

ABSTRACT
The group A Streptococcus (GAS; Streptococcus pyogenes) causes more than 700 million human infections each year. The success of this pathogen can be traced in part to the extensive arsenal of virulence factors that are available for expression in temporally and spatially specific manners. To modify the expression of these virulence factors, GAS use both protein- and RNA-based regulators, with the best-characterized RNA-based regulator being the small regulatory RNA (sRNA) FasX. FasX is a 205-nucleotide sRNA that contributes to GAS virulence by enhancing the expression of the thrombolytic secreted virulence factor streptokinase and by repressing the expression of the collagen-binding cell surface pili. Here, we have expanded the FasX regulon, showing that this sRNA also negatively regulates the expression of the adhesion- and internalization-promoting, fibronectin-binding proteins PrtF1 and PrtF2. FasX posttranscriptionally regulates the expression of PrtF1/2 through a mechanism that involves base pairing to the prtF1 and prtF2 mRNAs within their 5′ untranslated regions, overlapping the mRNA ribosome-binding sites. Thus, duplex formation between FasX and the prtF1 and prtF2 mRNAs blocks ribosome access, leading to an inhibition of mRNA translation. Given that FasX positively regulates the expression of the spreading factor streptokinase and negatively regulates the expression of the collagen-binding pili and of the fibronectin-binding PrtF1/2, our data are consistent with FasX functioning as a molecular switch that governs the transition of GAS between the colonization and dissemination stages of infection.
IMPORTANCE More than half a million deaths each year are a consequence of infections caused by GAS. Insights into how this pathogen regulates the production of proteins during infection may facilitate the development of novel therapeutic or preventative regimens aimed at inhibiting this activity. Here, we have expanded insight into the regulatory activity of the GAS small RNA FasX. In addition to identifying that FasX reduces the abundance of the cell surface-located fibronectin-binding proteins PrtF1/2, fibronectin is present in high abundance in human tissues, and we have determined the mechanism behind this regulation. Importantly, as FasX is the only mechanistically characterized regulatory RNA in GAS, it serves as a model RNA in this and related pathogens.
INTRODUCTION
The group A Streptococcus (GAS; Streptococcus pyogenes) is a Gram-positive obligate human pathogen. Infections caused by GAS range from mild and generally self-limiting pharyngitis (a.k.a. strep throat) or impetigo (a.k.a. school sores) to the serious and potentially life-threatening streptococcal toxic shock syndrome and necrotizing fasciitis (a.k.a. the flesh-eating disease) (1). In addition, postinfection sequelae can occur following untreated or poorly treated pharyngeal infection, including acute streptococcal glomerulonephritis and acute rheumatic fever (2). The ability of GAS to establish such a variety of disease states is in part a consequence of the regulated expression of distinct virulence factor profiles (3, 4).
GAS can colonize multiple anatomic sites, with this flexible tissue tropism being determined by the degree and type of adhesins expressed, which in turn is controlled by transcriptional and posttranscriptional regulatory mechanisms (5, 6). Fibronectin, a ubiquitous extracellular matrix (ECM) protein, is a common binding target for GAS adhesins, including proteins F1 (PrtF1; also known as SfbI) (7–9) and F2 (PrtF2; also known as FbaB or Pfbp) (10–12), and this functional redundancy is thought to emphasize the critical nature of this adhesive activity. However, during GAS dissemination the expression of fibronectin-binding proteins can be a stearic hindrance, as they may promote GAS entanglement in neutrophil extracellular traps (NETs) (13, 14) as well as the ECM itself, or other host defenses, such as platelet aggregations (15, 16). Thus, as with any dynamic system, the pathogen's ability to downregulate the expression of these virulence factors is as critical as the ability to upregulate their expression.
Small regulatory RNAs (sRNAs) are a key class of regulatory molecule that are used by most bacterial species to posttranscriptionally regulate gene expression (17). The vast majority of sRNAs are noncoding, with the RNA molecule itself being the regulatory factor, most commonly by base pairing to target mRNA molecules to modify their stability and/or translation (18, 19). To date only one bona fide sRNA has been described in GAS, the 205-nucleotide (nt) FasX sRNA (20). FasX has both positive and negative regulatory targets, and the regulation afforded by FasX contributes to the virulence of this pathogen in a mouse soft-tissue infection model (21). FasX positively regulates the expression of the thrombolytic agent streptokinase (SKA) by binding to the extreme 5′ end of ska mRNA, forming a secondary structure that leads to an enhancement in ska mRNA stability and, subsequently, SKA abundance (22). FasX negatively regulates the expression of GAS pili by, in a serotype-specific manner, binding to the extreme 5′ end of mRNAs encoding either a minor or the major pilus proteins and inhibiting their translation by blocking access to the ribosome binding site (21, 23).
In this work, we have investigated the posttranscriptional influence of FasX on the expression of the functionally similar, yet physically dissimilar, fibronectin-binding proteins PrtF1 and PrtF2. This study demonstrates that FasX negatively regulates PrtF1 and PrtF2 expression by directly base pairing to their respective mRNA ribosome binding sites, thereby occluding them from translation initiation, similar to the mechanism by which FasX negatively regulates pilus expression. Thus, FasX acts as a negative regulator of not only the collagen-binding pili but also the fibronectin-binding PrtF1 and PrtF2. We propose that FasX acts as a master regulator that controls the ability of GAS to transition between colonization and dissemination.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The GAS strains used in this study are listed in Table 1. Routine growth of liquid GAS cultures made use of Todd-Hewitt broth with 0.2% yeast extract (THY broth), and cultures were incubated statically at 37°C (5% CO2). Chloramphenicol (4 μg/ml) and/or spectinomycin (150 μg/ml) were added when required.
TABLE 1.
GAS strains used in this study
| Strain name | Description | Reference or source |
|---|---|---|
| MGAS6180 | Serotype M28 clinical GAS isolate that has been genome sequenced | 29 |
| M28 + pDCBB | MGAS6180 containing the empty shuttle vector pDCBB | 21 |
| M28ΔFasX + vector | MGAS6180 derivative in which the fasX gene has been replaced with a spectinomycin resistance cassette; also contains empty shuttle vector pDCBB | 21 |
| M28ΔFasX + pFasX | M28ΔFasX containing the pDCBB derivative pFasX, which contains a wild-type fasX allele downstream of the natural promoter | 21 |
| M28ΔFasX + pFasX.UC.loop | M28ΔFasX containing the pDCBB derivative pFX.C71/73G, in which the C nucleotides at positions 71 and 73 have been mutated to G | This work |
| M28ΔFasX + pFasX.ska.loop | M28ΔFasX containing the pDCBB derivative pFXC45/46G, in which the C nucleotides at positions 45 and 46 have been mutated to G | This work |
| M28ΔprtF1 + vector | MGAS6180 derivative in which the prtF1 gene has been replaced by a spectinomycin resistance cassette; also contains empty vector pDCBB | This work |
| M28ΔprtF2 + vector | MGAS6180 derivative in which the prtF2 gene has been replaced by a spectinomycin resistance cassette; also contains empty vector pDCBB | This work |
Creation of the MGAS6180 mutant derivatives M28ΔprtF1 and M28ΔprtF2.
Isogenic MGAS6180 prtF1 and prtF2 mutant strain derivatives were created by the replacement of these genes with a nonpolar spectinomycin resistance cassette. This was achieved via a PCR overlap extension-based approach, as described previously (22, 24). Primers used in the construction of the mutant strains are listed in Table 2. Confirmation that these mutants were correctly constructed was gained by PCR and targeted sequencing.
TABLE 2.
Primers and probes used in this study
| Primer name | Sequence (5′–3′) | Description |
|---|---|---|
| UNR50 | CTGGTTTGGGGAATAGTTACCGAAAATTG | Used in the creation of an M28 prtF1 mutant |
| UNR51 | CGTTATTAGTTATAGTTATTATAACATGTATTGTGTGCCAAAAAACCGGCTTCTTTATTC | Used in the creation of an M28 prtF1 mutant |
| UNR52 | CTATTTAAATAACAGATTAAAAAAATTATAACAAACAAAACAATAAAGTCTGACGTAAAAG | Used in the creation of an M28 prtF1 mutant |
| UNR53 | GCAATTCTATGACCTCTGACCAATAG | Used in the creation of an M28 prtF1 mutant |
| UNR56 | GAATAAAGAAGCCGGTTTTTTGGCACACAATACATGTTATAATAACTATAACTAATAACG | Used in the creation of an M28 prtF1 mutant |
| UNR57 | CTTTTACGTCAGACTTTATTGTTTTGTTTGTTATAATTTTTTTAATCTGTTATTTAAATAG | Used in the creation of an M28 prtF1 mutant |
| UNR238 | CTATTTAAATAACAGATTAAAAAAATTATAACAACTATGGAGTTGCGTGATTCATCTGGTAAAAC | Used in the creation of an M28 prtF2 mutant |
| UNR239 | ATGAGCATCATTATGGGAACGGTACAGTTG | Used in the creation of an M28 prtF2 mutant |
| UNR240 | CAAGTGCCATATAGGACAAGAGCTCTTC | Used in the creation of an M28 prtF2 mutant |
| UNR241 | CGTTATTAGTTATAGTTATTATAACATGTATTGACTGATACCTTTAGGATTGAG | Used in the creation of an M28 prtF2 mutant |
| UNR244 | GTTTTACCAGATGAATCACGCAACTCCATAGTTGTTATAATTTTTTTAATCTGTTATTTAAATAG | Used in the creation of an M28 prtF2 mutant |
| UNR245 | CTCAATCCTAAAGGTATCAGTCAATACATGTTATAATAACTATAACTAATAACG | Used in the creation of an M28 prtF2 mutant |
| M28.105TMF | ACTCCCGAGTTGGATGGTAGTC | TaqMan primer for M28 prtF1 |
| M28.105TMR | CTGGCGGCAATGTAGGTTCT | TaqMan primer for M28 prtF1 |
| M28.105TMP | ATTCCCGAAGACCCAAAACATCCAGATGAT | TaqMan probe for M28 prtF1 |
| M28.113TMF | CAAACACACAAGGAGAGGTTCATT | TaqMan primer for M28 prtF2 |
| M28.113TMR | TCACCTGCTGATAGCCTTTAGGT | TaqMan primer for M28 prtF2 |
| M28.113TMP | CCTGACCTCGGGCACCTACGACTTG | TaqMan probe for M28 prtF2 |
| tufATMF | AACTACTTTAACAGCTGCAATCACAACT | TaqMan primer for tufA |
| tufATMR | AGAAGCGTAATCTTTTGGTTGGTT | TaqMan primer for tufA |
| tufATMP | TATTGGCACGTCGCTTGCCTTCATC | TaqMan probe for tufA |
| prtF1.99 | CCATAGCATGAACTTTAAAAGTTTC | prtF1 gene-specific primer 1, 5′-RACE protocol |
| prtF1.GSPR1 | TTAAAATTCGGCACAGTCTTCTC | prtF1 gene-specific primer 2, 5′-RACE protocol |
| prtF2.98 | GCTTGAGGTAAAGGTCTGAACAGG | prtF2 gene-specific primer 1, 5′-RACE protocol |
| prtF2.GSPR1 | CTTTGGTGTCAGTGAAAAAGTT | prtF2 gene-specific primer 2, 5′-RACE protocol |
| UNR66 | TCGTAAAAGGTCGAGGATAG | RT-PCR R primer to characterize prtF1 TSS |
| UNR114 | ATGAATAACAAAATATTTTTGAATAAAG | RT-PCR F1 primer to characterize prtF1 TSS |
| UNR115 | GTTTTTGAGAGGAGAGAAAATG | RT-PCR F2 primer to characterize prtF1 TSS |
| UNR116 | AATAACGTGGTAAGCTCATATAT | RT-PCR F3 primer to characterize prtF1 TSS |
| UNR67 | GTCCCTGGTTGGAGATTTTGAG | RT-PCR R primer to characterize prtF2 TSS |
| UNR120 | ATGACACAAAAAAATAGCTATAAG | RT-PCR F1 primer to characterize prtF2 TSS |
| UNR121 | TGACAGTTGTCCTGTAGTCTTTAG | RT-PCR F2 primer to characterize prtF2 TSS |
| UNR122 | GACAACTGAAAATGGTAAAATAACTATT | RT-PCR F3 primer to characterize prtF2 TSS |
| UNR124 | CTTAATACGACTCACTATAGGTTTTTGAGAGGAGAGAAAATGAATAAC | Primer used with UNR125 for prtF1 EMSA template and with UNR215 for prtF1.FLAG in vitro translation template |
| UNR125 | CCTTTTTCTTTTTGTGTGTGC | Primer to amplify the 5′ end of prtF1 and place downstream of T7 promoter for use in in vitro transcription |
| UNR128 | CTTAATACGACTCACTATAGGTGACAGTTGTCCTGTAGTCTTTAG | Primer used with UNR129 for prtF2 EMSA template and with UNR216 for prtF2.FLAG in vitro translation template |
| UNR129 | CAGGAAGCTTAACTTATAGC | Primer to amplify the 5′ end of prtF2 and place downstream of T7 promoter for use in in vitro transcription |
| UNR215 | ATATGAGTAAACTTGGTCTGACAGTCATTTATCATCGTCATCTTTATAATCATTATTGCCGTCATTAGGGTAGCCGTTATAC | Used with UNR124 to amplify the 5′ end of prtF1 to encode a T7 promoter upstream and FLAG tag downstream for use in in vitro transcription and translation assays |
| UNR216 | ATATGAGTAAACTTGGTCTGACAGTCATTTATCATCGTCATCTTTATAATCCATTGTTGATTTTTCCAACTG | Used with UNR128 to amplify the 5′ end of prtF2 to encode a T7 promoter upstream and FLAG tag downstream for use in in vitro transcription and translation assays |
| SPDTRN | ATATGAGTAAACTTGGTCTGACAGCTATTTATCATCGTCATCTTTATAATCTCCGACATAAGATAGACCATTTAACC | Primer to create template for in vitro transcription of spd.FLAG RNA |
| T7SPD | CTTAATACGACTCACTATAGGGTTAGTGAGCGAAATTAGAAAAGAGG | Primer to create template for in vitro transcription of spd.FLAG RNA |
| FASEND9 | AAAAAACCCGGCAAGCCGGGCT | Primer to create template for in vitro transcription of FasX and mutant FasX |
| UNR123 | CTTAATACGACTCACTATAGGTAAATAAAGATTTACGAAGTC | Used with FASEND9 to amplify fasX with a T7 polymerase site at the 5′ end to enable in vitro transcription of FasX |
| DC123 ECORV | CCTTATTAACATTCACAAC | Primer used in conjunction with DC123 BglII for sequencing plasmid-carried mutant fasX alleles |
| DC123 BGLII | TATCATCCACTCAAGACTTTTGAC | Primer used in conjunction with DC123 EcoRV for sequencing plasmid-carried mutant fasX alleles |
Complementation of strain M28ΔFasX with pFasX.ska.loop and pFasX.UC.loop.
To investigate whether we could separate the streptokinase-regulating effects of FasX from the adhesin-regulating effects, we created two derivatives of the FasX-complementing plasmid pFasX. Plasmid pFasX.ska.loop contains two single-nucleotide polymorphisms (SNPs) which alter two of the C nucleotides that base pair with ska mRNA, changing them into G nucleotides. Plasmid pFasX.UC.loop also contains two SNPs, altering two of the C nucleotides that base pair with the adhesin-encoding mRNAs (prtF1, prtF2, and pilus), changing them into G nucleotides. These plasmids were transformed into the isogenic MGAS6180 fasX mutant strain M28ΔFasX (21) for comparison.
Quantitative RT-PCR analysis.
RNA was isolated from GAS cultures grown to an optical density at 600 nm (OD600) of 0.5 (exponential phase) as previously described (22). Isolated RNA was triple DNase treated (TURBO-DNase; Life Technologies) and used in cDNA synthesis reactions using the reverse transcriptase Superscript III (Life Technologies) per the manufacturer's instructions. Using the CFX Connect real-time system (Bio-Rad), TaqMan quantitative reverse transcription-PCR (RT-PCR) was performed via the ΔΔCT method (24), where CT is threshold cycle. Primers and TaqMan probes for the genes of interest, and the internal control gene tufA, are listed in Table 2.
Isolation of secreted protein fractions.
Aliquots (10 ml) were recovered from GAS strains grown in THY broth to an OD600 of 0.5 (midexponential phase), and the cells were pelleted by centrifugation (5,000 × g for 10 min). The supernatant then was collected through a 0.22-μm filter into 35 ml of 100% ethanol and precipitated overnight at −20°C. This solution subsequently was pelleted as described above, the supernatant discarded, and the pellet resuspended in 500 μl of SDS-PAGE buffer (containing 2-mercaptoethanol) for analysis by Western blotting.
Western blot analysis of GAS secreted proteins.
Protein samples were electrophoresed through 12% SDS-PAGE gels and transferred by semidry electroblotting to nitrocellulose membranes. Western blot analyses were performed using polyclonal sheep antistreptokinase and rabbit anti-SpeC as the primary antibodies (used at 1:2,000 dilution). SpeC is a GAS secreted protein unregulated by FasX; therefore, its expression was used as a loading control. After washing, blots were probed with anti-sheep (streptokinase) or anti-rabbit (SpeC) horseradish peroxidase (HRP)-conjugated secondary antibody at a concentration of 1:20,000. Blots were developed using the SuperSignal west femto maximum sensitivity kit (ThermoFisher), and chemiluminescence was captured using a G-Box Chemi XT4 (Syngene).
Isolation of cell wall protein fractions.
Forty-milliliter aliquots were recovered from GAS strains grown in THY broth to an OD600 of 0.5 (exponential phase), and the cells were pelleted by centrifugation (5,000 × g for 11 min). The cells were washed once with 10 ml TE buffer before resuspending in 1 ml of TE-sucrose buffer (48 mM Tris-HCl, pH 7.5, 1 mM EDTA, 0.55 M sucrose, 1 mg/ml lysozyme, 250 μg/ml mutanolysin, 250 μg/ml hyaluronidase). Samples were incubated at 37°C with end-to-end rotation in 1.5-ml tubes for 2 h and centrifuged at 15,000 × g for 5 min to pellet the protoplasts, and the supernatants were removed to clean 1.5-ml tubes. The centrifugation was repeated to pellet any insoluble material carried over, and the final supernatant was analyzed by Western blotting.
Western blot analysis of GAS cell wall proteins.
Protein samples were separated on 6% SDS-PAGE gels. Membranes were stained using the MemCode reversible protein stain kit (Pierce) for use as a loading control. Subsequently, Western blot analyses were performed using polyclonal rabbit anti-PrtF1 or anti-PrtF2 as the primary antibody. After washing, blots were probed with Alexa Fluor 680-conjugated donkey anti-rabbit IgG antibody (1:10,000 dilution; Molecular Probes), and the fluorescent signal was detected using the Odyssey system (Li-Cor).
5′-RACE analysis of the prtF1 and prtF2 TSS.
The transcriptional start sites (TSS) of prtF1 and prtF2 were identified by utilizing the 5′ system for rapid amplification of cDNA ends kit (5′-RACE; v2; Invitrogen) according to the manufacturer's specifications. Briefly, 3 μg of total RNA was used for first-strand cDNA synthesis, generated by reverse transcriptase, using the gene-specific primers for prtF1 and prtF2 that are listed in Table 2. These cDNAs were RNase treated and column purified, and their 3′ ends were tagged using terminal deoxynucleotidyl transferase and dCTP to create homopolymeric tails of C nucleotides. These products then were amplified using one primer complementary to the dC tail and one primer specific to the prtF1 and prtF2 genes; amplicons were gel extracted and cloned into E. coli (TOPO TA; Invitrogen) for sequencing analysis to identify the transcriptional start sites.
RT-PCR analysis of the prtF1 and prtF2 transcriptional start sites.
Total RNA was isolated, treated with TURBO-DNase (Life Technologies), and used to generate cDNA with the reverse transcriptase SuperScript III (Invitrogen). The generated cDNA was used in RT-PCR analysis to verify the putative transcriptional start sites of prtF1 and prtF2 that were identified by the 5′-RACE method (see Fig. 4A and B). For both prtF1 and prtF2, a single reverse primer was designed that is located within each gene (primer R in Fig. 4C). The first forward primer (F1 in Fig. 4C) begins at the AUG start codon of prtF1 and prtF2, which should produce an amplicon in both our cDNA and genomic DNA (gDNA) reactions. The second forward primer (F2 in Fig. 4C) was designed to begin at the putative transcriptional start site identified by 5′-RACE, and as before, this reaction would be anticipated to produce an amplicon in both cDNA and gDNA reactions. The third forward primer (F3 in Fig. 4C) is designed to end immediately upstream of the identified transcriptional start site; therefore, it would only be expected to amplify the gDNA template. Water and the product of the cDNA synthesis reaction performed without reverse transcriptase were used as negative PCR controls.
FIG 4.
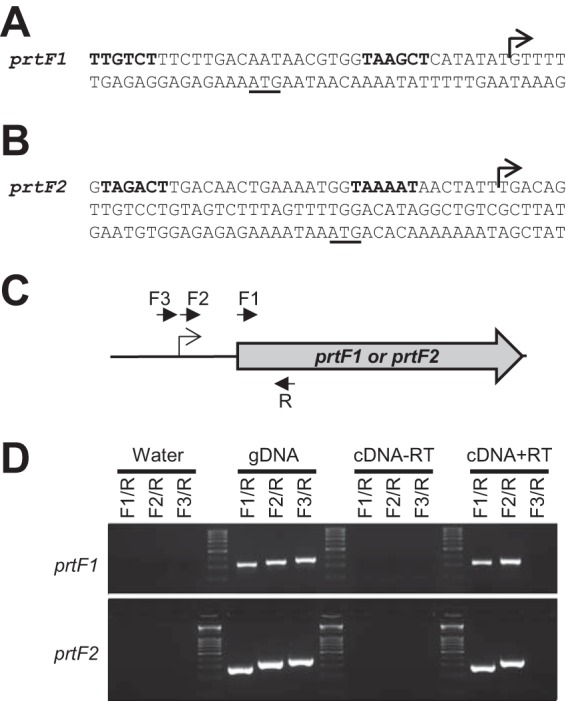
Determination of the transcriptional start sites of prtF1 and prtF2. The transcriptional start sites of prtF1 and prtF2 were determined through 5′-RACE (A and B) and confirmed through RT-PCR analysis (C and D). The transcriptional start sites identified for prtF1 and prtF2 are highlighted with bent arrows in panels A and B, with putative −10 and −35 promoter sequences in boldface and the ATG start codon underlined. Panel C shows a schematic of the relative locations of the PCR primers used in the RT-PCR analysis depicted in panel D. The R primers (one per gene) are located within the prtF1 and prtF2 genes. The F1 primers are located at the translational start site of each gene. The 5′ ends of the F2 primers are located at the putative transcriptional start sites identified by 5′-RACE. The 3′ ends of the F3 primers are located immediately upstream of the F2 primers. Panel D shows the RT-PCR data gained using the primers depicted in panel C in conjunction with water (negative control), gDNA (positive control), cDNA generated from a no-reverse-transcriptase reaction (cDNA-RT; to control against contaminating DNA in the isolated RNA), and a plus reverse transcriptase cDNA synthesis reaction (cDNA+RT; our test conditions). Amplification in the cDNA+RT reaction mixtures containing primers F2/R but not primers F3/R is consistent with the 5′-RACE analysis correctly identifying the prtF1 and prtF2 transcriptional start sites.
In vitro transcription reactions.
In vitro-transcribed RNAs for use in the RNA-RNA electrophoretic mobility assays (EMSAs) (see Fig. 5 and 6) and the in vitro translation experiments (see Fig. 8) were created using the MEGAshortscript (FasX and the deletion mutant derivative FasXΔ72-77) and MEGAscript (prtF1.FLAG, prtF2.FLAG, and spd.FLAG mRNAs) kits (Life Technologies) in accordance with the manufacturer's recommendations. Template DNAs for use in the in vitro transcription reactions were generated by PCR and encode a T7 promoter sequence designed in the 5′ primer to allow transcription using T7 RNA polymerase (Table 2 lists primer sequences). The template DNA was removed using TURBO DNase (Life Technologies) following the in vitro transcription reactions, and the RNA products were purified using the RNA Clean & Concentrator-25 kit (Zymo Research). The RNA6000 Nano chip on the Agilent Bioanalyzer 2100 system was used to qualitatively and quantitatively evaluate the purified RNA.
FIG 5.
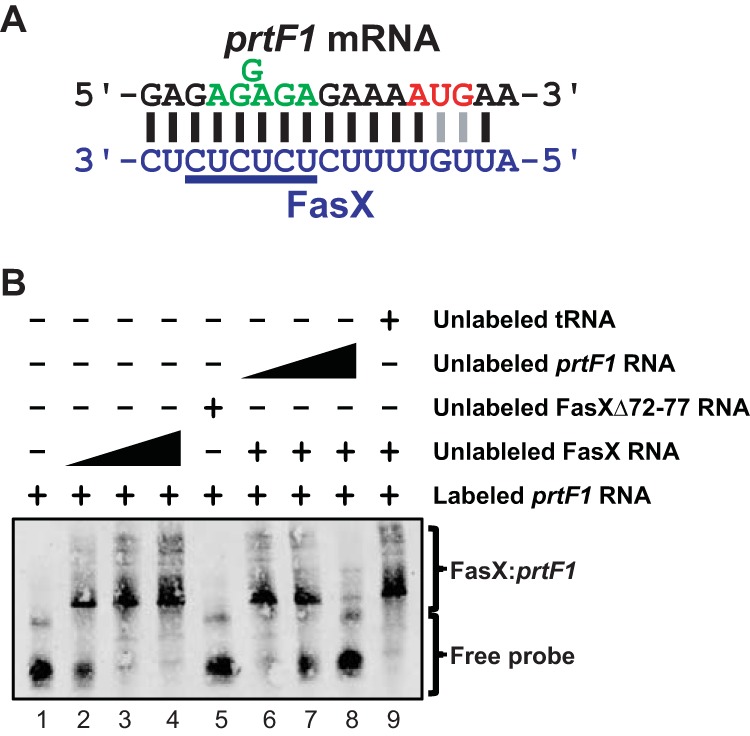
FasX base pairs to the 5′-UTR of prtF1. (A) Identified region of complementarity between FasX and the 5′ region of prtF1 mRNA. The mRNA start codon (red) and ribosome-binding site (green) are highlighted. Lines between nucleotides of the mRNA and FasX highlight standard (Watson-Crick; black) and nonstandard (G-U wobble; gray) base-pairing interactions. (B) RNA-RNA electrophoretic mobility shift assays verifying base pairing between FasX and the 5′ end of prtF1 mRNA in M28 GAS. A biotin-labeled RNA probe was incubated with wild-type FasX (FasX RNA; lanes 2 to 4 and 6 to 9), a FasX mutant in which six of the complementary nucleotides had been deleted (FasXΔ72-77 RNA; lane 5), unlabeled probe RNA (lanes 6 to 8), and/or unlabeled yeast tRNA (lane 9).
FIG 6.
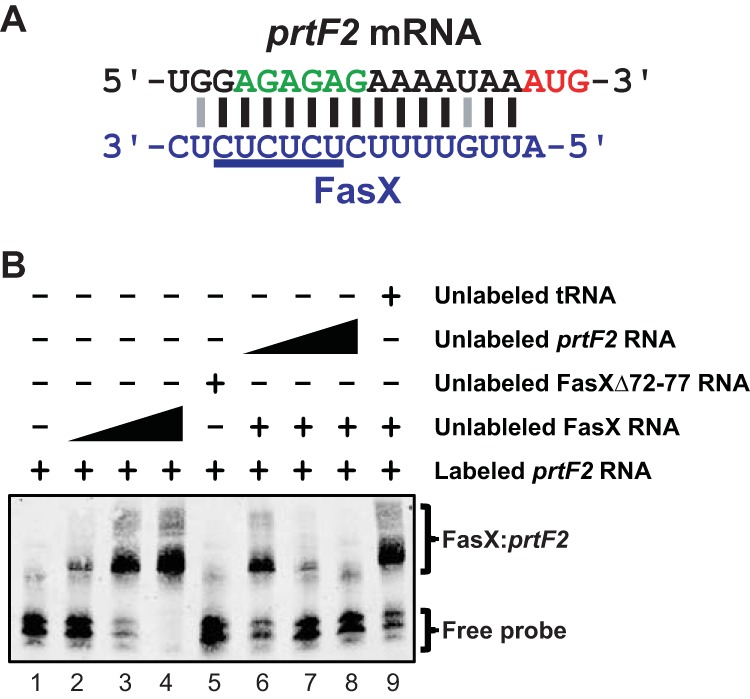
FasX base pairs to the 5′-UTR of prtF2. (A) Identified region of complementarity between FasX and the 5′ region of prtF2 mRNA. The mRNA start codon (red) and ribosome-binding site (green) are highlighted. Lines between nucleotides of the mRNA and FasX highlight standard (Watson-Crick; black) and nonstandard (G-U wobble; gray) base-pairing interactions. (B) RNA-RNA electrophoretic mobility shift assays verifying base pairing between FasX and the 5′ end of prtF2 mRNA in M28 GAS. A biotin-labeled RNA probe was incubated with wild-type FasX (FasX RNA; lanes 2 to 4 and 6 to 9), a FasX mutant in which six of the complementary nucleotides had been deleted (FasXΔ72-77 RNA; lane 5), unlabeled RNA probe (lanes 6 to 8), and/or unlabeled yeast tRNA (lane 9).
FIG 8.
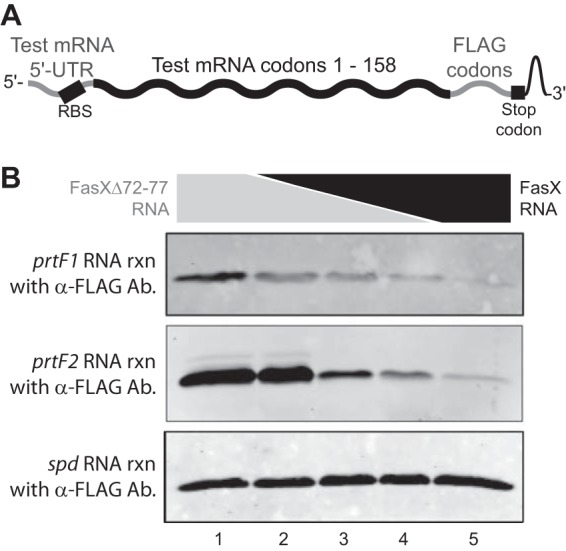
FasX inhibits PrtF1 and PrtF2 expression at the level of mRNA translation. (A) Schematic showing the individual components of each of the three constructed chimeric mRNAs. (B) Western blot analyses of in vitro translation reactions (rxn). A total of 10 pmol of prtF1.FLAG, prtF2.FLAG, or spd.FLAG mRNA was preincubated with 50 pmol of RNA composed of different ratios of FasX and the deletion mutant derivative FasXΔ72-77. Following the in vitro translation reactions, the products were used in Western blot analysis. In lanes 1 to 5, the ratios of FasXΔ72-77 to FasX (in picomoles) present in the reaction mixtures were 50:0, 40:10, 25:25, 10:40, and 0:50, respectively. The spd.FLAG mRNA was used as a negative control for FasX-mediated regulation. Ab, antibody.
RNA-RNA EMSA.
In vitro-transcribed RNAs consisting of the first ∼150 nucleotides of the prtF1 and prtF2 mRNAs (the untranslated regions [UTRs] plus >100 nucleotides of coding sequence), as well as all of FasX (FasX RNA; 205 nt) and FasXΔ72-77 (FasXΔ72-77 RNA; 199 nt), were produced. Aliquots of the in vitro-transcribed prtF1 and prtF2 mRNAs were biotin labeled using the Pierce RNA 3′ end biotinylation kit (Thermo Scientific) in order to generate the probes for these assays and then cleaned up using an RNA Clean & Concentrator-5 column (Zymo Research). The RNA6000 Nano chip on the Agilent Bioanalyzer 2100 system was used to qualitatively evaluate and quantify the labeled mRNA probes. To perform the EMSA reactions, labeled mRNAs (15 nM) were incubated in the presence or absence of FasX RNA (0, 0.84, 8.4, and 84 nM), FasXΔ72-77 RNA (0 or 31 nM), unlabeled mRNA (0, 6, 63, 630 nM), and/or unlabeled yeast RNA (0 or 1000 nM). Reactions were performed as previously described (23), the products of which were electrophoresed through a 5% Tris-borate-EDTA (TBE) minigel, transferred by semidry electroblotting to a positively charged nylon membrane, and then cross-linked by UV. The RNA-bound membrane then was blocked for 1 h (Odyssey blocking buffer; Li-Cor), incubated with Streptavidin IRDye (Molecular Probes) at room temperature for 20 min, and washed, and the labeled RNA was detected using the Odyssey system (Li-Cor).
In vitro translation.
Chimeric mRNAs were in vitro transcribed, consisting of ∼550 nucleotides that contain the 5′-UTRs and 5′ coding regions of the spd, prtF1, and prtF2 genes. The mRNAs were designed so as to incorporate a FLAG tag at the C terminus of the encoded proteins, allowing for the examination of the in vitro translation products via Western blot analysis using an anti-FLAG antibody. Briefly, 10 pmol of prtF1.FLAG, prtF2.FLAG, or spd.FLAG mRNA was incubated with 50 pmol of RNA composed of different ratios of FasX and FasXΔ72-77. The ratio of FasXΔ72-77 to FasX present in the reaction mixtures were 5:0, 4:1, 1:1, 1:4, and 0:5 in lanes one to five, respectively. We then completed in vitro translation assays using the E. coli S30 extract system for linear templates as previously described (23). Proteins were electrophoresed through 12% SDS-PAGE gels, transferred to nitrocellulose membranes in a Trans-Blot semi-dry transfer cell (Bio-Rad), and probed with rabbit anti-FLAG antibodies (1:2,000 dilution; Sigma). An Alexa Fluor 680–donkey anti-rabbit secondary antibody (1:10,000; Life Technologies) was used for fluorescent visualization, and the signal was captured using the Odyssey system (Li-Cor).
RESULTS
Genes encoding the fibronectin-binding proteins PrtF1 and PrtF2 are located within the FCT pathogenicity island in serotype M28 GAS.
Previously, we reported that GAS uses the FasX sRNA to negatively regulate the expression of the collagen-binding pilus (23). The pilus biosynthesis genes are located in a variable region of the genome termed the fibronectin-/collagen-binding/T-antigen (FCT) region. Currently, nine distinct FCT regions have been described among the different GAS serotypes, with these differing in both gene content and gene order (26). Serotype M28 GAS strains harbor FCT-4, which, in addition to encoding the pilus biosynthesis genes, also encodes PrtF1 and PrtF2 (Fig. 1). The prtF1 and/or prtF2 genes are encoded by approximately half of all GAS serotypes (27, 28). Given that FasX negatively regulates the expression of pili from the FCT region, we hypothesized that FasX also negatively regulates, via a posttranscriptional mechanism, the expression of PrtF1 and PrtF2. This hypothesis is consistent with the initial FasX study where it was identified that, through unknown mechanisms, a FasX mutant strain bound fibronectin at a higher level than the parental strain (20).
FIG 1.

Schematic of the FCT region from the serotype M28 GAS strain MGAS6180. Genes are represented by arrows facing the direction of transcription. Genes encoding transcription factors are colored red. Genes encoding pilus biosynthesis proteins are colored blue. Genes encoding fibronectin-binding proteins are colored green.
FasX negatively regulates the abundance of prtF1 and prtF2 mRNA transcripts.
To begin to test the hypothesis that FasX negatively regulates PrtF1 and PrtF2 expression, we performed quantitative RT-PCR analysis of prtF1 and prtF2 mRNAs. For this analysis we used the parental serotype M28 GAS isolate MGAS6180 containing empty vector (termed M28 + vector), its fasX mutant derivative M28ΔfasX containing empty vector (M28ΔfasX + vector), and the complemented mutant derivative containing a plasmid-carried wild-type fasX allele (M28ΔfasX + pFasX). We investigated mRNA levels as, for other FasX-regulated targets, changes in mRNA abundance have been observed regardless of whether the main mechanism of regulation is the modulation of mRNA stability (i.e., regulation of streptokinase) (22) or of mRNA translation (i.e., regulation of pilus) (21, 23). Relative to the parental strain, the fasX mutant had a small but statistically significant increase in the abundance of prtF2, but not prtF1, mRNA (Fig. 2). The complemented mutant strain, which overexpresses FasX due to pFasX being a multicopy plasmid, resulted in a statistically significant decrease of both transcripts (Fig. 2). The small but reproducible change in prtF1 and prtF2 mRNA abundance by FasX mirrors that observed for the pilus mRNAs (21, 23); this points to the primary mode of regulation of PrtF1 and PrtF2 expression by FasX as potentially also being at the level of mRNA translation.
FIG 2.
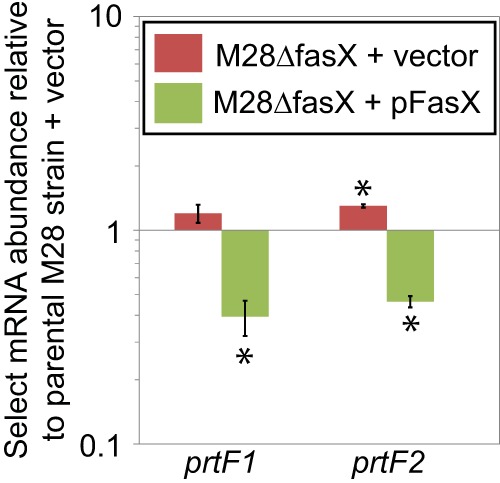
FasX negatively regulates the abundance of mRNAs encoding the fibronectin-binding proteins PrtF1 and PrtF2. Parental, fasX mutant, and complemented mutant strains of the serotype M28 GAS strain MGAS6180 were grown to mid-exponential phase, and RNA was isolated and used in quantitative RT-PCR analysis. Statistical significance was tested by t test with significant data points being highlighted by asterisks (P < 0.05 relative to the parental strain). Shown are the averages (±standard deviations) from three independent experiments.
FasX inhibits cell surface expression of PrtF1 and PrtF2.
To definitively identify whether FasX regulates PrtF1 and/or PrtF2 expression, we performed Western blot analyses of GAS cell wall protein fractions. In addition to using the parental, fasX mutant, and complemented mutant strains, we also created and utilized prtF1 (M28ΔprtF1 + vector) and prtF2 (M28ΔprtF2 + vector) mutant strains (as negative controls for the two antibodies). For both PrtF1 (Fig. 3A) and PrtF2 (Fig. 3B), the fasX mutant expressed higher levels of these proteins than the parental strain, while the FasX-overexpressing complemented strain reduced PrtF1 and PrtF2 expression below that observed in the parental strain. Thus, the data are consistent with FasX negatively regulating the expression of these fibronectin-binding proteins.
FIG 3.
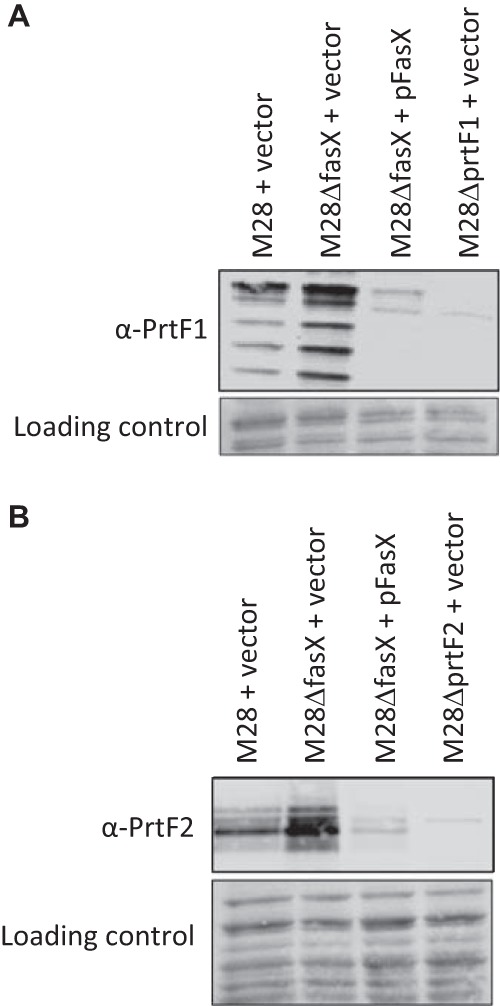
FasX negatively regulates the abundance of PrtF1 and PrtF2 on the GAS cell surface. Western blot analyses of cell wall protein fractions from the indicated GAS strains. Western blot analyses were performed using anti-PrtF1 (A) and anti-PrtF2 (B) antibodies. The protein-bound membranes were stained after transfer but prior to Western analysis for use as loading controls. Images shown are representative of five different experiments. Note that the laddering pattern of reactivity observed for PrtF1 is similar to that gained from a previous, unrelated analysis of this protein (59).
Determination of the prtF1 and prtF2 transcriptional start sites.
FasX downregulates the expression of other FCT region (pilus) genes via direct base pairing to target gene transcripts (21, 23). Hence, we hypothesized that a similar mechanism underlies the FasX-mediated negative regulation of PrtF1 and PrtF2. To facilitate testing of whether FasX base pairs to prtF1 and/or prtF2 mRNAs, we first required information as to the location of the transcriptional start sites of the prtF1 and prtF2 genes. Through the use of 5′-RACE (Fig. 4A and B) and RT-PCR (Fig. 4C and D) analyses, we identified that the transcriptional start site for prtF1 is nucleotide 116824 in the annotated MGAS6180 genome (the translational start site is nt 116843) (29), while the transcriptional start site for prtF2 is nucleotide 126920 (the translational start site is nt 126989). Note that our data regarding the prtF1 transcriptional start site differs by two nucleotides from a previously reported start site for this gene (30).
Putative FasX binding sites are located in the 5′-UTRs of prtF1 and prtF2 mRNAs.
To date, all FasX-regulated target mRNAs have been shown to bind to FasX through nucleotides contained within their 5′-UTRs (21–23). To investigate whether the same was true for prtF1 and prtF2, we used the transcriptional start site data described above to manually curate these sequences, searching for regions of complementarity to FasX. Strong regions of complementarity were observed in both the prtF1 (Fig. 5A) and prtF2 (Fig. 6A) mRNAs, and in both cases FasX binding would overlap the Shine-Dalgarno ribosome-binding sites, consistent with FasX potentially inhibiting the translation of these mRNAs. Of interest, the putative base pairing between FasX and prtF1 mRNA is identical to that observed between FasX and the FasX-regulated cpa (pilus) mRNA in M1 GAS (23), giving further credence to the hypothesis that FasX binds to prtF1 mRNA.
FasX forms an sRNA-mRNA complex with both prtF1 and prtF2 mRNAs.
To determine whether FasX directly base pairs to prtF1 and/or prtF2 mRNAs, we used RNA-RNA electrophoretic mobility shift assays (EMSAs). Biotin-labeled target RNAs corresponding to the 5′ ends of either prtF1 or prtF2 were incubated with increasing concentrations of FasX RNA, resulting in an increasing shift of the probe to higher-molecular-weight complexes (Fig. 5B and 6B, lanes 1 to 4). To verify that the FasX nucleotides highlighted in Fig. 5A and 6A were contributing to this sRNA-mRNA interaction, we also used a derivative of FasX in which six of the putative hybridizing nucleotides (those underlined in Fig. 5A and 6A) had been deleted (FasXΔ72-77) (23). FasXΔ72-77 failed to shift either of the probes (Fig. 5B and 6B, lanes 5), as expected. Finally, to confirm that the interactions between FasX and prtF1 and prtF2 mRNAs were specific, we identified that increasing concentrations of unlabeled probe RNAs could compete FasX away from the labeled probe RNAs (Fig. 5B and 6B, lanes 6 to 8), while high concentrations of an unrelated RNA could not (yeast tRNA) (Fig. 5B and 6B, lanes 9). These data are consistent with FasX selectively base pairing with both prtF1 and prtF2 mRNAs at their 5′-UTRs.
The streptokinase- and adhesin-regulating properties of FasX can be separated. The data shown here, in association with those previously published (21, 22, 23), is consistent with there being two distinct loops of the FasX molecule that are involved in the regulation of GAS virulence factors. One loop positively regulates streptokinase expression (Fig. 7A, red nucleotides), while a second loop negatively regulates the expression of PrtF1, PrtF2, and the GAS pilus (Fig. 7A, blue nucleotides). To test whether we could separate the streptokinase-regulating activity of FasX from the adhesin-regulating activity, we created two plasmid-encoded FasX derivatives that each were mutated for one of the two regulatory loops. These plasmids, pFasX.ska.loop and pFasX.UC.loop, were transformed into M28ΔFasX and compared with the parental M28 isolate, along with the fasX mutant and complemented mutant derivatives. The analysis was performed at the protein level via Western blot analyses using either secreted (SKA and SpeC) or cell wall (PrtF1 and PrtF2) protein fractions. As expected, the mutations present within pFasX.ska.loop prevented the positive regulation of streptokinase expression but did not prevent the negative regulation of adhesin expression (Fig. 7B). Similarly, the mutations present within pFasX.UC.loop prevented the negative regulation of adhesin expression but did not prevent the positive regulation of streptokinase expression (Fig. 7B). Thus, the regulatory activities of FasX can be separated, and our data also provide additional support for the nucleotides of the UC loop being important for regulation of PrtF1 and PrtF2 expression.
FIG 7.
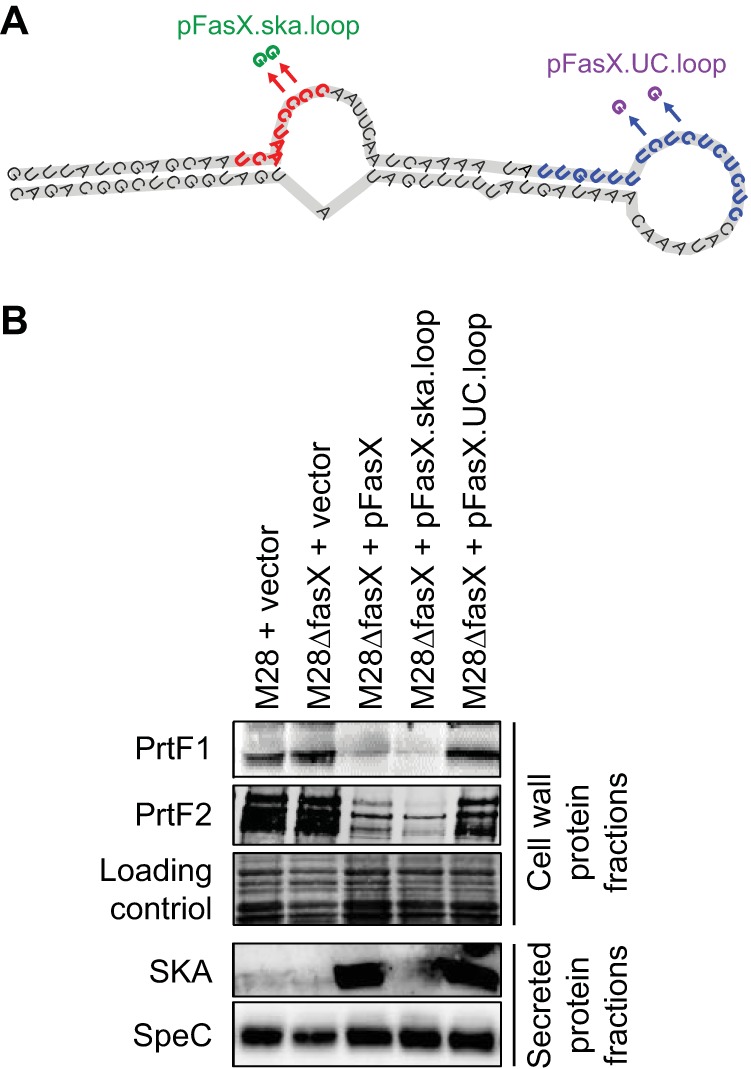
Site-specific mutation of fasX can separate the streptokinase- and adhesin-regulating activities of FasX. (A) Schematic showing the locations of two C-to-G SNPs present in plasmids pFasX.ska.loop (green) and pFasX.UC.loop (purple). The red nucleotides are those that base pair to ska mRNA to regulate streptokinase expression. The blue nucleotides are those that base pair to the adhesin-encoding mRNAs (prtF1, prtF2, and pilus). (B) Western blot analyses assaying the ability of the mutant fasX alleles present within plasmids pFasX.ska.loop and pFasX.UC.loop to complement PrtF1, PrtF2, and SKA expression in the mutant derivative M28ΔFasX. A stained membrane was used as a loading control for the cell wall fraction in Western blot analyses, while expression of the non-FasX-regulated protein SpeC was used as a control for the secreted fraction in Western blot analyses.
FasX-mRNA duplex formation inhibits the translation of prtF1 and prtF2 mRNAs.
The regions of base pairing between FasX and the prtF1 and prtF2 mRNAs include the putative mRNA ribosome binding sites (Fig. 5A and 6A, green nucleotides). Thus, similar to the FasX-mediated negative regulation of the collagen-binding pilus (21, 23), we hypothesized that FasX negatively regulates PrtF1 and PrtF2 expression by blocking ribosome access and inhibiting translation of the encoding mRNAs. To test our hypothesis, we performed in vitro translation assays utilizing RNA substrates consisting of the 5′-UTRs and first ∼158 codons of each of the tested mRNAs (prtF1, prtF2, and spd mRNA as a negative control), to which the sequence for a FLAG tag had been fused at the 3′ end to enable detection of translated proteins with an anti-FLAG antibody (Fig. 8A). For the chimeric prtF1 and prtF2 mRNAs but not the chimeric control spd mRNA, the inclusion of FasX RNA in the in vitro translation reaction inhibited mRNA translation in a dose-dependent manner (Fig. 8B). In contrast, the inclusion of the FasX mutant RNA FasXΔ72-77 did not inhibit translation for any of the three tested chimeric mRNAs. Thus, FasX negatively regulates the expression of the fibronectin-binding proteins PrtF1 and PrtF2 by binding to, and inhibiting the translation of, the prtF1 and prtF2 mRNAs, respectively.
DISCUSSION
The ability to modulate virulence factor expression is critical for bacterial pathogens to undergo productive infections. Factors that enhance virulence during one stage of infection (e.g., adhesins during colonization) can inhibit virulence if expressed during a different stage of infection (e.g., adhesins during dissemination). To modulate GAS virulence factor expression, this pathogen uses an array of two-component regulatory systems (31, 32), quorum-sensing systems (33), and stand-alone transcription factors (34). In addition, GAS uses an unknown number of sRNAs (our current estimate is ∼60) (35, 36), with FasX being the only characterized GAS sRNA. We and others previously identified that FasX positively regulates the expression of the important virulence factor streptokinase (20, 22). In addition, we identified that FasX also negatively regulates the expression of the collagen-binding pilus (21, 23). Here, we discovered that FasX negatively regulates not only pilus expression but also the expression of the fibronectin-binding proteins PrtF1 and PrtF2. This finding provides an explanation for previously published data, in which it was identified that FasX, through unknown mechanisms, reduced the ability of GAS to bind fibronectin (20). Given the regulatory activity of FasX, we propose that this sRNA functions at the interphase between colonization and dissemination, repressing the expression of collagen (pilus) and fibronectin (PrtF1 and PrtF2) binding adhesins and enhancing the expression of streptokinase, which promotes GAS dissemination via blood clot and tissue barrier degradation (Fig. 9).
FIG 9.

Model of how the FasX sRNA acts as a master regulator of GAS dissemination. The inset is the putative secondary structure of the FasX molecule, with the portion of the molecule involved in mRNA binding highlighted by the green box. The remainder of the figure shows the FasX nucleotides that are involved in the regulation of ska mRNA (red) (22), fctA (pilus) mRNA (blue) (21, 23), prtF1 mRNA (blue), and prtF2 mRNA (blue). Only the mRNA 5′ ends are shown, with the ribosome binding sites (green) and AUG start codons (red) highlighted. The consequences of these interactions also are shown.
FasX reduces PrtF1 and PrtF2 expression by base pairing with the prtF1 and prtF2 mRNAs at sites that overlap their ribosome-binding sites. Duplex formation between FasX and these mRNAs occludes the ribosome-binding sites, inhibiting translation. This posttranscriptional mechanism of regulation is identical to that observed for the FasX-mediated regulation of pilus expression (21, 23). Due to the variability of the FCT region, the pilus biosynthesis gene targeted by FasX for inhibition differs in an FCT-type/serotype-dependent manner (21). Whether FasX targets mRNAs encoding the major (fctA, FCT-1 [e.g., serotype M6 GAS] and FCT-4 [e.g., serotype M28 GAS]) or a minor (cpa, FCT-2 [e.g., serotype M1 GAS] and FCT-6 [e.g., serotype M2 GAS]) pilus protein, FasX binds to these mRNAs within their 5′-UTRs to block ribosome binding and inhibit their translation. Importantly, nucleotides from within the same hairpin loop of FasX are responsible for base pairing to the pilus, prtF1, and prtF2 mRNAs (Fig. 9). Thus, key to the ability of FasX to regulate these distinct mRNAs is its tolerance for small numbers of mismatches, nucleotide insertions/deletions, and non-Watson-Crick base pairing. We propose that the five-times-repeated UC dinucleotide repeat that is present in single-stranded form in the repressor loop of FasX is well suited to bind to the ribosome-binding sites of target mRNAs, which typically are A/G rich. Further, we propose that specificity is determined by the surrounding nucleotides, providing FasX the ability to selectively target only a subset of GAS mRNAs for binding and, as a result, regulation.
While FasX mutation and complementation result in relatively minor changes at the mRNA level (Fig. 2), they result in larger changes at the protein level (Fig. 3), consistent with our finding that the inhibition of target mRNA translation is the major mechanism by which FasX regulates PrtF1 and PrtF2 expression (Fig. 8). While an increase in PrtF1 and PrtF2 expression is observed in the fasX mutant strain relative to that of the parental strain, it is possible that the relative difference could be enhanced in other serotypes. This is due to our previous finding that our parental serotype M28 GAS isolate used, MGAS6180 (Table 1), only moderately expresses FasX (35). That expressing FasX in higher abundance will result in a greater degree of regulation is observed from the complemented strain (Fig. 3), which has greatly enhanced FasX abundance due to the multicopy nature of the complementation plasmid (23). While the amount of FasX produced by the complemented strain likely is not physiologically relevant, and although we cannot rule out that FasX abundance is not enhanced in vivo relative to that in vitro, this does not take away from the key finding that the removal of FasX (via mutation) results in enhanced PrtF1 and PrtF2 expression and, hence, that FasX negatively regulates the expression of these virulence factors.
How FasX expression is regulated has not been fully elucidated. What is known is that fasX is the fourth gene of a four-gene locus, and the other genes encode proteins homologous to membrane-spanning sensor kinases (fasBC) or cytoplasmic response regulators (fasA). The fasBCA genes all are required for high-level FasX expression (P. Sumby, unpublished data) (20), and this sRNA is produced in increasing abundance from early exponential phase to early stationary phase and then rapidly declines, such that it is undetectable at late stationary phase during growth in THY broth (20, 35). How FasBCA function, how they interact with one another, what they sense, and how they modify FasX abundance is under investigation by our laboratory.
PrtF1/2 expression also is controlled at the transcriptional level. The transcriptional regulator MsmR, which is present in only ∼50% of GAS serotypes that express PrtF2, binds to the prtF2 promoter to enhance transcription 5-fold (37). In addition, the orthologous regulatory proteins RofA/Nra, one of which is expressed by all GAS serotypes, also have regulatory activity on the prtF1 and/or prtF2 genes, although whether these regulators function in a positive or negative manner differs in a serotype-specific manner (38–40). That PrtF1/2 expression is subject to control by multiple independent regulators highlights the importance of finely tuning their production when environmental conditions dictate. Note that we hypothesize that the small but reproducible increase in prtF1 and prtF2 mRNA abundance following fasX mutation (Fig. 2) is a consequence of the increased stability of these transcripts, which in turn is a consequence of higher ribosome occupancy on these mRNAs, as there is no FasX-mediated blocking of ribosome binding, providing increased protection against RNase activity.
Collagen (bound by pili) and fibronectin (bound by PrtF1/2) are major constituents of the host ECM, and the importance of pili and PrtF1/2 to GAS adherence has been well documented (7, 8, 13, 41–44). Thus, by inhibiting pilus and PrtF1/2 expression, FasX reduces GAS adherence, which is consistent with the data from our previous tissue culture assays that examined the contribution of FasX to the ability of GAS to bind human keratinocytes (21, 23). Importantly, in addition to PrtF1/2 expression promoting GAS adherence, they also may promote the ability of GAS to invade host cells, as surface-bound fibronectin can act as a bridge to bind host integrins (7, 45, 46). Indeed, the fibronectin-binding domains of PrtF1 have been shown to form a tandem beta-zipper with fibronectin, and this rearranges the conformation of the fibronectin, providing access to the Arg-Gly-Asp (RGD) domain by α5β1 integrins (47). Ligand-bound integrin clustering-induced signal transduction is sufficient for GAS internalization into epithelial and endothelial cells (10, 12, 48–50), providing protection against host defenses as well as antimicrobial therapies (46, 51, 52). Interestingly, the number of fibronectin-binding domains found in PrtF1 and PrtF2 can differ in a strain-dependent fashion (12, 45, 53, 54), and this may at least in part explain strain-specific differences seen in internalization and adhesion efficiency (7, 45, 55), as different numbers of binding repeats presumably alter the conformation of the bound ligand.
Here, we have expanded the known regulon of the GAS sRNA FasX. We have shown that this sRNA posttranscriptionally negatively regulates the expression of the adherence- and internalization-promoting fibronectin-binding proteins PrtF1 and PrtF2, in addition to the previously characterized negative regulation of the collagen-binding pili and the positive regulation of the thrombolytic agent streptokinase. Thus, our data are consistent with our hypothesis that FasX promotes GAS dissemination in response to one or more as-yet-unknown extracellular signals. The nature of these signals, and whether FasX has additional regulatory targets, will be investigated as part of ongoing research. Finally, given that GAS pili and PrtF1/2 are under investigation as possible candidate vaccine antigens (56, 57), that FasX negatively regulates the expression of these virulence factors (21, 23), and that GAS serotypes are variable in their level of FasX abundance (and of FasX-mediated regulation) (35, 58), our data may inform with respect to the suitability of using these surface proteins as antigens.
ACKNOWLEDGMENTS
We thank Debra Bessen (New York Medical College, Valhalla, NY) for providing anti-PrtF2 antibodies.
This research was funded by grant R01 AI087747 from the National Institute of Allergy and Infectious Diseases (to P.S.). J.L.D. is a Hitchcock Scholar.
REFERENCES
- 1.Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev 13:470–511. doi: 10.1128/CMR.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cunningham MW. 2008. Pathogenesis of group A streptococcal infections and their sequelae. Adv Exp Med Biol 609:29–42. doi: 10.1007/978-0-387-73960-1_3. [DOI] [PubMed] [Google Scholar]
- 3.Cole JN, Barnett TC, Nizet V, Walker MJ. 2011. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol 9:724–736. doi: 10.1038/nrmicro2648. [DOI] [PubMed] [Google Scholar]
- 4.Kreikemeyer B, McIver KS, Podbielski A. 2003. Virulence factor regulation and regulatory networks in Streptococcus pyogenes and their impact on pathogen-host interactions. Trends Microbiol 11:224–232. doi: 10.1016/S0966-842X(03)00098-2. [DOI] [PubMed] [Google Scholar]
- 5.Hasty DL, Ofek I, Courtney HS, Doyle RJ. 1992. Multiple adhesins of streptococci. Infect Immun 60:2147–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hasty DL, Courtney HS. 1996. Group A streptococcal adhesion. All of the theories are correct. Adv Exp Med Biol 408:81–94. doi: 10.1007/978-1-4613-0415-9_10. [DOI] [PubMed] [Google Scholar]
- 7.Schwarz-Linek U, Hook M, Potts JR. 2004. The molecular basis of fibronectin-mediated bacterial adherence to host cells. Mol Microbiol 52:631–641. doi: 10.1111/j.1365-2958.2004.04027.x. [DOI] [PubMed] [Google Scholar]
- 8.Talay SR, Valentin-Weigand P, Jerlstrom PG, Timmis KN, Chhatwal GS. 1992. Fibronectin-binding protein of Streptococcus pyogenes: sequence of the binding domain involved in adherence of streptococci to epithelial cells. Infect Immun 60:3837–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Towers RJ, Fagan PK, Talay SR, Currie BJ, Sriprakash KS, Walker MJ, Chhatwal GS. 2003. Evolution of sfbI encoding streptococcal fibronectin-binding protein I: horizontal genetic transfer and gene mosaic structure. J Clin Microbiol 41:5398–5406. doi: 10.1128/JCM.41.12.5398-5406.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaffe J, Natanson-Yaron S, Caparon MG, Hanski E. 1996. Protein F2, a novel fibronectin-binding protein from Streptococcus pyogenes, possesses two binding domains. Mol Microbiol 21:373–384. doi: 10.1046/j.1365-2958.1996.6331356.x. [DOI] [PubMed] [Google Scholar]
- 11.Kreikemeyer B, Oehmcke S, Nakata M, Hoffrogge R, Podbielski A. 2004. Streptococcus pyogenes fibronectin-binding protein F2: expression profile, binding characteristics, and impact on eukaryotic cell interactions. J Biol Chem 279:15850–15859. doi: 10.1074/jbc.M313613200. [DOI] [PubMed] [Google Scholar]
- 12.Ramachandran V, McArthur JD, Behm CE, Gutzeit C, Dowton M, Fagan PK, Towers R, Currie B, Sriprakash KS, Walker MJ. 2004. Two distinct genotypes of prtF2, encoding a fibronectin binding protein, and evolution of the gene family in Streptococcus pyogenes. J Bacteriol 186:7601–7609. doi: 10.1128/JB.186.22.7601-7609.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crotty Alexander LE, Maisey HC, Timmer AM, Rooijakkers SH, Gallo RL, von Kockritz-Blickwede M, Nizet V. 2010. M1T1 group A streptococcal pili promote epithelial colonization but diminish systemic virulence through neutrophil extracellular entrapment. J Mol Med 88:371–381. doi: 10.1007/s00109-009-0566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Kockritz-Blickwede M, Nizet V. 2009. Innate immunity turned inside-out: antimicrobial defense by phagocyte extracellular traps. J Mol Med 87:775–783. doi: 10.1007/s00109-009-0481-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Svensson L, Baumgarten M, Morgelin M, Shannon O. 2014. Platelet activation by Streptococcus pyogenes leads to entrapment in platelet aggregates, from which bacteria subsequently escape. Infect Immun 82:4307–4314. doi: 10.1128/IAI.02020-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng S, Lu X, Resendiz JC, Kroll MH. 2006. Pathological shear stress directly regulates platelet alphaIIbbeta3 signaling. Am J Physiol Cell Physiol 291:C1346–C1354. doi: 10.1152/ajpcell.00559.2005. [DOI] [PubMed] [Google Scholar]
- 17.Gripenland J, Netterling S, Loh E, Tiensuu T, Toledo-Arana A, Johansson J. 2010. RNAs: regulators of bacterial virulence. Nat Rev Microbiol 8:857–866. doi: 10.1038/nrmicro2457. [DOI] [PubMed] [Google Scholar]
- 18.Brantl S, Bruckner R. 2014. Small regulatory RNAs from low-GC Gram-positive bacteria. RNA Biol 11:443–456. doi: 10.4161/rna.28036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller EW, Cao TN, Pflughoeft KJ, Sumby P. 2014. RNA-mediated regulation in Gram-positive pathogens: an overview punctuated with examples from the group a Streptococcus. Mol Microbiol 94:9–20. doi: 10.1111/mmi.12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreikemeyer B, Boyle MD, Buttaro BA, Heinemann M, Podbielski A. 2001. Group A streptococcal growth phase-associated virulence factor regulation by a novel operon (Fas) with homologies to two-component-type regulators requires a small RNA molecule. Mol Microbiol 39:392–406. doi: 10.1046/j.1365-2958.2001.02226.x. [DOI] [PubMed] [Google Scholar]
- 21.Danger JL, Cao TN, Cao TH, Sarkar P, Trevino J, Pflughoeft KJ, Sumby P. 2015. The small regulatory RNA FasX enhances group A Streptococcus virulence and inhibits pilus expression via serotype-specific targets. Mol Microbiol 96:249–262. doi: 10.1111/mmi.12935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramirez-Pena E, Trevino J, Liu Z, Perez N, Sumby P. 2010. The group A Streptococcus small regulatory RNA FasX enhances streptokinase activity by increasing the stability of the ska mRNA transcript. Mol Microbiol 78:1332–1347. doi: 10.1111/j.1365-2958.2010.07427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Z, Trevino J, Ramirez-Pena E, Sumby P. 2012. The small regulatory RNA FasX controls pilus expression and adherence in the human bacterial pathogen group A Streptococcus. Mol Microbiol 86:140–154. doi: 10.1111/j.1365-2958.2012.08178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 25.Reference deleted.
- 26.Bessen DE, Kalia A. 2002. Genomic localization of a T serotype locus to a recombinatorial zone encoding extracellular matrix-binding proteins in Streptococcus pyogenes. Infect Immun 70:1159–1167. doi: 10.1128/IAI.70.3.1159-1167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodfellow AM, Hibble M, Talay SR, Kreikemeyer B, Currie BJ, Sriprakash KS, Chhatwal GS. 2000. Distribution and antigenicity of fibronectin binding proteins (SfbI and SfbII) of Streptococcus pyogenes clinical isolates from the northern territory, Australia. J Clin Microbiol 38:389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baldassarri L, Creti R, Imperi M, Recchia S, Pataracchia M, Orefici G. 2007. Detection of genes encoding internalization-associated proteins in Streptococcus pyogenes isolates from patients with invasive diseases and asymptomatic carriers. J Clin Microbiol 45:1284–1287. doi: 10.1128/JCM.02119-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green NM, Zhang S, Porcella SF, Nagiec MJ, Barbian KD, Beres SB, LeFebvre RB, Musser JM. 2005. Genome sequence of a serotype M28 strain of group a streptococcus: potential new insights into puerperal sepsis and bacterial disease specificity. J Infect Dis 192:760–770. doi: 10.1086/430618. [DOI] [PubMed] [Google Scholar]
- 30.Granok AB, Parsonage D, Ross RP, Caparon MG. 2000. The RofA binding site in Streptococcus pyogenes is utilized in multiple transcriptional pathways. J Bacteriol 182:1529–1540. doi: 10.1128/JB.182.6.1529-1540.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ichikawa M, Minami M, Isaka M, Tatsuno I, Hasegawa T. 2011. Analysis of two-component sensor proteins involved in the response to acid stimuli in Streptococcus pyogenes. Microbiology 157:3187–3194. doi: 10.1099/mic.0.050534-0. [DOI] [PubMed] [Google Scholar]
- 32.Sitkiewicz I, Musser JM. 2006. Expression microarray and mouse virulence analysis of four conserved two-component gene regulatory systems in group a streptococcus. Infect Immun 74:1339–1351. doi: 10.1128/IAI.74.2.1339-1351.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jimenez JC, Federle MJ. 2014. Quorum sensing in group A Streptococcus. Front Cell Infect Microbiol 4:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patenge N, Fiedler T, Kreikemeyer B. 2013. Common regulators of virulence in streptococci. Curr Top Microbiol Immunol 368:111–153. [DOI] [PubMed] [Google Scholar]
- 35.Perez N, Trevino J, Liu Z, Ho SCM, Babitzke P, Sumby P. 2009. A genome-wide analysis of small regulatory RNAs in the human pathogen group A Streptococcus. PLoS One 4:e7668. doi: 10.1371/journal.pone.0007668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patenge N, Billion A, Raasch P, Normann J, Wisniewska-Kucper A, Retey J, Boisguerin V, Hartsch T, Hain T, Kreikemeyer B. 2012. Identification of novel growth phase- and media-dependent small non-coding RNAs in Streptococcus pyogenes M49 using intergenic tiling arrays. BMC Genomics 13:550. doi: 10.1186/1471-2164-13-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakata M, Podbielski A, Kreikemeyer B. 2005. MsmR, a specific positive regulator of the Streptococcus pyogenes FCT pathogenicity region and cytolysin-mediated translocation system genes. Mol Microbiol 57:786–803. doi: 10.1111/j.1365-2958.2005.04730.x. [DOI] [PubMed] [Google Scholar]
- 38.Kreikemeyer B, Beckert S, Braun-Kiewnick A, Podbielski A. 2002. Group A streptococcal RofA-type global regulators exhibit a strain-specific genomic presence and regulation pattern. Microbiology 148:1501–1511. doi: 10.1099/00221287-148-5-1501. [DOI] [PubMed] [Google Scholar]
- 39.Luo F, Lizano S, Bessen DE. 2008. Heterogeneity in the polarity of Nra regulatory effects on streptococcal pilus gene transcription and virulence. Infect Immun 76:2490–2497. doi: 10.1128/IAI.01567-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Podbielski A, Woischnik M, Leonard BA, Schmidt KH. 1999. Characterization of nra, a global negative regulator gene in group A streptococci. Mol Microbiol 31:1051–1064. doi: 10.1046/j.1365-2958.1999.01241.x. [DOI] [PubMed] [Google Scholar]
- 41.Danne C, Dramsi S. 2012. Pili of gram-positive bacteria: roles in host colonization. Res Microbiol 163:645–658. doi: 10.1016/j.resmic.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 42.Lizano S, Luo F, Bessen DE. 2007. Role of streptococcal T antigens in superficial skin infection. J Bacteriol 189:1426–1434. doi: 10.1128/JB.01179-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manetti AG, Zingaretti C, Falugi F, Capo S, Bombaci M, Bagnoli F, Gambellini G, Bensi G, Mora M, Edwards AM, Musser JM, Graviss EA, Telford JL, Grandi G, Margarit I. 2007. Streptococcus pyogenes pili promote pharyngeal cell adhesion and biofilm formation. Mol Microbiol 64:968–983. doi: 10.1111/j.1365-2958.2007.05704.x. [DOI] [PubMed] [Google Scholar]
- 44.Anderson EL, Cole JN, Olson J, Ryba B, Ghosh P, Nizet V. 2014. The fibrinogen-binding M1 protein reduces pharyngeal cell adherence and colonization phenotypes of M1T1 group A Streptococcus. J Biol Chem 289:3539–3546. doi: 10.1074/jbc.M113.529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edwards ML, Fagan PK, Currie BJ, Sriprakash KS. 2004. The fibronectin-binding capacity and host cell adherence of Streptococcus pyogenes strains are discordant with each other. Microbes Infect 6:1156–1162. doi: 10.1016/j.micinf.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 46.Kreikemeyer B, Klenk M, Podbielski A. 2004. The intracellular status of Streptococcus pyogenes: role of extracellular matrix-binding proteins and their regulation. Int J Med Microbiol 294:177–188. doi: 10.1016/j.ijmm.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 47.Schwarz-Linek U, Werner JM, Pickford AR, Gurusiddappa S, Kim JH, Pilka ES, Briggs JA, Gough TS, Hook M, Campbell ID, Potts JR. 2003. Pathogenic bacteria attach to human fibronectin through a tandem beta-zipper. Nature 423:177–181. doi: 10.1038/nature01589. [DOI] [PubMed] [Google Scholar]
- 48.Jadoun J, Ozeri V, Burstein E, Skutelsky E, Hanski E, Sela S. 1998. Protein F1 is required for efficient entry of Streptococcus pyogenes into epithelial cells. J Infect Dis 178:147–158. doi: 10.1086/515589. [DOI] [PubMed] [Google Scholar]
- 49.Ozeri V, Rosenshine I, Mosher DF, Fassler R, Hanski E. 1998. Roles of integrins and fibronectin in the entry of Streptococcus pyogenes into cells via protein F1. Mol Microbiol 30:625–637. doi: 10.1046/j.1365-2958.1998.01097.x. [DOI] [PubMed] [Google Scholar]
- 50.Talay SR, Zock A, Rohde M, Molinari G, Oggioni M, Pozzi G, Guzman CA, Chhatwal GS. 2000. Co-operative binding of human fibronectin to Sfbl protein triggers streptococcal invasion into respiratory epithelial cells. Cell Microbiol 2:521–535. doi: 10.1046/j.1462-5822.2000.00076.x. [DOI] [PubMed] [Google Scholar]
- 51.Medina E, Goldmann O, Toppel AW, Chhatwal GS. 2003. Survival of Streptococcus pyogenes within host phagocytic cells: a pathogenic mechanism for persistence and systemic invasion. J Infect Dis 187:597–603. doi: 10.1086/373998. [DOI] [PubMed] [Google Scholar]
- 52.Thulin P, Johansson L, Low DE, Gan BS, Kotb M, McGeer A, Norrby-Teglund A. 2006. Viable group A streptococci in macrophages during acute soft tissue infection. PLoS Med 3:e53. doi: 10.1371/journal.pmed.0030053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baldassarri L, Recchia S, Imperi M, Creti R, Alfarone G, Pataracchia M, Orefici G. 2006. Fibronectin binding protein genes and cell invasion ability of Streptococcus pyogenes isolated from different sources. Int Congress Ser 1289:243–245. doi: 10.1016/j.ics.2005.09.100. [DOI] [Google Scholar]
- 54.Gorton D, Norton R, Layton R, Smith H, Ketheesan N. 2005. Presence of fibronectin-binding protein gene prtF2 in invasive group A streptococci in tropical Australia is associated with increased internalisation efficiency. Microbes Infect 7:421–426. doi: 10.1016/j.micinf.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 55.Schwarz-Linek U, Pilka ES, Pickford AR, Kim JH, Hook M, Campbell ID, Potts JR. 2004. High affinity streptococcal binding to human fibronectin requires specific recognition of sequential F1 modules. J Biol Chem 279:39017–39025. doi: 10.1074/jbc.M405083200. [DOI] [PubMed] [Google Scholar]
- 56.Olive C, Schulze K, Sun HK, Ebensen T, Horvath A, Toth I, Guzman CA. 2007. Enhanced protection against Streptococcus pyogenes infection by intranasal vaccination with a dual antigen component M protein/SfbI lipid core peptide vaccine formulation. Vaccine 25:1789–1797. doi: 10.1016/j.vaccine.2006.11.031. [DOI] [PubMed] [Google Scholar]
- 57.Guzman CA, Talay SR, Molinari G, Medina E, Chhatwal GS. 1999. Protective immune response against Streptococcus pyogenes in mice after intranasal vaccination with the fibronectin-binding protein SfbI. J Infect Dis 179:901–906. doi: 10.1086/314655. [DOI] [PubMed] [Google Scholar]
- 58.Cao TN, Liu Z, Cao TH, Pflughoeft KJ, Treviño J, Beres SB, Musser JM, Sumby P. 2014. Natural disruption of two regulatory networks in M3 group A Streptococcus isolates contributes to the virulence factor profile of this hypervirulent serotype. Infect Immun 82:1744–1754. doi: 10.1128/IAI.01639-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hyland KA, Wang B, Cleary PP. 2007. Protein F1 and Streptococcus pyogenes resistance to phagocytosis. Infect Immun 75:3188–3191. doi: 10.1128/IAI.01745-06. [DOI] [PMC free article] [PubMed] [Google Scholar]