

ABSTRACT
Methamphetamine (METH) is a major drug of abuse in the United States and worldwide. Furthermore, Staphylococcus aureus infections and METH use are coemerging public health problems. S. aureus is the single most important bacterial pathogen in infections among injection drug users, with skin and soft tissue infections (SSTI) being extremely common. Notably, the incidence of SSTI, especially in drug users, is difficult to estimate because such infections are often self-treated. Although there is substantial information on the behavioral and cognitive defects caused by METH in drug users, there is a dearth of knowledge regarding its impact on bacterial infections and immunity. Therefore, we hypothesized that METH exacerbates S. aureus skin infection. Using a murine model of METH administration and wound infection, we demonstrated that METH reduces wound healing and facilitates host-mediated collagen degradation by increased expression and production of matrix metalloproteinase-2 (MMP-2). Additionally, we found that METH induces S. aureus biofilm formation and leads to detrimental effects on the functions of human and murine phagocytic cells, enhancing susceptibility to S. aureus infection. Our findings provide empirical evidence of the adverse impact of METH use on the antimicrobial efficacy of the cells that comprise innate immunity, the initial host response to combat microbial infection.
IMPORTANCE
METH is an extremely addictive central nervous system stimulant that is frequently administered by injection. SSTI, common problems among injection drug users, result in serious morbidity for patients and costly hospitalizations for treatment of superficial wounds and incision and drainage of abscesses; however, there has been little etiologic or preventive epidemiological research on this problem. In addition, the evasive nature of injection drug users toward medical care complicates our ability to accurately predict the prevalence of these infections. Hence, this study investigated the impact of METH use on S. aureus skin infection. Our findings demonstrate that this drug of abuse promotes biofilm formation and negatively impacts the wound healing process and innate immune function, exacerbating susceptibility to S. aureus infection. The findings may translate into new knowledge and development of therapeutic and public health strategies to deal with the devastating complications of METH abuse.
INTRODUCTION
Staphylococcus aureus is an immotile Gram-positive coccus that frequently colonizes human nasal membranes and skin. It is responsible for the majority of superficial and invasive skin infections, resulting in over 12,000,000 outpatient/emergency room (ER) visits (1) and 400,000 hospital admissions annually in the United States (2). Notably, in a study performed with multiple ERs across the United States, methicillin-resistant S. aureus (MRSA) strains were isolated from 61% of abscesses and 53% of purulent wounds (3). Also, certain S. aureus clinical strains have recently evolved with resistance to vancomycin, an antibiotic to which staphylococci had previously been uniformly susceptible. Although the vancomycin-resistant strains remain rare, MRSA infections are increasingly common (4), and the incidence of community-acquired MRSA strains has increased severalfold over the past several years (5).
Methamphetamine (METH) is an extremely addictive central nervous system stimulant abused by individuals worldwide, and the drug is a major threat in many developed countries. The intoxicating effects of METH alter judgment and reduce inhibitions, leading people to engage in unsafe activities that put them at risk for acquiring transmissible microbes (6, 7). S. aureus, including community-acquired MRSA, is the most important bacterial pathogen in skin and soft tissue infections (SSTI) among drug users (8–10). SSTI incidence is difficult to assess among drug users because such infections are often self-treated (11). However, crystal METH injection is associated with frequent visits to the ER due to abscesses, cellulitis, and other skin infections (12). Additionally, MRSA, particularly USA300 strains, causes most SSTI in METH users (13).
METH is associated with several socioeconomic and behavioral risk factors that may predispose individuals to MRSA SSTI (11). Notably, even noninjection use is associated with MRSA SSTI, as smoking METH is an independent risk factor for MRSA, including primary skin abscesses and invasive infections (13, 14). In addition, skin picking is associated with MRSA SSTI. METH use causes formication, a sensation of something crawling on the body or under the skin, which can lead to skin-picking behavior and skin breakdown (10, 15). Furthermore, METH injection is also associated with the introduction of bacteria into the skin, especially if the user fails to clean injection sites or shares drug paraphernalia (16).
The effects of METH on host responses have not been extensively described. However, limited studies about the effects of METH on immune function show that its abuse has profound implications for host immunity. The injection of 25 mg/kg of body weight of METH into rats induces apoptotic death in thymic and splenic lymphocytes (17). METH also reduces thymic and splenic cellularity and alters peripheral T lymphocyte populations in treated mice (18). METH exposure results in mitochondrial oxidative damage and causes dysfunction of primary human T cells (19). Also, METH is an immunosuppressive agent; it alkalizes normally acidic organelles within immune cells, inhibits antigen presentation, and impairs phagocytosis (20). Moreover, METH negatively alters antibody and cytokine production (21).
Although there is a clear clinical association of METH use and MRSA disease, there has not been a defined biological link between the immune response and wound healing capacity of a METH user with an increased susceptibility to S. aureus. However, animal studies demonstrate that METH suppresses both innate and adaptive immunity (22, 23) and alters immune cell gene expression (24). Here, we investigated whether METH facilitates MRSA skin infections and provide evidence that the drug has negative effects on human-derived phagocytic cells.
RESULTS
METH alters wound healing.
METH administration decreased the wound healing rate significantly compared with results in untreated animals (Fig. 1A). Moreover, METH administration and MRSA infection (METH-MRSA) induced visible inflammation and decreased wound healing compared with all other conditions (Fig. 1A). In fact, the wounds in infected or METH-treated mice demonstrated a significant increase in size relative to the initial wound, which did not occur in the untreated, uninfected animals. At day 3, eschars in untreated wounds were ~4.4 mm in diameter, whereas eschars of animals treated with METH alone or untreated MRSA-infected or METH-MRSA wounds were ~7 mm (P < 0.001, compared with untreated). At day 7 (Fig. 1B), eschars in the untreated group were ~2.4 mm, whereas the eschars of wounds in the MRSA, METH, or METH-MRSA treatment groups were ~3.9 mm (P < 0.05), ~6.5 mm (P < 0.001), and ~6.9 mm (P < 0.001), respectively. At day 13 (Fig. 1B), the wounds of untreated mice showed complete closure, whereas eschars of MRSA, METH, or METH-MRSA wounds were ~1.7 mm (P < 0.01), ~2.1 mm (P < 0.01), and ~4.9 mm (P < 0.001), respectively. Complete wound closure was reached by day 17 for MRSA- or METH-treated mice, whereas complete wound healing in the METH-MRSA group took 19 days. Mice treated with METH or MRSA alone had similar resolution of wounds during days 9 to 17, although METH-exposed mice had slower healing at day 7.
FIG 1 .

METH decreases wound healing in mice infected with MRSA. (A) Wounds of BALB/c mice (n = 5 per group) uninfected and untreated (untreated), infected with MRSA, treated with methamphetamine (METH), or treated with METH and infected with MRSA (METH + MRSA), at days 3, 5, 7, 9, 11, 13, 15, and 17. Bar, 5 mm. (B) Wound size analysis of BALB/c mouse skin lesions. Wounds were uninfected and untreated, infected with MRSA, treated with METH, or treated with METH and infected with MRSA. Symbols are the averages of the results for six measurements at each time interval, and error bars denote standard deviations. P values of <0.05 were considered significant, calculated by ANOVA. ϕ, χ, and π indicate significantly higher eschar sizes in METH + MRSA mice than sizes for untreated, MRSA-infected, or METH-treated groups, respectively. This experiment was performed twice and similar results were obtained. The results shown are representative of an individual experiment.
METH enhances MRSA burden.
Qualitative histological examinations revealed that wounds of untreated, uninfected mice and of METH-treated uninfected mice had less inflammation along with increased fibrin deposition than MRSA-infected tissues, and no evidence of bacteria (Fig. 2A). MRSA-infected mice showed localized epidermal inflammation (Fig. 2A). However, wounds of METH-treated MRSA-infected mice had intense inflammatory infiltrates in both the epidermal and dermal layers, along with extensive cell necrosis (Fig. 2A). Tissue Gram stains of MRSA-infected and of METH-treated MRSA-infected samples displayed large numbers of Gram-positive cocci (Fig. 2A, insets), with larger bacterial clusters in biofilm-like arrangements evident in the sections from METH-treated animals.
FIG 2 .
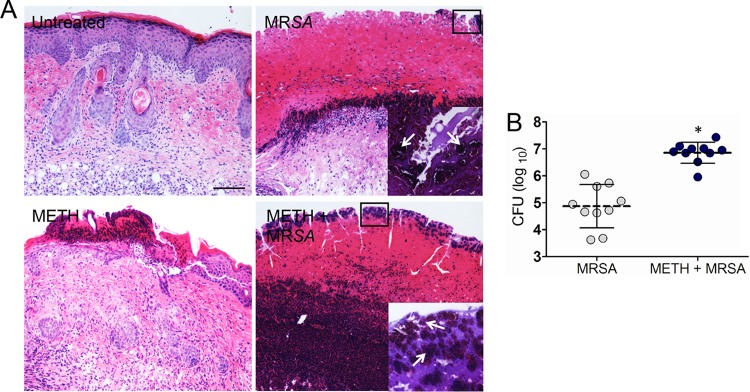
METH enhances MRSA burden in superficial skin lesions. (A) Histological analysis of BALB/c mice uninfected and untreated (untreated), infected with MRSA (MRSA), treated with METH (METH), or treated with METH and infected with MRSA (METH + MRSA), at day 7. Mice were infected with 107 MRSA bacterial cells. Representative H&E-stained sections of the skin lesions are shown. Bars, 25 µm. The insets depict Gram stain results for the boxed areas, and the panels reveal MRSA cells (shown in purple). The arrows in these insets indicate bacterial clusters in biofilm-like arrangements. (B) Wound bacterial burdens (CFU) in METH-treated mice infected with 107 MRSA cells were significantly higher than those in MRSA-infected, untreated mice at 7 days after infection (n = 10 per group). Each symbol represents one animal; dashed bars are the averages for each group, and error bars denote standard deviations. Asterisks denote P values of <0.05, calculated by using Student’s t test. These experiments were performed twice, and similar results were obtained. The results shown are representative of an individual experiment.
MRSA-infected mice treated with METH had significantly higher, by 2 logs, microbial burdens than untreated MRSA-infected mice at day 7 postinfection (P < 0.05) (Fig. 2B). Less than 30 bacterial CFU were detected in the skin of uninfected mice, and MRSA was not identified.
METH impairs wound healing by mediating host matrix metalloproteinase-2 collagen degradation.
The mechanisms through which METH impairs wound healing were explored by examining whether METH decreased collagen deposition in wound tissue (Fig. 3). Collagen content, in both the epidermal and dermal layers, was lowest in METH-treated MRSA-infected wounds (Fig. 3A). The lack of blue stain indicated lower tissue collagen formation in METH-treated MRSA-infected wounds, suggesting that METH administration augmented MRSA dermal collagen degradation. Figure 3B presents the results of a morphometric analysis of the data shown in Fig. 3A.
FIG 3 .

METH impairs wound healing by enhancing the host’s MMP-2-mediated collagen degradation. (A) Histological analysis of untreated, MRSA, METH, or METH + MRSA treatment groups of BALB/c mice at day 7 after wounding. The blue stain indicates collagen. Bar, 25 µm. (B) Quantitative measurement of collagen intensity in tissues from untreated, MRSA, METH, and METH + MRSA mice were measured using ImageJ software. Bars are the averages of the results, and error bars denote standard deviations. *, #, and & indicate significantly lower collagen intensites than in untreated, MRSA-infected, or METH-treated groups, respectively. (C) Gene expression analysis of collagen type I and III in cutaneous lesions. ϕ, χ, and π indicate significantly higher fold changes than in untreated, MRSA-infected, or METH-treated groups, respectively. (D) Collagen degradation by MRSA staphopain A. FITC-conjugated type I collagen was incubated with 10 µM staphopain A and METH (25 or 50 µM) for 3 h at 37°C. The fluorescence of the supernatant was measured. Heat-inactivated staphopain A was utilized as a control. Dashed lines represent the averages for three measurements per condition, and error bars denote standard deviations. *, #, and & indicate significantly lower relative fluorescence than in the untreated or 25 µM METH groups after cells were incubated with active staphopain A, respectively. (E) Gene expression analysis of MMP-2 in murine wounds. ϕ, χ, and π indicate significantly higher fold changes than in untreated, MRSA-infected, or METH-treated groups, respectively. (F) Collagen degradation by MMP-2. FITC-conjugated type I collagen was incubated with 10 µM MMP-2 and METH (25 or 50 µM) for 3 h at 37°C. ϕ and π indicate significantly higher relative fluorescence values than in the untreated or 25 µM METH groups, respectively. *, #, and & indicate significantly lower relative fluorescence than in cells from untreated or the 25 or 50 µM METH groups incubated with active MMP-2, respectively. (G) Western blot analysis of MMP-2 in wounded tissue (7 days) of untreated, MRSA, METH, or METH + MRSA treatment in BALB/c mice. GAPDH was used as a loading control. For panels B to F, P values (with significance defined as P < 0.05) were calculated by using an ANOVA. All the experiments in the figure were performed twice, and similar results were obtained. The results shown are representative of an individual experiment.
To determine whether METH affects collagen deposition during wound healing, we assessed mRNA expression of collagen type I and III in murine wound tissue (Fig. 3C). METH-treated MRSA-infected samples had significantly increased mRNA collagen type I and III expression, ~18-fold higher, compared to untreated, uninfected control tissues (P < 0.001). Notably, tissues from MRSA-infected mice had an 8-fold increase in collagen type III expression compared to tissues from untreated, uninfected animals (P < 0.001) (Fig. 3C), but there was no difference in collagen I expression.
S. aureus staphopains are cysteine proteases involved in tissue destruction and collagen degradation. Therefore, we examined whether METH enhances collagen degradation by staphopain A (Fig. 3D). Surprisingly, increased concentrations of METH significantly reduced the degradation of collagen by staphopain A (P < 0.05). Similarly, untreated and METH-treated heat-inactivated staphopain A showed lower collagen degradation than active protease.
To define the host’s contribution in collagen degradation after METH administration and MRSA infection, we analyzed the expression of matrix metalloproteinase-2 (MMP-2) in mouse wounded tissue (Fig. 3E). METH-treated or METH-treated, MRSA-infected tissues had significantly increased mRNA MMP-2 expression levels, by ~4- and 6-fold, respectively, above those in untreated, uninfected control tissues (P < 0.001). Then, we investigated whether METH increased collagen degradation by MMP-2 (Fig. 3F). METH exacerbated collagen degradation by MMP-2 relative to that in untreated tissue (25 µM [P < 0.05]; 50 µM [P < 0.001]). No differences were observed between untreated tissues and those in the METH treatment group that received heat-inactivated MMP-2. Western blot analysis was performed to confirm the upregulation of MMP-2 protein levels in METH-treated tissues (Fig. 3G). We found that METH increased the expression of MMP-2 in both uninfected and MRSA-infected wounds. Together, these results suggest that wound healing in METH-treated animals is reduced, at least in part, by an increase in the host’s MMP-2 levels.
METH decreases the number of phagocytic cells in the blood of treated BALB/c mice.
We examined whether METH administration depleted phagocytes in the blood of BALB/c mice by using differential leukocyte staining. Light microscopy images showed reduced numbers of phagocytic cells in the blood of METH-treated mice compared with numbers in untreated mice (Fig. 4A). Cell count analysis showed that METH-treated animals had significantly lower numbers of circulating phagocytes in blood compared to numbers in controls (P < 0.05) (Fig. 4B).
FIG 4 .

METH-treated animals displayed fewer phagocytes in blood. (A) Light microscopy images of blood smears from untreated or METH-treated mice preinfection. Arrows indicate phagocytic cells (purple). Bar, 20 µm. (B) Numbers of phagocytic cells per field in blood of untreated or METH-treated mice. Each black circle or red square represents the number of neutrophils per individual field. Dashed lines and error bars denote averages and standard deviations of 40 counts, respectively. Asterisks denote significant P values (P < 0.05), calculated using Student’s t test. The experiment was performed twice with similar results obtained. The results shown are a combination of two independent experiments.
METH does not inhibit S. aureus growth.
MRSA growth with and without exposure to METH was determined in real time for 24 h (Fig. 5). METH did not alter bacterial growth during a 24-h coincubation with 25 or 50 µM METH compared with untreated S. aureus controls.
FIG 5 .
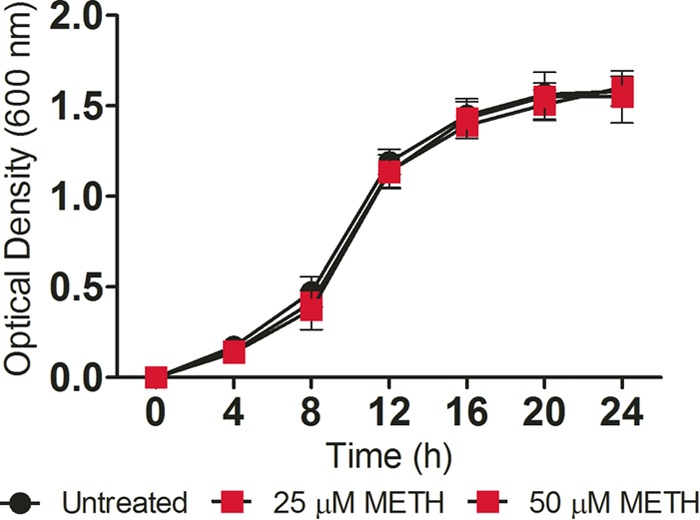
METH does not affect S. aureus viability in vitro. The effect of METH on S. aureus growth kinetics was determined via Bioscreen C analysis. S. aureus was grown in the absence (untreated) or presence of METH (25 or 50 µM). Each point represents the average of three measurements. The experiment was performed twice with similar results obtained. The results shown are representative of an individual experiment.
METH enhances S. aureus biofilm formation.
We evaluated the effect of METH on biofilm formation by six clinical S. aureus isolates to confirm the results obtained in the murine model. S. aureus biofilms on 96-well plates were incubated with METH (25 or 50 µM) or phosphate-buffered saline (PBS) for 24 h. Cell viability and metabolic activity were determined by CFU and 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazolium hydroxide (XTT) reduction assays, respectively (Fig. 6A and B). On average, METH promoted S. aureus biofilm formation compared to untreated controls. S. aureus biofilms incubated with METH displayed higher CFU counts (P < 0.0001) (Fig. 6A), and cells within biofilms were more metabolically active (P < 0.0001) (Fig. 6B) than untreated controls.
FIG 6 .

METH promotes biofilm formation by S. aureus clinical isolates (n = 6), based on CFU counts (A) and XTT reduction (B) after 24 h. Dashed lines and error bars represent the averages and standard deviations of six measurements (each symbol represents a single strain) from three plates per well per strain. (C) Confocal microscopy of MRSA 6498 biofilms after treatment with METH. Representative three-dimensional images of biofilms show viable (green [SYTO 9]) and dead (red [propidium iodide]) cells. The thickness and morphology of the bacterial biofilms can be observed in the Z-stack reconstruction. The pictures were taken at a magnification of ×63. Bar, 50 µm. (D) Expression analysis of the ica gene in S. aureus strains after incubation with METH. For panels A, B, and D, significance (P < 0.05) was calculated by ANOVA. ϕ indicates significantly higher fold change than untreated group. These experiments were performed twice with similar results obtained. The results shown are representative of an individual experiment.
Confocal microscopic examination was used to correlate the decreases in CFU and the XTT reduction assay results with the visual effects on biofilm structure (Fig. 6C). In the figure, regions of green fluorescence represent viable cells; the red fluorescence indicates metabolically inactive or nonviable cells. Untreated MRSA strain 6498 biofilms were ~21 µm thick, whereas biofilms grown in the presence of 25 µM METH showed a more robust architecture with a thickness of ~71 µm (Fig. 6C). The METH-treated biofilm metabolic activity observed corresponded with the XTT results.
The S. aureus gene ica encodes an intercellular adhesion protein, and disruption of this gene leads to decreased biofilm formation (25). All isolates in the present study were found to carry the ica gene (data not shown). Hence, we investigated the impact of METH on the expression of the ica gene by biofilm-associated cells via quantitative reverse transcription-PCR (qRT-PCR) (Fig. 6D). S. aureus biofilm-associated cells exhibited significantly increased ica expression after exposure to METH (25 µM [P < 0.0001] and 50 µM [P < 0.0001]).
METH affects murine neutrophil functions.
We investigated the effect of METH on murine neutrophil effector functions in MRSA infection. METH significantly inhibited neutrophil migration (25 µM, P < 0.05; 50 µM, P < 0.01) in a concentration-dependent manner compared to untreated or chloroquine (Chlq)-treated cells (Fig. 7A). As expected, cytochalasin D (CytD)-treated cells (25 µM) displayed a significant reduction in migration (P < 0.01) compared to untreated cells.
FIG 7 .

METH modifies neutrophil effector functions. (A) Percentages of neutrophils (polymorphonuclear lymphocytes [PMNs]) migrating at various time points (20, 40, and 60 min) after exposure of uninfected cells to the chemoattractant fMLP. Neutrophils were isolated from BALB/c mice and treated with PBS (untreated), Chlq (25 µM), CytD (25 µM), or METH (25 or 50 µM) and coincubated with MRSA. (B and C) METH interferes with neutrophil-mediated killing (NMK) (B) and phagocytosis of MRSA cells (C). For panel C, fluorescence intensity (in arbitrary units [A.U.]) refers to blue-labeled bacteria inside neutrophils after their phagocytosis. (D) Confocal microscopy of untreated or CytD- or METH-treated neutrophils interacting with MRSA. Alexa Fluor 350 (blue) was used to label bacterial cells. Actin-specific (red) staining was used to label cell bodies of neutrophils. 4′,6-Diamidino-2-phenylindole (blue) was used to label the nuclei. Bar, 10 µm. (E) NO production in infected neutrophils was quantified using the Griess method. (F) MPO released by neutrophils was measured using the EnzChek MPO activity assay. For panels A to C and E to F, the values presented are averages and standard deviations. All experiments were performed three times with similar results obtained. Each point or bar represents the average of three measurements per condition. The results shown are representative of an individual experiment. Significance (P < 0.05) was calculated by an ANOVA. *, #, and & indicate results significantly lower than in untreated, Chlq, or 25 µM METH-treated groups, respectively.
We examined whether METH interfered with neutrophil-mediated killing (NMK) of MRSA strain 6498. When untreated neutrophils and MRSA 6498 were coincubated, the majority of bacteria were killed within 80 min (Fig. 7B). Similarly, Chlq-treated neutrophils showed an increase in bacterial killing over time, albeit to a lesser extent than untreated neutrophils. In contrast, METH more significantly impaired bacterial killing, in a concentration-dependent manner (P < 0.05, 60 and 80 min) by stationary neutrophils, and likewise, CytD interfered with phagocytic killing (Fig. 7B).
Confocal microscopic examination was used to correlate the killing assay results with the visual effects on the impact of METH on neutrophil morphology (red fluorescence [actin]) and phagocytosis of MRSA (blue fluorescence [Alexa-labeled S. aureus]) (Fig. 7C). METH-treated neutrophils (25 and 50 µM; P < 0.05) and CytD-treated neutrophils (25 µM; P < 0.05) significantly reduced phagocytosis of MRSA, compared to that of untreated or Chlq-treated cells 30 min after coincubation (Fig. 7C). Untreated neutrophils displayed multiple protrusions and actin polymerization, engaged in phagocytic and killing interactions with S. aureus (Fig. 7D), and contained an increased number of internalized bacteria. In contrast, METH- or CytD-treated neutrophils displayed a large rounded morphology, fewer protrusions, and reduced interactions with bacteria, and the cells were surrounded by multiple bacterial cells (Fig. 7D).
Neutrophils treated with METH (50 µM; P < 0.001) or CytD (25 µM; P < 0.001) showed significantly lower levels of NO production after MRSA infection than did untreated neutrophils (Fig. 7E). Both METH (25 µM) and Chlq (25 µM) showed a trend toward a reduction in NO production. Similarly, the release of myeloperoxidase (MPO) by neutrophils was significantly decreased by METH (25 µM; P < 0.01; 50 µM, P < 0.001) or CytD (25 µM; P < 0.001) (Fig. 7F).
METH impairs the effector functions of human neutrophils.
We analyzed the effects of achievable physiological METH doses on phagocytosis and killing of six clinical S. aureus isolates by human neutrophils via fluorescence-activated cell sorting (FACS) analysis. METH significantly reduced phagocytosis of S. aureus by neutrophils, compared with the untreated or Chlq‐treated (25 µM Chlq) cells (25 µM METH, P < 0.01; 50 µM METH, P < 0.01; compared to untreated and Chlq groups) (Fig. 8A). Our results showed 52.5% and 46% S. aureus phagocytosis by cells treated with METH (25 or 50 µM) compared to untreated (77%) or 25 µM Chlq-treated cells (75.5%). As expected, CytD-treated cells displayed a significant reduction in phagocytosis compared to cells in all the other groups, with the exception of 50 µM METH (P < 0.001).
FIG 8 .

METH reduces human neutrophil phagocytosis, respiratory burst, and killing of S. aureus. (A) Phagocytosis (percentage) of FITC-labeled S. aureus clinical strains (n = 6) by human neutrophils was determined using FACS analysis after a 60-min incubation with METH. (B) Killing of S. aureus by neutrophils was determined from CFU counts. For panels A and B, dashed lines and error bars represent the averages and standard deviation of six measurements (each symbol represents a single strain). Significance (P < 0.05) was calculated by an ANOVA and adjusted by use of the Bonferroni correction. For panel A, *, #, and & indicate a lower phagocytosis percentage than in untreated, 25 µM Chlq, or 25 µM METH groups, respectively. For panel B, ϕ, χ, @, and $ indicate a higher survival percentage than in untreated, 25 µM Chlq, 25 µM METH, and 50 µM METH groups, respectively. (C) Oxidative burst was quantified for 60 min based on luminol chemiluminescence after untreated or Chlq-, CytD-, or METH-treated neutrophils were coincubated with MRSA strain 6498. Untreated and uninfected neutrophils were also used as controls. Symbols and error bars denote means and standard deviations. Significance (P < 0.05) was calculated by an ANOVA and adjusted by use of the Bonferroni correction at each time point. This experiment was performed twice and similar results were obtained. The results shown are representative of an individual experiment.
We again also examined whether METH interfered with neutrophil-mediated killing of S. aureus cells. CytD and METH significantly reduced bacterial killing by human neutrophils (P < 0.0001) compared to untreated cells (Fig. 8B). These phagocytic cells mostly kill bacteria via NADPH oxidase-derived reactive oxygen species (ROS). As a result, we evaluated the impact of METH on the neutrophil oxidative burst by measuring luminol chemiluminescence intensity. METH significantly decreased ROS production compared to untreated or Chlq-treated, S. aureus-exposed neutrophils (Fig. 8C) (P < 0.05; 25 to 60 min). Minimal production of ROS was observed in untreated and unstimulated controls or CytD-treated cells (P < 0.05; 30 to 60 min).
METH alters macrophage function.
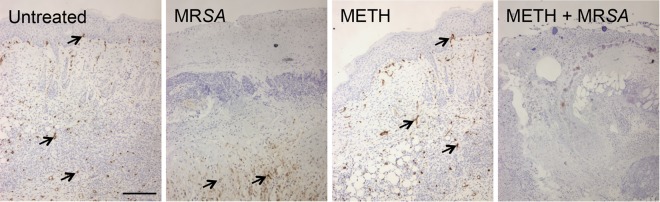
We identified macrophage-like infiltration in wound tissue by measuring the expression of Iba-1, which is mostly expressed and upregulated during the activation of these cells. Tissue sections from murine wounds in the METH-treated (uninfected) group showed only scattered macrophage-like cells, whereas untreated or MRSA-infected mice displayed massive macrophage-like cell infiltrations throughout the wound area (Fig. 9A). Additionally, METH-treated MRSA-infected wounds demonstrated minimal macrophage-like cell infiltration.
FIG 9 .

METH alters macrophage effector functions. (A) Histological analysis of untreated, MRSA, METH, and METH-MRSA wounded BALB/c mice at day 7. The brown staining indicates macrophage infiltration. Representative Iba1-immunostained sections of the skin lesions are shown. Bars, 25 µm. (B to F) Primary macrophages were isolated from BALB/c mice and treated with PBS (untreated), Chlq (25 µM), CytD (25 µM), or METH (25 or 50 µM) and coincubated with MRSA (C to F). (B) Uninfected macrophage migration (as a percentage) after exposure to the chemoattractant fMLP. Significance (P < 0.05) was calculated by an ANOVA. *, #, and & indicate a significantly lower percentage of macrophages than in untreated or Chlq- or 25 µM METH-treated groups, respectively. (C) Phagocytosis (percentage of cells) was analyzed by FACS analysis after a 30-min incubation. (D) CFU determinations were performed after the 30-min incubation. (E) NO production was quantified using the Griess method. Significance (P < 0.05) was calculated by an ANOVA. Asterisks indicate results significantly lower than in untreated groups. (F) The pH levels in phagosomes of primary macrophages that contained MRSA labeled with pH-sensitive and pH-insensitive probes were measured using a spectrofluorimeter. For panels B to F, the values presented are averages and standard deviations. For panels C, D, and F, Significance (P < 0.05) was calculated by an ANOVA. ϕ, χ, π, and λ indicate a significant increase compared to results in untreated or Chlq-, CytD-, or 25 µM METH-treated groups, respectively. All the experiments were performed twice with similar results obtained. Each bar represents the average of three measurements per condition. The results shown are representative of an individual experiment.
METH (25 µM, P < 0.05; 50 µM, P < 0.01) significantly inhibited macrophage migration in a concentration-dependent manner compared to untreated or Chlq-treated cells (Fig. 9B). CytD-treated cells (25 µM; P < 0.01) displayed a significant reduction compared to untreated cells.
METH (25 and 50 µM) significantly stimulated phagocytosis of bacterial cells by murine macrophages, compared with the untreated, Chlq‐treated (25 µM), or CytD-treated (25 µM) cells (P < 0.01 for 25 µM METH; P < 0.001 for 50 µM METH) (Fig. 9C). Furthermore, METH decreased the eradication of bacteria within macrophages (Fig. 9D).
After phagocytosis, macrophages produce NO in response to pathogens within the phagosome. Macrophages treated with METH (50 µM; P < 0.001) or CytD (25 µM; P < 0.001) showed significantly lower levels of NO production after infection with MRSA strain 6498 than did untreated macrophages (Fig. 9E). Both the METH (25 µM) and Chlq (25 µM) treatment groups showed a trend toward a decrease in NO production.
Phagosome acidification was studied to elucidate the mechanism by which METH interferes with MRSA 6498 killing after phagocytosis. The acidification of phagosomes of primary macrophages containing MRSA labeled with pH-sensitive and pH-insensitive probes was measured using a spectrofluorimeter. Standard curves were generated with fluorophores. METH induced the alkalization of macrophage phagosomes that contained MRSA 6498 cells. The pH of phagosomes in macrophages infected with the bacteria for 2 h was analyzed. In macrophages treated with METH (25 or 50 µM) or Chlq (25 µM), the phagosomal pH was significantly elevated (pH 5.6, 6.4, and 6.8, respectively) compared with the pH in untreated control or CytD-treated (25 µM) macrophages (pH 4.8 and 4.4; P < 0.001) (Fig. 9F).
METH-treated MRSA-infected mice displayed decrease angiogenesis.
We investigated the effect of METH on angiogenesis in wounded skin infected with MRSA by measuring the expression of CD34 (Fig. 10). Tissue sections from untreated or METH-treated uninfected murine wounds displayed dense vascularization, whereas wounds of MRSA-infected mice showed localized formation of blood vessels in dermal tissue. Moreover, METH-treated MRSA-infected wounds demonstrated minimal vessel formation in the epidermis and dermis. Therefore, the combination of METH and MRSA infection visually reduces vascularization in murine cutaneous wounds, and this qualitative observation is associated with decreased healing.
FIG 10 .
METH-treated MRSA-infected mice displayed decreased angiogenesis in wounded skin. Tissue sections are shown for untreated, MRSA, METH, or METH-MRSA wounded BALB/c mice at day 7. Brown staining indicates vascularization. Representative CD34-immunostained sections of the skin lesions are shown. Black arrows denote dense vascularization. Bars, 25 µm.
METH alters cytokine expression.
Twenty-four hours after infection, wound tissue of mice infected with MRSA contained significantly higher quantities of gamma interferon (IFN-γ), interleukin-1β (IL-1β), IL-12, monocyte chemoattractant protein 1 (MCP-1), and tumor necrosis factor alpha (TNF-α) than those under all other conditions (Table 1). Transforming growth factor β (TGF-β) was significantly elevated in MRSA-infected mice (MRSA and METH-MRSA) groups. There was a significant increase in IL-6 and IL-10 production in METH-treated animals (METH and METH-MRSA) compared to untreated conditions (uninfected or MRSA groups). There were significant decreases in vascular endothelial growth factor (VEGF) and TNF-α for the MRSA and METH-MRSA groups, respectively, compared to all other conditions. IL-6 and TGF-β levels were significantly reduced in untreated animals relative to the other groups. There were no differences in IL-4 production between any of the experimental groups.
TABLE 1 .
Cytokine levels in wounds of BALB/c mice 24 h after wounding
| Treatment | Cytokine levela (pg/ml) (avg ±SD) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IFN-γ | IL-1β | IL-4 | IL-6 | IL-10 | IL-12 | MCP-1 | TGF-β | TNF-α | VEGF | |
| Untreated | 1,951.74±16.62 | 791.80±11.57 | 81.25±10.74 | 452.93±8.91 | 398.26±6.61 | 367.91±6.45 | 374.08±11.13 | 1,996.55±5.02 | 3,852.01±15.12 | 4,623.52±15.09 |
| MRSA | 2,339.10±10.65ϕ | 859.25±6.47ϕ | 91.52±5.32 | 614.37±4.63ϕ | 321.83±7.36* | 414.53±5.37ϕ | 421.54±4.02ϕ | 2,459.32±4.46ϕ | 4,157.19±8.12ϕ | 2,071.90±23.61* |
| METH | 1,398.46±5.68*# | 645.32±6.04*# | 75.26±5.97 | 897.25±7.98ϕχ | 497.21±6.54ϕχ | 362.83±8.81# | 200.87±9.09*# | 2,085.31±16.25# | 2,736.14±11.95*# | 3,027.11±6.72*χ |
| METH-MRSA | 1,765.44±4.21*#π | 701.33±9.26*#π | 79.69±5.97 | 811.85±8.23ϕχ& | 505.21±9.22ϕχ | 388.24±8.65# | 398.08±7.12#π | 2,369.84±6.35ϕ | 1,977.25±12.81χ#& | 2,545.15±7.01*χπ |
ϕ, χ, and π indicate a significantly higher cytokine level (P < 0.05) than in the untreated, MRSA, or METH group, respectively. *, #, and & indicate a significantly lower cytokine level (P < 0.05) than in the untreated, MRSA, or METH group, respectively.
At day 7 after infection, wound tissue of METH-treated mice infected with MRSA contained significantly higher quantities of IFN-γ, TNF-α, IL-4, and TGF-β than tissues of uninfected or MRSA-treated mice (Table 2). There was a significant increase in IL-12 and IL-10 production in MRSA-infected animals compared to all the other treatment groups. Significant increases in IL-6 and IL-1β and decreases in VEGF were measured under all the other conditions, compared to results in uninfected mice. MCP-1 and VEGF levels were significantly reduced in MRSA-treated animals relative to the other groups.
TABLE 2 .
Cytokine levels in wounds of BALB/c mice 7 days after wounding
| Treatment | Cytokine levela (pg/ml) (avg ±SD) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IFN-γ | IL-1β | IL-4 | IL-6 | IL-10 | IL-12 | MCP-1 | TGF-β | TNF-α | VEGF | |
| Untreated | 2,321.58±15.84 | 2,598.36±10.89 | 251.26±10.28 | 2,245.35±8.65 | 474.36±13.47 | 936.32±6.47 | 4,932.21±10.58 | 10,365.9±5.47 | 1,901.26±14.74 | 1,5369.7±16.56 |
| MRSA | 4,531.08±9.45ϕ | 3,426.65±7.21ϕ | 384.08±5.05 | 5,057.15±9.14ϕ | 4,197.85±7.05ϕ | 1,682.09±12.75ϕ | 1,173.07±21.65* | 16,658.95±16.71ϕ | 1,383.62±7.81* | 6,312.28±12.43* |
| METH | 5,987.21±6.01ϕχ | 3,501.98±5.97ϕ | 1,223.36±11.71ϕ | 8,569.61±12.58ϕχ | 625.36±6.99ϕ# | 1,256.12±8.58ϕ# | 4,864.36±9.71χ | 16,723.1±13.89ϕ | 3,821.36±11.32ϕχ | 1,2154.7±10.06*χ |
| METH-MRSA | 15,806.1±4.66ϕχπ | 3,568.59±8.17ϕ | 1,462.59±6.18ϕχπ | 9,841.99±15.04ϕχπ | 592.4±9.54ϕ# | 1,145.13±9.16ϕ# | 5,215.42±19.14χ | 32,454.7±22.27ϕχπ | 5,121.88±5.81ϕχπ | 1,0172.2±9.28*χ& |
ϕ, χ, and π indicate a significantly higher cytokine level (P < 0.05) than in the untreated, MRSA, or METH group, respectively. *, #, and & indicate a significantly lower cytokine level (P < 0.05) than in the untreated, MRSA, or METH group, respectively.
DISCUSSION
We used a murine model to investigate the effects of METH on MRSA superficial skin infection. Our findings showed that METH-treated mice displayed reduced wound healing in the presence or absence of MRSA infection compared with untreated, uninfected mice, with the longest delay in wound closure occurring in the METH-MRSA group. In this regard, our results from the measurement of collagen deposition in wound tissue revealed increased collagen degradation in the METH-MRSA treatment group compared with the controls. Collagen plays a crucial role in the proliferative phase of the wound healing process, along with angiogenesis, granulation tissue formation, and epithelialization (26). The amount of collagen present in the wound during the healing process reflects a balance between new collagen production by fibroblasts and degradation by collagenases and other factors. In the early phases of healing of simple, uninfected wounds, synthesis exceeds degradation, so collagen levels in the wound rise; later, production and degradation become equal (26). This balance was disrupted by METH and MRSA. The expression levels of genes regulating collagen type I and III production were found to be significantly increased in the METH-MRSA group as well; however, the accelerated degradation process led to reduced collagen content in the wound and thus impaired healing. Notably, another contributing factor to the delayed wound closure was the decreased angiogenesis under the influence of METH-MRSA, as qualitatively demonstrated by the reduced expression of CD34, a marker for blood vessel formation. For instance, histological analyses of tissues from METH-MRSA mice showed high infiltration of inflammatory cells confined to the dermal area of the cutaneous tissue, but the cells were largely unable to reach the epidermal layer, where most of the bacteria were located. This observation may explain the 2-log-higher bacterial counts in the skin samples of the METH-treated group relative to the untreated mice. Likewise, in vitro and in vivo studies demonstrated that METH impairs phagocytic cell migration and antimicrobial activity as well as reduces phagocytes in the bloodstream. The prolonged presence of an elevated number of macrophages and neutrophils in the skin tissue may be associated with increased free radical accumulation (27), which can inhibit wound healing.
We investigated whether the difference in collagen deposition was due to METH-induced bacterial or host breakdown of collagen. We originally thought that S. aureus staphopain collagenolytic activity might be implicated in cutaneous tissue destruction in the setting of METH exposure (28). However, in vitro experiments determined that these bacterial cysteine proteases did not contribute to collagen breakdown. We cannot completely rule out the possibility that other virulence factors produced by S. aureus may be detrimental for the healing process. More research is needed to better identify specific MRSA virulence factors involved in dysregulating wound healing in the setting of drug abuse.
However, we successfully identified a host factor involved in collagen degradation after METH administration and MRSA infection. METH increased the expression of MMP-2, a metalloprotease that can interfere with wound healing and enhance susceptibility to bacterial infections (29, 30). In HIV-associated dementia, the HIV-1-encoded protein Tat and METH increase MMP-2 levels in supernatants of human neuron/astrocyte cultures (31). Previous studies have shown that MMP-2 plays crucial roles in METH-induced behavioral sensitization and reward by regulating METH-induced dopamine release and uptake via dopamine transporters in the brain (32). Dopamine has been reported to decrease mitosis of keratinocytes (33), the most abundant cells in skin tissue, whereas D2-like receptors (D2, D3, and D4) are involved in epidermal barrier homeostasis (34). For instance, the D2 receptor is present in the basal epidermis, and the D4 receptor is in the uppermost layer of the epidermis (34). These receptors have been implicated in the pathogenesis of skin inflammatory diseases (35, 36) and regulation of wound healing (37, 38). Although further studies are warranted, METH may induce dopamine production and D2-like receptor expression by keratinocytes, which could substantially reduce the wound healing rate. Additionally, METH promoted the collagenolytic activity of MMP-2 in a concentration-dependent manner, providing another possible explanation for the negative impact of this drug in wound healing. The late (day 7 postinjury) augmented production of inflammatory cytokines like TNF-α, IFN-γ, IL-6, and TGF-β observed in METH-treated groups promotes the activation of MMP-2 via NF-κB signaling in dermal fibroblasts embedded in collagen type I (39). Our study is important because self-management of injection-related wounds is common among injection drug users (IDUs) (11). Specifically, IDUs are more likely to engage in potentially harmful self-management behaviors, increasing their susceptibility to MRSA infections due to a combination of aggressive management of their wounds and an underlying slow healing rate. In a previous study, IDUs who had ever injected amphetamines were more likely to engage in potentially harmful self-management behaviors (11). Furthermore, the inability of phagocytic cells to be recruited and/or proactively fight invading microbes at the site of infection could exacerbate disease in IDUs.
Another important finding of this study is the increase in inflammation and cell necrosis in the wounds of METH-treated and infected mice compared with control groups. For instance, CFU analysis showed an increased bacterial burden in the wound tissues in the METH-MRSA group. Chronic wounds on METH users are an ideal environment for S. aureus biofilm formation. The necrotic tissue and debris allow bacterial attachment, and wounds are susceptible to infection due to an impaired host immune response. Evidence suggests that biofilms play a significant role in the inability of chronic wounds to heal. Biofilms are present in only 6% of acute wounds but over 90% of chronic wounds (40). We found that METH enhanced biofilm formation by multiple clinical S. aureus strains. The presence and persistence of biofilms on chronic skin wounds can affect cellular function (leukocytes, keratinocytes, endothelial cells, and fibroblasts), the inflammatory cellular response, the cutaneous innate immune response, and the repair phase of wound healing (angiogenesis and fibrogenesis). For example, keratinocytes produce more MMPs in chronic biofilm-challenged wounds (41). These infections develop gradually and may be slow to produce explicit symptoms. In addition, the vast majority of bacteria reside in the eschar above the wound bed (42). Once established, however, biofilm infections often persist. Understanding the impact of METH on S. aureus pathogenesis in IDUs warrants the pursuit of further studies addressing the influence of the drug on the multitude of virulence factors expressed by this microbe during infection, including its toxins.
Another possible explanation for S. aureus persistence in wounded tissue of drug users is that METH alters the effector functions of neutrophils and macrophages. The initial and most obvious function of inflammatory cells at the site of injury is to provide specific and nonspecific defenses against pathogens. The first cells that infiltrate a wound are neutrophils, which remove foreign particles and bacteria. In the skin, neutrophils appear in the wound bed within minutes after injury. We demonstrated that METH inhibits neutrophil chemotaxis and the production of NO and MPO in a concentration-dependent fashion, thus reducing the antimicrobial armamentarium of activated murine neutrophils. Notably, METH compromised S. aureus phagocytosis, the respiratory burst, and killing by human neutrophils, suggesting that the detrimental effects of the drug observed in murine-derived cells can be similarly achieved in human cells, which validates the mouse as a good model for these types of studies. METH-mediated phagocytosis dysfunction by neutrophils may be associated with reduced expression of GTPase-RhoA, a key regulator of the actin polymerization signaling cascade, given that CytD-treated cells displayed a similar deficiency in engulfing the microbe. Also, impaired phagocytosis may be associated with immobilization of the complement receptor 3 (CR-3) on the surface of neutrophils and deregulation of cytokines (21). Neutrophils are nevertheless a major source of several inflammatory cytokines, such as TNF-α and IL-1β, which stimulate attracted monocytes to differentiate into M1 (classical activation) macrophages (43, 44). We observed that METH reduces the early (24 h postinjury) production of these two cytokines in METH-treated groups. Moreover, elevated levels of IL-10 in homogenates of METH-treated groups during the early inflammatory response suggest an important role of these factors in reducing the recruitment of phagocytic cells to the site of injury and infection.
METH inhibited the migration of macrophages to the infection site as observed in vitro and in the histological analysis. Interestingly, we found that primary macrophages exposed to METH displayed increased MRSA phagocytosis; however, these macrophages showed impaired intracellular bacterial killing leading to increased pathogen survival. As in our prior work, METH disrupted macrophage effector functions (21), including alkalization of the phagosomal pH and inhibition of NO and superoxide production. One study using a mouse model of catheter-associated biofilm infection showed that S. aureus biofilms caused changes in macrophage function by suppressing microbicidal activity, altering gene expression toward M2 phenotype cells, decreasing migration, and increasing cell death (45). Those investigators showed that macrophages were able to phagocytose planktonic cells but not biofilm-associated cells, suggesting that the in vitro experiments do not necessarily reflect in vivo situations. During the proliferative phase, macrophages stimulate proliferation of connective, endothelial, and epithelial tissues directly and indirectly. In particular, fibroblasts, keratinocytes, and endothelial cells are stimulated by macrophages during this phase to induce and complete extracellular matrix formation, reepithelialization, and neovascularization. Once recruited to the wound, macrophages induce angiogenesis. This is predominantly exerted by macrophages releasing TNF-α and VEGF. TNF-α may in turn induce VEGF expression by keratinocytes and fibroblasts (46). Depletion of macrophages in the wound results in reduced vascularization (47) and also leads to severe hemorrhage and fibrin and serum exudates in macrophage-depleted granulation tissue (48). This has been reported to be mainly caused by withdrawal of macrophage-derived TGF-β and VEGF (47, 48), supporting an important role for macrophages in promoting angiogenesis via their secretory products.
METH alters cytokine expression, as indicated by our measurements of cytokine levels in the wounds. The late (day 7 postinjury) increased levels of inflammatory cytokines in the METH-MRSA group correlated with our histological observations of increased local inflammation in their wounds. Nevertheless, the disabled immune machinery in the METH-treated mice failed to contain the infection, and the initial inflammatory cascade led to detrimental local outcomes with increased cell necrosis and impaired healing. METH has been linked to a shift in the immune response from a dominant Th1 to a mixed Th1-Th2 phenotype (49). In our infection model, we found high levels of endogenous TNF-α and IFN-γ in METH-treated mice. However, the increased levels of the two Th1 cytokines were not sufficient to rescue mice from the lethal effects of MRSA. Thus, METH might play a role in inducing a dominant nonprotective Th2 response to MRSA.
In conclusion, this is the first study that has empirically addressed the effect of METH on immunity during infection by S. aureus. These findings should raise awareness of the negative impact of METH use on the overall community health status, as alarming data in this regard have been published on the high incidence of MRSA skin and soft tissue infections among METH consumers (13). Although wound debridement remains a proven cornerstone for wound management, perhaps in part because a microbial biofilm is removed from the scab, it is plausible to think that wounds in METH users become chronic and difficult to manage because of a combination of bacterial persistence, drug-related immune deficiency, and the IDU behaviors, which include stereotypical formication and lack of hygiene during injections, making these individuals highly susceptible for S. aureus reinfection. A possible limitation of the study is that the in vitro and in vivo METH treatments were not equivalent, since in vitro the phagocytes were exposed for a few hours to METH, whereas in vivo they were exposed for 21 days. However, METH doses used in both animals and tissue cultures were similar or closely related to those measured in people that chronically abuse the drug. Further research is needed to understand the biological and management complexity of infected wounds in METH users and to best focus prevention efforts to reduce morbidity and improve wound care.
MATERIALS AND METHODS
S. aureus.
A total of six S. aureus clinical isolates were used in this study. The characteristics of each strain are described in Table 3. The MRSA 6498 isolate used in the majority of the experiments in this report is a USA300 strain collected from a patient’s wound and has been extensively utilized in wound healing studies (49, 50). The strains were stored at −80°C in brain heart infusion (BHI) broth with 40% glycerol until use. Test organisms were grown in tryptic soy broth (TSB) overnight at 37°C on a rotary shaker set at 150 rpm. Growth was monitored by measuring the optical density at 600 nm and use of a microtiter plate reader.
TABLE 3 .
Characteristics of S. aureus clinical strains tested in this studya
| Strain | Source | mecA | SCC | ACME | MLST | Toxin genes |
|---|---|---|---|---|---|---|
| 038 | Blood | + | IV | − | CC 8 | seb, sek, sep, seq, ser, seu |
| 067 | Wound | − | MSSA | − | CC 8 | sea, seb, sek, sep, seq, ser, seu |
| 085b | Wound | + | IV | + | CC 8 | pvl, sek, sep, ser |
| 112 | Wound | + | IV | + | CC 8 | sek, ser, seu |
| 132 | Wound | + | IV | − | NDc | seb, sek, ser |
| 6498b | Wound | + | IV | + | CC 8 | pvl, sek, sep, ser |
Toxin profiles were determined by PCR amplification with primer sets for 19 different toxins. Abbreviations (corresponding gene): staphylococcal enterotoxin a (sea), B (seb), K (sek), P (sep), Q (seq), R (ser), U (seu), and Panton-Valentine leukocidin (pvl). ACME, arginine catabolic mobile element; MLST, multilocus sequence type; CC, clonal complex; ND, not determined.
USA300 strain.
METH.
Most high-dose METH abusers initially use small amounts of the drug intermittently before progressively increasing the dose (51). To simulate this pattern, we used increasing daily doses (2.5, 5, and 10 mg/kg/day on weeks 1, 2, and 3, respectively) of METH that were intraperitoneally (i.p.) administered to female BALB/c mice (6 to 8 weeks old) over 21 days, as described previously (21). As controls, mice received equivalent volumes of PBS.
Wound and infection model.
At day 21, METH- and PBS-treated BALB/c mice were anesthetized (100 mg/kg ketamine, 10 mg/kg xylazine), their back hair was removed, and skin was disinfected with iodine. Five-millimeter-diameter full-thickness excision wounds in the center of the backs were achieved using surgical punches. A suspension containing 107 S. aureus strain 6498 in PBS was inoculated onto each wound. Uninfected METH- or PBS-treated mice were used as additional controls. Photographs of the wounds were taken on days 3, 5, 7, 9, 11, 13, 15, and 17. The wound dimensions were measured every other day by two different operators using dial calipers in a blinded fashion. Although mice experience formication after METH administration, this behavior did not influence the results obtained, given that each animal was isolated in their own cage, eliminating the possibility of mice scratching other mice; in addition, the location of the wound made it difficult for the animals to scratch their own wounds. For histology, CFU determinations, and gene expression analysis, animals were euthanized at day 7 and wound tissues were excised. Animals were otherwise euthanized at day 21, by which time all wounds had healed.
Ethics statement.
Animal experiments were performed according to the guide published by the Institute of Laboratory Animal Resources of the National Research Council. Animal care for this study was approved by the Animal Welfare and Research Ethics Committee at the Albert Einstein College of Medicine (protocol number 20110402).
Histology.
Skin tissues were excised and fixed in 4% paraformaldehyde (PFA) for 24 h. Tissues were processed and embedded in paraffin, and 4-µm vertical sections were fixed to glass slides. Tissue sections were stained with hematoxylin and eosin (H&E), Gram stain, and Gomori’s trichrome, and for CD34 or Iba-1, to examine tissue morphology, bacteria, collagen deposition, vascularization, and macrophage infiltration, respectively. Slides were visualized using an Axiovert 40CFL inverted microscope (Carl Zeiss), and images were captured with an AxioCam MrC digital camera using the Zen 2011 digital imaging software.
Collagen deposition determinations.
Collagen deposition was measured based on intensity, using ImageJ software (National Institutes of Health, USA) (52). The software possesses threshold filters to discriminate between the stain colors (white, red, and blue), providing an accurate measurement of each color’s intensity. Ten 40× fields were evaluated per section.
CFU determinations.
Excised wound tissues were homogenized in PBS and plated on tryptic soy agar (TSA) for CFU determinations. The results were normalized by tissue weights.
Real-time (RT)-PCR.
For RT-PCR analysis of host gene expression, we analyzed col1, col3, and mmp2 expression. col1 encodes collagen type I, and col3 encodes collagen type III, whereas mmp2 encodes matrix metalloproteinase-2. At day 7, excised tissues were homogenized, cells were collected and washed, and then RNA was isolated using an RNeasy kit. col1, col3, and mmp2 expression levels were analyzed by RT-PCR as described elsewhere (52).
In S. aureus, ica encodes the intercellular adhesion locus and is required for S. aureus biofilm formation. Twenty-four hours after incubation with METH, bacterial RNA was isolated and analyzed by RT-PCR as described previously (53).
For an internal mRNA control, we used primers specific for glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Phagocytic cell counts in blood.
Twenty-one days after METH administration, phagocytic cell counts were performed by differential leukocyte count for samples from all experimental animals by using a Hema 3 Stat Pack kit and light microscopy.
Rationale for METH doses used in in vitro studies.
Controlled studies indicated that a single 260-mg dose peaks at a level of 7.5 µM (54). Thus, a single dose of 260 mg/g would be expected to produce 7.5 to 28.8 µM blood METH levels. IDUs tend to self-administer METH in binges, and as the drug exhibits a half-life of 11.4 to 12 h (55, 56), this can lead to higher drug levels. Published studies modeling binge patterns of use in individuals have shown that the fourth administration of 260 mg during a single day produced blood levels of 17 µM and could reach 20 µM on the second day of such a binge (54). Thus, binge doses of 260 to 1,000 mg produce 17 to 80 µM blood METH levels and levels in the micromolar range of hundreds in organs, including the brain and the spleen (57). Therefore, we selected ~25 to 50 µM METH to perform our in vitro experiments.
Bacterial viability assay.
To determine the impact of the METH on S. aureus growth, 1 ml of TS broth (TSB) was inoculated with a 24-h colony of the bacterium grown on TSA. One hundred microliters of S. aureus broth suspension was inoculated in each well of a 200-well plate, containing 100 µl of TSB with either 25 or 50 µM METH. The inoculated plate was then incubated for 24 h at 37°C. Controls included wells containing bacterial suspension alone (untreated). Growth was assessed every 15 min using a microplate reader set at an optical density (OD) of 600 nm.
Collagenolytic activity assay.
Collagenolytic activity was measured using murine fluorescein isothiocyanate (FITC)-conjugated type I collagen (28). FITC-conjugated type I collagen was incubated with a 10 nM concentration of either staphopain A or MMP-2 for 3 h at 37°C. Then, 100 µl of the solution was mixed with 100 µl ethanol (70% ethanol–0.17 M Tris-HCl buffer, 0.67 M NaCl) and centrifuged for 10 min at 4°C. To assess collagen degradation, 70 µl of the supernatant was withdrawn, and fluorescence was measured with a multilabel plate reader with emission at 520 nm and excitation at 495 nm.
Western blot analysis.
To further characterize the role of METH in wound healing, we assessed MMP-2 expression in the wounds of BALB/c mice via Western blot analysis, as described previously (52).
Biofilm formation.
For each strain, 100 µl of a suspension with bacterial cells in RPMI 1640 medium supplemented with 1% Casamino Acids was added into individual wells of polystyrene 96-well plates, and the plates were incubated at 37°C without shaking as previously described (58). The biofilms grown with PBS or METH (25 and 50 µM) were allowed to form for 24 h.
Quantitation of biofilms.
Measurement of biofilm formation and viability was done by CFU determinations and assessment of the metabolic activity of the attached cells in an XTT reduction assay, as previously described (59).
Biofilm architecture visualization.
The structural integrity of biofilms formed in the absence or presence of METH was examined by using the LIVE/DEAD biofilm viability kit and confocal microscopy, as previously described (60).
Isolation of peripheral blood neutrophils from mice and humans.
Retroorbital puncture was used to obtain blood from BALB/c mice, whereas whole human blood was purchased from the Interstate Blood Bank, Inc. (Memphis, TN). Erythrocytes were removed by hypotonic lysis, and neutrophils were separated from the remaining cells by centrifugation over discontinuous Percoll gradients. Neutrophils were >95% viable, as determined by Wright-Giemsa staining. Recovered neutrophils (~98% as determined by FACS, using Ly-6G as a marker) were briefly maintained (<30 min; 37°C, 10% CO2) in RPMI 1640 supplemented with 10 mM HEPES (pH 7.4) and 10% fetal calf serum (FCS) prior to use.
Controls.
Chloroquine is a weak base and a well-known inhibitor of endosomal acidification (61), whereas CytD is a potent inhibitor of actin polymerization. Both reagents were used as controls in the in vitro studies of S. aureus-phagocyte interactions.
Murine neutrophil killing assay.
For killing assays (62), 3 × 105 neutrophils in 200 µl of RPMI 1640 with 10% FCS were incubated with METH (25 or 50 µM), Chlq (25 µM), CytD (25 µM), or PBS in microcentrifuge tubes with 8 rpm rotation for 2 h at 37°C, 5% CO2. S. aureus 6498 cultures were washed twice in PBS, diluted to a concentration of 4.5 × 106 CFU in 100 µl RPMI 1640 plus 10% FCS (feeding medium), and mixed with washed neutrophils in the same medium (ratio, 15 bacteria:1 neutrophil), centrifuged at 1,200 rpm for 5 min, and then incubated at 37°C in a 5% CO2 incubator. Gentamicin (final concentration of 400 µg/ml for S. aureus) was added after 10 min to kill extracellular bacteria. At specified time points (20 to 80 min), the contents of sample wells were withdrawn, centrifuged to pellet the neutrophils, and washed to remove the antibiotic medium. Neutrophils were then lysed in 0.02% Triton X-100, and CFU were calculated after plating on TSA.
Macrophages.
Primary macrophages were isolated from untreated BALB/c mice by lavage of the abdominal cavity with Hanks’ balanced salt solution with 1 mM EGTA (21).
Phagocytosis.
For neutrophils, phagocytosis was determined by using confocal microscopy. Primary murine neutrophils were incubated with METH (25 or 50 µM), Chlq (25 µM), CytD (25 µM), or PBS for 90 min at 37°C and 5% CO2, prior to incubation for phagocytosis in the presence of Alexa Fluor 350 (Alexa)-labeled S. aureus 6498 cells for 30 min. Gentamicin (400 µg/ml) was added to kill extracellular bacteria. Similarly, extracellular bacteria were quenched with trypan blue to prevent interference with the assay. Microscopic examinations of neutrophils were performed with a Leica TCS SP5 confocal laser scanning microscope. Z-stack images and measurements were corrected by utilizing Bio-Rad LaserSharp 2000 software in deconvolution mode. The fluorescent intensity of bacteria inside neutrophils was analyzed using Adobe Photoshop software (63).
For macrophages, phagocytosis was determined by using FACS analysis. Primary macrophages were incubated with METH (25 or 50 µM), Chlq (25 µM), CytD (25 µM), or PBS for 2 h at 37°C and 5% CO2, prior to incubation for phagocytosis in the presence of Alexa-labeled S. aureus 6498 for 30 min. Gentamicin (400 µg/ml) was added to kill extracellular bacteria. Similarly, extracellular bacteria were quenched with trypan blue to prevent interference with the assay. Samples were processed (10,000 events per condition) on an LSRII flow cytometer (BD) and were analyzed using FlowJo software.
Macrophage killing assay.
Primary macrophages were first allowed to phagocytize S. aureus 6498 cells for 30 min. Each well containing interacting cells was gently washed with feeding medium and incubated with feeding medium supplemented with gentamicin (400 µg/ml) and either PBS, METH (25 or 50 µM), Chlq (25 µM), or CytD (25 µM) for 30 to 120 min. Quantification of viable S. aureus cells was determined by measuring CFU after lysis of macrophages (64).
Phagosomal pH.
The pH of S. aureus 6498-containing phagosomes in primary macrophages was determined as described elsewhere (21).
Nitric oxide and myeloperoxidase production.
NO production was quantified using a Griess method kit (Promega). Similarly, MPO produced by N-formylmethionyl-leucyl-phenylalanine (fMLP)-stimulated neutrophils in culture after coincubation with S. aureus was measured using the myeloperoxidase assay kit.
Chemotaxis.
Chemotaxis was measured using a transwell chamber with 6.5-mm-diameter polycarbonate filters (3-µm pore size). Immediately after isolation, cells were incubated in the absence or presence of METH (25 or 50 µM) for 2 h. Chlq (25 µM) and CytD (25 µM) were used as positive controls. Cells were transferred to RPMI 1640 with heat-inactivated FCS, cultivated on filters, and allowed to migrate toward the chemoattractant fMLP or cultured in medium alone at 37°C, 5% CO2. After various time points (20, 40, and 60 min), the filters were removed, and the cells that migrated through the membrane were fixed, stained, and counted by light microscopy (400×) as described elsewhere (65). Ten nonoverlapping and randomized fields were counted by one scorer.
Human neutrophil phagocytic and killing assays.
Phagocytosis and killing assays were performed as previously described (66). For the phagocytosis assay, FACS analysis was employed. Human neutrophils (106 cells) were incubated on six-well plates with feeding medium supplemented with METH (25 or 12.5 µM) or PBS for 2 h at 37°C and 5% CO2. FITC-labeled S. aureus cells were incubated with 25% human serum for 30 min to allow complement proteins to opsonize S. aureus cells. Bacterial cells were washed and then 107 were added to the 106 neutrophils for 60 min. Similarly, extracellular bacteria were quenched with trypan blue to prevent interference with the assay. Samples were processed on an LSRII flow cytometer and were analyzed using FlowJo software.
Since METH reduces S. aureus phagocytosis by neutrophils, for the killing assays leukocytes were first allowed to phagocytize S. aureus cells for 0.5 h to determine initial uptake. Each well containing interacting cells was gently washed with feeding medium and incubated with feeding medium supplemented with gentamicin (400 µg/ml, to kill extracellular bacteria) and either PBS or METH (25 or 50 µM) for 4 h. Viable bacteria were released from neutrophils following 0.5 or 4 h of host-cell interaction by forcibly subjecting cultures to 27-gauge needle passage 5 to 7 times for efficient lysis. Four microtiter wells per condition were used to ascertain CFU. For each well, serial dilutions were plated in triplicates onto TSA plates, which were incubated at 37°C for 24 h prior to CFU tallying.
Luminol chemiluminescence assay.
ROS signals were made chemiluminescent (CL) by using luminol (1 mM). CL was monitored for 30 min with an automatic luminescence analyzer (SpectraMax L; Molecular Devices) at 37°C.
Cytokine determinations.
Cutaneous tissues from mice were excised at 24 h and 7 days after wounding and homogenized in PBS with protease inhibitors. Cell debris was removed from homogenates by centrifugation at 6,000 × g for 10 min. Samples were stored at −80°C until tested. Supernatants were tested for IFN-γ, IL-1β, IL-4, IL-6, IL-10, IL-12p70, MCP-1, TGF-β, TNF-α, and VEGF in an enzyme-linked immunosorbent assay (OptEIA kits; Becton, Dickinson Biosciences).
Statistical analysis.
All data were analyzed using Prism (GraphPad, La Jolla, CA). Analyses of CFU (Fig. 2B) and phagocytic cell count (Fig. 4B) data were performed using Student’s t test. Analysis of all the other data in the study was performed using analysis of variance (ANOVA) and adjusted by use of the Bonferroni correction. P values of <0.05 were considered significant.
ACKNOWLEDGMENTS
E.A.E. is supported by the National Institutes of Health (NIH MH096625) and PHRI internal awards. A.J.G. is supported by Fundação de Apoio a Pesquisa do Estado do Rio de Janeiro and Conselho Nacional de Pesquisa e Desenvolvimento (CNPq). B.C.F. is supported by NIH 2R01AI059681-06 and R21AI087564-01. J.D.N. is supported in part by an Irma T. Hirschl/Monique Weill-Caulier Trust Research award. L.R.M. was supported by NIH NIAID award 5K22A1087817-02 and NYIT COM start-up funds.
M.R.M. and J.R.-S. performed CFU, cytokine, and histological analyses. B.P.S. performed the phagocytic cell counts and MMP-2-related experiments. A.K.V. and B.C.F. performed the in vivo wound healing experiments. L.N.N. performed the collagen gene expression analysis. A.J.G. and J.D.N. performed chemotaxis, phagocytosis, nitric oxide production, and killing assays. E.A.E. performed the confocal microscopy analysis. J.D.N. assisted in data analysis and editing of the manuscript. H.H.L. performed the experiments involving human neutrophils. L.R.M. directed the overall design of the experiments, analysis of the data, and writing of the manuscript.
Footnotes
Citation Mihu MR, Roman-Sosa J, Varshney AK, Eugenin EA, Shah BP, Ham Lee H, Nguyen LN, Guimaraes AJ, Fries BC, Nosanchuk JD, Martinez LR. 2015. Methamphetamine alters the antimicrobial efficacy of phagocytic cells during methicillin-resistant Staphylococcus aureus skin infection. mBio 6(6):e01622-15. doi:10.1128/mBio.01622-15.
REFERENCES
- 1.McCaig L, McDonald L, Mandal S, Jernigan D. 2006. Staphylococcus aureus-associated skin and soft tissue infections in ambulatory care. Emerg Infect Dis 12:1715–1723. doi: 10.3201/eid1211.060190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noskin GA, Rubin RJ, Schentag JJ, Kluytmans J, Hedblom EC, Jacobson C, Smulders M, Gemmen E, Bharmal M. 2007. National trends in Staphylococcus aureus infection rates: impact on economic burden and mortality over a 6-year period (1998–2003). Clin Infect Dis 45:1132–1140. doi: 10.1086/522186. [DOI] [PubMed] [Google Scholar]
- 3.Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, Talan DA. 2006. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med 355:666–674. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 4.Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover FC. 1997. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother 40:135–136. [DOI] [PubMed] [Google Scholar]
- 5.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 6.Ellis R, Childers M, Cherner M, Lazzaretto D, Letendre S, Grant I. 2003. Increased human immunodeficiency virus loads in active methamphetamine users are explained by reduced effectiveness of antiretroviral therapy. J Infect Dis 188:1820–1826. doi: 10.1086/379894. [DOI] [PubMed] [Google Scholar]
- 7.Gonzales R, Marinelli-Casey P, Shoptaw S, Ang A, Rawson RA. 2006. Hepatitis C virus infection among methamphetamine-dependent individuals in outpatient treatment. J Subst Abus Treat 31:195–202. doi: 10.1016/j.jsat.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 8.Salgado CD, Farr BM, Calfee DP. 2003. Community-acquired methicillin-resistant Staphylococcus aureus: a meta-analysis of prevalence and risk factors. Clin Infect Dis 36:131–139. doi: 10.1086/345436. [DOI] [PubMed] [Google Scholar]
- 9.Ebright JR, Pieper B. 2002. Skin and soft tissue infections in injection drug users. Infect Dis Clin North Am 16:697–712. doi: 10.1016/S0891-5520(02)00017-X. [DOI] [PubMed] [Google Scholar]
- 10.Gordon RJ, Lowy FD. 2005. Bacterial infections in drug users. N Engl J Med 353:1945–1954. doi: 10.1056/NEJMra042823. [DOI] [PubMed] [Google Scholar]
- 11.Roose RJ, Hayashi AS, Cunningham CO. 2009. Self-management of injection-related wounds among injecting drug users. J Addict Dis 28:74–80. doi: 10.1080/10550880802545200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kerr T, Wood E, Grafstein E, Ishida T, Shannon K, Lai C, Montaner J, Tyndall MW. 2005. High rates of primary care and emergency department use among injection drug users in Vancouver. J Public Health 27:62–66. doi: 10.1093/pubmed/fdh189. [DOI] [PubMed] [Google Scholar]
- 13.Cohen AL, Shuler C, McAllister S, Fosheim GE, Brown MG, Abercrombie D, Anderson K, McDougal LK, Drenzek C, Arnold K, Jernigan D, Gorwitz R. 2007. Methamphetamine use and methicillin-resistant Staphylococcus aureus skin infections. Emerg Infect Dis 13:1707–1713. doi: 10.3201/eid1311.070148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pollini RA, Gallardo M, Hasan S, Minuto J, Lozada R, Vera A, Zúñiga ML, Strathdee SA. 2010. High prevalence of abscesses and self-treatment among injection drug users in Tijuana, Mexico. Int J Infect Dis 14(Suppl 3):e117–e122. doi: 10.1016/j.ijid.2010.02.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cespedes C, Saïd-Salim B, Miller M, Lo S, Kreiswirth B, Gordon R, Vavagiakis P, Klein R, Lowy F. 2005. The clonality of Staphylococcus aureus nasal carriage. J Infect Dis 191:444–452. doi: 10.1086/427240. [DOI] [PubMed] [Google Scholar]
- 16.Vlahov D, Sullivan M, Astemborski J, Nelson KE. 1992. Bacterial infections and skin cleaning prior to injection among intravenous drug users. Public Health Rep 107:595–598. [PMC free article] [PubMed] [Google Scholar]
- 17.Iwasa M, Maeno Y, Inoue H, Koyama H, Matoba R. 1996. Induction of apoptotic cell death in rat thymus and spleen after a bolus injection of methamphetamine. Int J Leg Med 109:23–28. doi: 10.1007/BF01369597. [DOI] [PubMed] [Google Scholar]
- 18.Freire-Garabal M, Balboa J, Núñez MJ, Castaño MT, Llovo J, Fernández-Rial J, Belmonte A. 1991. Effects of amphetamine on T-cell immune response in mice. Life Sci 49:PL107–PL112. doi: 10.1016/0024-3205(91)90570-2. [DOI] [PubMed] [Google Scholar]
- 19.Potula R, Hawkins BJ, Cenna JM, Fan S, Dykstra H, Ramirez SH, Morsey B, Brodie MR, Persidsky Y. 2010. Methamphetamine causes mitrochondrial oxidative damage in human T lymphocytes leading to functional impairment. J Immunol 185:2867–2876. doi: 10.4049/jimmunol.0903691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tallóczy Z, Martinez J, Joset D, Ray Y, Gácser A, Toussi S, Mizushima N, Nosanchuk J, Goldstein H, Loike J, Sulzer D, Santambrogio L. 2008. Methamphetamine inhibits antigen processing, presentation, and phagocytosis. PLoS Pathog 4:e28. doi: 10.1371/journal.ppat.0040028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez L, Mihu M, Gácser A, Santambrogio L, Nosanchuk J. 2009. Methamphetamine enhances histoplasmosis by immunosuppression of the host. J Infect Dis 200:131–141. doi: 10.1086/599328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu Q, Zhang D, Walston M, Zhang J, Liu Y, Watson RR. 2002. Chronic methamphetamine exposure alters immune function in normal and retrovirus-infected mice. Int Immunopharmacol 2:951–962. doi: 10.1016/S1567-5769(02)00047-4. [DOI] [PubMed] [Google Scholar]
- 23.In SW, Son EW, Rhee DK, Pyo S. 2005. Methamphetamine administration produces immunomodulation in mice. J Toxicol Environ Health A 68:2133–2145. doi: 10.1080/15287390500177156. [DOI] [PubMed] [Google Scholar]
- 24.Mahajan SD, Hu Z, Reynolds JL, Aalinkeel R, Schwartz SA, Nair MPN. 2006. Methamphetamine modulates gene expression patterns in monocyte derived mature dendritic cells: implications for HIV-1 pathogenesis. Mol Diagn Ther 10:257–269. doi: 10.1007/BF03256465. [DOI] [PubMed] [Google Scholar]
- 25.Magnet S, Courvalin P, Lambert T. 2001. Resistance-nodulation-cell division-type efflux pump involved in aminoglycoside resistance in Acinetobacter baumannii strain BM4454. Antimicrob Agents Chemother 45:3375–3380. doi: 10.1128/AAC.45.12.3375-3380.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rangaraj A, Harding K, Leaper D. 2011. Role of collagen in wound management. Wounds UK 7:54–63. http://www.wounds-uk.com/pdf/content_10039.pdf. [Google Scholar]
- 27.Goularte TA, Craven DE. 1986. Results of a survey of infection control practices for respiratory therapy equipment. Infect Control 7:327–330. [DOI] [PubMed] [Google Scholar]
- 28.Ohbayashi T, Irie A, Murakami Y, Nowak M, Potempa J, Nishimura Y, Shinohara M, Imamura T. 2011. Degradation of fibrinogen and collagen by staphopains, cysteine proteases released from Staphylococcus aureus. Microbiology 157:786–792. doi: 10.1099/mic.0.044503-0. [DOI] [PubMed] [Google Scholar]
- 29.Kanangat S, Postlethwaite A, Hasty K, Kang A, Smeltzer M, Appling W, Schaberg D. 2006. Induction of multiple matrix metalloproteinases in human dermal and synovial fibroblasts by Staphylococcus aureus: implications in the pathogenesis of septic arthritis and other soft tissue infections. Arthritis Res Ther 8:R176. doi: 10.1186/ar2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang JH, Kwon HJ, Jang YJ. 2010. Staphylococcus aureus increases cytokine and matrix metalloproteinase expression in nasal mucosae of patients with chronic rhinosinusitis and nasal polyps. Am J Rhinol Allergy 24:422–427. doi: 10.2500/ajra.2010.24.3509. [DOI] [PubMed] [Google Scholar]
- 31.Conant K, St Hillaire CS, Anderson C, Galey D, Wang J, Nath A. 2004. Human immunodeficiency virus type 1 Tat and methamphetamine affect the release and activation of matrix-degrading proteinases. J Neurovirol 10:21–28. doi: 10.1080/13550280490261699. [DOI] [PubMed] [Google Scholar]
- 32.Mizoguchi H, Yamada K, Nabeshima T. 2011. Matrix metalloproteinases contribute to neuronal dysfunction in animal models of drug dependence, Alzheimer’s disease, and epilepsy. Biochem Res Int 2011:681385. doi: 10.1155/2011/681385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harper RA, Flaxman BA. 1975. Effect of pharmacological agents on human keratinocyte mitosis in vitro. II. Inhibition by catecholamines. J Cell Physiol 86:293–299. doi: 10.1002/jcp.1040860213. [DOI] [PubMed] [Google Scholar]
- 34.Fuziwara S, Suzuki A, Inoue K, Denda M. 2005. Dopamine D2-like receptor agonists accelerate barrier repair and inhibit the epidermal hyperplasia induced by barrier disruption. J Invest Dermatol 125:783–789. doi: 10.1111/j.0022-202X.2005.23873.x. [DOI] [PubMed] [Google Scholar]
- 35.Grando SA, Pittelkow MR, Schallreuter KU. 2006. Adrenergic and cholinergic control in the biology of epidermis: physiological and clinical significance. J Invest Dermatol 126:1948–1965. doi: 10.1038/sj.jid.5700151. [DOI] [PubMed] [Google Scholar]
- 36.Sivamani RK, Lam ST, Isseroff RR. 2007. Beta adrenergic receptors in keratinocytes. Dermatol Clin 25:643–653. doi: 10.1016/j.det.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shome S, Rana T, Ganguly S, Basu B, Chaki Choudhury S, Sarkar C, Chakroborty D, Dasgupta PS, Basu S. 2011. Dopamine regulates angiogenesis in normal dermal wound tissues. PLoS One 6:e25215. doi: 10.1371/journal.pone.0025215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sivamani RK, Pullar CE, Manabat-Hidalgo CG, Rocke DM, Carlsen RC, Greenhalgh DG, Isseroff RR. 2009. Stress-mediated increases in systemic and local epinephrine impair skin wound healing: potential new indication for beta blockers. PLoS Med 6:e12. doi: 10.1371/journal.pmed.1000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han YP, Tuan TL, Wu H, Hughes M, Garner WL. 2001. TNF-alpha stimulates activation of pro-MMP2 in human skin through NF-κB mediated induction of MT1-MMP. J Cell Sci 114:131–139. http://jcs.biologists.org/content/114/1/131.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.James GA, Swogger E, Wolcott R, Pulcini E, Secor P, Sestrich J, Costerton JW, Stewart PS. 2008. Biofilms in chronic wounds. Wound Repair Regen 16:37–44. doi: 10.1111/j.1524-475X.2007.00321.x. [DOI] [PubMed] [Google Scholar]
- 41.Zhao G, Usui ML, Underwood RA, Singh PK, James GA, Stewart PS, Fleckman P, Olerud JE. 2012. Time course study of delayed wound healing in a biofilm-challenged diabetic mouse model. Wound Repair Regen 20:342–352. doi: 10.1111/j.1524-475X.2012.00793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao G, Hochwalt PC, Usui ML, Underwood RA, Singh PK, James GA, Stewart PS, Fleckman P, Olerud JE. 2010. Delayed wound healing in diabetic (dB/dB) mice with Pseudomonas aeruginosa biofilm challenge: a model for the study of chronic wounds. Wound Repair Regen 18:467–477. doi: 10.1111/j.1524-475X.2010.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hübner G, Werner S. 1996. Serum growth factors and proinflammatory cytokines are potent inducers of activin expression in cultured fibroblasts and keratinocytes. Exp Cell Res 228:106–113. doi: 10.1006/excr.1996.0305. [DOI] [PubMed] [Google Scholar]
- 44.Werner S, Grose R. 2003. Regulation of wound healing by growth factors and cytokines. Physiol Rev 83:835–870. doi: 10.1152/physrev.00032.2002. [DOI] [PubMed] [Google Scholar]
- 45.Thurlow LR, Hanke ML, Fritz T, Angle A, Aldrich A, Williams SH, Engebretsen IL, Bayles KW, Horswill AR, Kielian T. 2011. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J Immunol 186:6585–6596. doi: 10.4049/jimmunol.1002794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frank S, H bner G, Breier G, Longaker MT, Greenhalgh DG, Werner S. 1995. Regulation of vascular endothelial growth factor expression in cultured keratinocytes. Implications for normal and impaired wound healing. J Biol Chem 270:12607–12613. doi: 10.1074/jbc.270.21.12607. [DOI] [PubMed] [Google Scholar]
- 47.Mirza R, DiPietro LA, Koh TJ. 2009. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol 175:2454–2462. doi: 10.2353/ajpath.2009.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Muller W, Roers A, Eming SA. 2010. Differential roles of macrophages in diverse phases of skin repair. J Immunol 184:3964–3977. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 49.Martinez LR, Han G, Chacko M, Mihu MR, Jacobson M, Gialanella P, Friedman AJ, Nosanchuk JD, Friedman JM. 2009. Antimicrobial and healing efficacy of sustained release nitric oxide nanoparticles against Staphylococcus aureus skin infection. J Invest Dermatol 129:2463–2469. doi: 10.1038/jid.2009.95. [DOI] [PubMed] [Google Scholar]
- 50.Han G, Martinez LR, Mihu MR, Friedman AJ, Friedman JM, Nosanchuk JD. 2009. Nitric oxide releasing nanoparticles are therapeutic for Staphylococcus aureus abscesses in a murine model of infection. PLoS One 4:e7804. doi: 10.1371/journal.pone.0007804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simon SL, Richardson K, Dacey J, Glynn S, Domier CP, Rawson RA, Ling W. 2001. A comparison of patterns of methamphetamine and cocaine use. J Addict Dis 21:35–44. doi: 10.1300/J069v21n01_04. [DOI] [PubMed] [Google Scholar]
- 52.Han G, Nguyen LN, Macherla C, Chi Y, Friedman JM, Nosanchuk JD, Martinez LR. 2012. Nitric oxide-releasing nanoparticles accelerate wound healing by promoting fibroblast migration and collagen deposition. Am J Pathol 180:1465–1473. doi: 10.1016/j.ajpath.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 53.Conlon KM, Humphreys H, O’Gara JP. 2002. Regulation of icaR gene expression in Staphylococcus epidermidis. FEMS Microbiol Lett 216:171–177. doi: 10.1111/j.1574-6968.2002.tb11432.x. [DOI] [PubMed] [Google Scholar]
- 54.Melega WP, Cho AK, Harvey D, Laćan G. 2007. Methamphetamine blood concentrations in human abusers: application to pharmacokinetic modeling. Synapse 61:216–220. doi: 10.1002/syn.20365. [DOI] [PubMed] [Google Scholar]
- 55.Cho AK, Melega WP, Kuczenski R, Segal DS. 2001. Relevance of pharmacokinetic parameters in animal models of methamphetamine abuse. Synapse 39:161–166. doi:. [DOI] [PubMed] [Google Scholar]
- 56.Harris D, Boxenbaum H, Everhart ET, Sequeira G, Mendelson JE, Jones RT. 2003. The bioavailability of intranasal and smoked methamphetamine. Clin Pharmacol Ther 74:475–486. doi: 10.1016/j.clpt.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 57.Riviere GJ, Gentry WB, Owens SM. 2000. Disposition of methamphetamine and its metabolite amphetamine in brain and other tissues in rats after intravenous administration. J Pharmacol Exp Ther 292:1042–1047. http://jpet.aspetjournals.org/content/292/3/1042.long. [PubMed] [Google Scholar]
- 58.Scherr TD, Hanke ML, Huang O, James DBA, Horswill AR, Bayles KW, Fey PD, Torres VJ, Kielian T. 2015. Staphylococcus aureus biofilms induce macrophage dysfunction through leukocidin AB and alpha-toxin. mBio 6:e0102-15. doi: 10.1128/mBio.01021-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chandra J, Mukherjee PK, Ghannoum MA. 2008. In vitro growth and analysis of Candida biofilms. Nat Protoc 3:1909–1924. doi: 10.1038/nprot.2008.192. [DOI] [PubMed] [Google Scholar]
- 60.Anderson MJ, Lin Y, Gillman AN, Parks PJ, Schlievert PM, Peterson ML. 2012. Alpha-toxin promotes Staphylococcus aureus mucosal biofilm formation. Front Cell Infect Microbiol 2:64. doi: 10.3389/fcimb.2012.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zwart W, Griekspoor A, Kuijl C, Marsman M, van Rheenen J, Janssen H, Calafat J, van Ham M, Janssen L, van Lith M, Jalink K, Neefjes J. 2005. Spatial separation of HLA-DM/HLA-DR interactions within MIIC and phagosome-induced immune escape. Immunity 22:221–233. doi: 10.1016/j.immuni.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 62.Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, Fierer J, Nizet V. 2005. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med 202:209–215. doi: 10.1084/jem.20050846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Silveira CP, Piffer AC, Kmetzsch L, Fonseca FL, Soares DA, Staats CC, Rodrigues ML, Schrank A, Vainstein MH. 2013. The heat shock protein (Hsp) 70 of Cryptococcus neoformans is associated with the fungal cell surface and influences the interaction between yeast and host cells. Fungal Genet Biol 60:53–63. doi: 10.1016/j.fgb.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 64.Steenbergen JN, Shuman HA, Casadevall A. 2001. Cryptococcus neoformans interactions with amoebae suggest an explanation for its virulence and intracellular pathogenic strategy in macrophages. Proc Natl Acad Sci U S A 98:15245–15250. doi: 10.1073/pnas.261418798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McHugh D, Tanner C, Mechoulam R, Pertwee RG, Ross RA. 2008. Inhibition of human neutrophil chemotaxis by endogenous cannabinoids and phytocannabinoids: evidence for a site distinct from CB1 and CB2. Mol Pharmacol 73:441–450. doi: 10.1124/mol.107.041863. [DOI] [PubMed] [Google Scholar]
- 66.Gandhi JA, Ekhar VV, Asplund MB, Abdulkareem AF, Ahmadi M, Coelho C, Martinez LR. 2014. Alcohol enhances Acinetobacter baumannii-associated pneumonia and systemic dissemination by impairing neutrophil antimicrobial activity in a murine model of infection. PLoS One 9:e95707. doi: 10.1371/journal.pone.0095707. [DOI] [PMC free article] [PubMed] [Google Scholar]