnke MS, Khan A, Sibley LD. 2015. Genetic mapping reveals that sinefungin resistance in Toxoplasma gondii is controlled by a putative amino acid transporter locus that can be used as a negative selectable marker. Eukaryot Cell 14:140–148. doi: 10.1128/EC.00229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Behnke MS, Khan A, Wootton JC, Dubey JP, Tang K, Sibley LD. 2011. Virulence differences in Toxoplasma mediated by amplification of a family of polymorphic pseudokinases. Proc Natl Acad Sci U S A 108:9631–9636. doi: 10.1073/pnas.1015338108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor S, Barragan A, Su C, Fux B, Fentress SJ, Tang K, Beatty WL, Hajj HE, Jerome M, Behnke MS, White M, Wootton JC, Sibley LD. 2006. A secreted serine–threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314:1776–1780. doi: 10.1126/science.1133643. [DOI] [PubMed] [Google Scholar]
- 45.Pernas L, Adomako-Ankomah Y, Shastri AJ, Ewald SE, Treeck M, Boyle JP, Boothroyd JC. 2014. Toxoplasma effector MAF1 mediates recruitment of host mitochondria and impacts the host response. PLoS Biol 12:e1001845. doi: 10.1371/journal.pbio.1001845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klausberger T, Somogyi P. 2008. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, Baekkeskov S. 1997. Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A 94:14060–14065. doi: 10.1073/pnas.94.25.14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. 1996. Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem Biophys Res Commun 229:891–895. doi: 10.1006/bbrc.1996.1898. [DOI] [PubMed] [Google Scholar]
- 49.Asada H, Kawamura Y, Maruyama K, Kume H, Ding R-, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. 1997. Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A 94:6496–6499. doi: 10.1073/pnas.94.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krook-Magnuson E, Armstrong C, Oijala M, Soltesz I. 2013. On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Commun 4:1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luft BJ, Remington JS. 1992. Toxoplasmic encephalitis in AIDS. Clin Infect Dis 15:211–222. doi: 10.1093/clinids/15.2.211. [DOI] [PubMed] [Google Scholar]
- 52.Ngoungou EB, Bhalla D, Nzoghe A, Darde ML, Preux PM. 2015. Toxoplasmosis and epilepsy—systematic review and meta analysis. PLoS Negl Trop Dis 9:e0003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koshy AA, Dietrich HK, Christian DA, Melehani JH, Shastri AJ, Hunter CA, Boothroyd JC. 2012. Toxoplasma co-opts host cells it does not invade. PLoS Pathog 8:e1002825. doi: 10.1371/journal.ppat.1002825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sutterland AL, Fond G, Kuin A, Koeter MWJ, Lutter R, van Gool T, Yolken R, Szoke A, Leboyer M, de Haan L. 2015. Beyond the association. Toxoplasma gondii in schizophrenia, bipolar disorder, and addiction: systematic review and meta-analysis. Acta Psychiatr Scand 132:161–179 doi: 10.1111/acps.12423. [DOI] [PubMed] [Google Scholar]
- 55.Ingram WM, Goodrich LM, Robey EA, Eisen MB. 2013. Mice infected with low-virulence strains of Toxoplasma gondii lose their innate aversion to cat urine, even after extensive parasite clearance. PLoS One 8:e75246. doi: 10.1371/journal.pone.0075246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vyas A, Kim S-, Giacomini N, Boothroyd JC, Sapolsky RM. 2007. Behavioral changes induced by Toxoplasma infection of rodents are highly specific to aversion of cat odors. Proc Natl Acad Sci U S A 104:6442–6447. doi: 10.1073/pnas.0608310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flegr J, Lenochova P, Hodny Z, Vondrova M. 2011. Fatal attraction phenomenon in humans: cat odour attractiveness increased for Toxoplasma-infected men while decreased for infected women. PLoS Negl Trop Dis 5:e1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wiley M, Sweeney KR, Chan DA, Brown KM, McMurtrey C, Howard EW, Giaccia AJ, Blader IJ. 2010. Toxoplasma gondii activates hypoxia inducible factor by stabilizing the HIF-1alpha subunit via type I activin like receptor kinase receptor signaling. J Biol Chem 285:26976–26986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hammer S, Carrillo GL, Govindaiah G, Monavarfeshani A, Bircher JS, Su J, Guido W, Fox MA. 2014. Nuclei-specific differences in nerve terminal distribution, morphology, and development in mouse visual thalamus. Neural Dev 9:16. doi: 10.1186/1749-8104-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Su J, Gorse K, Ramirez F, Fox MA. 2010. Collagen XIX is expressed by interneurons and contributes to the formation of hippocampal synapses. J Comp Neurol 518:229–253. doi: 10.1002/cne.22228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Singh R, Su J, Brooks J, Terauchi A, Umemori H, Fox MA. 2012. Fibroblast growth factor 22 contributes to the development of retinal nerve terminals in the dorsal lateral geniculate nucleus. Front Mol Neurosci 4:61. doi: 10.3389/fnmol.2011.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phelps ED, Sweeney KR, Blader IJ. 2008. Toxoplasma gondii rhoptry discharge correlates with activation of the EGR2 host cell transcription factor. Infect Immun 76:4703–4712. doi: 10.1128/IAI.01447-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takahashi H, Katayama K, Sohya K, Miyamoto H, Prasad T, Matsumoto Y, Ota M, Yasuda H, Tsumoto T, Aruga J, Craig AM. 2012. Selective control of inhibitory synapse development by Slitrk3-PTPdelta trans-synaptic interaction. Nat Neurosci 15:389–398. doi: 10.1038/nn.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Type II ME49 Toxoplasma alters GAD67 distribution in the dorsal lateral geniculate nucleus (dLGN). Retinal and inhibitory terminals were labeled in mock-infected and type II ME49-infected dLGN (A and B, respectively) with antibodies against VGluT2 and GAD67, respectively. As in the hippocampus, GAD67 appeared diffusely localized in type II ME49-infected dLGN, in contrast to its normal punctate appearance in controls. To quantify this phenotype, single-channel fluorescent images were subjected to binarizing in ImageJ (shown in black and white in panels A and B). (C) The area of binary signal was measured in ImageJ and revealed significant increases in the GAD67 immunoreactivity distribution in type II ME49-infected dLGN (P < 0.0001 by Student’s t test; n = 4 mice). No differences were observed in the area occupied by VGluT2 immunoreactivity. Scale bars in panels A and B, 20 µm. Download
Type II ME49 Toxoplasma alters GAD67 distribution in the ventral horn of the thoracic spinal cord. (A) Excitatory and inhibitory terminals were labeled in mock-infected and type II ME49-infected spinal cord with antibodies against VGluT2 and GAD67, respectively. Examples of spinal cords from two type II ME49-infected mice are shown. As in the hippocampus and dLGN, GAD67 appeared diffusely localized in ME49-infected spinal cord, in contrast to its normal punctate appearance in the controls. (B) High-magnification images of the changes in GAD67 immunoreactivity in the ventral horn of mock-infected and 2 type II ME49-infected mice. Download
GAD67 is not expressed in macrophages or astrocytes in Toxoplasma-infected brains. Hippocampal CA3 regions from mock-infected, type II ME49-infected, and type III CEP-infected mice 30 days postinfection were stained with anti-GAD67, anti-Iba1 (macrophages), or anti-GFAP (astrocytes). Note the lack of GAD67 expression in Iba1-positive (Iba1+) or GFAP+ cells. Download