


Abstract
Olfactory receptor (Olfr) genes comprise the largest gene family in mice. Despite their importance in olfaction, how most Olfr mRNAs are regulated remains unexplored. Using RNA-seq analysis coupled with analysis of pre-existing databases, we found that Olfr mRNAs have several atypical features suggesting that post-transcriptional regulation impacts their expression. First, Olfr mRNAs, as a group, have dramatically higher average AU-content and lower predicted secondary structure than do control mRNAs. Second, Olfr mRNAs have a higher density of AU-rich elements (AREs) in their 3′UTR and upstream open reading frames (uORFs) in their 5 UTR than do control mRNAs. Third, Olfr mRNAs have shorter 3′ UTR regions and with fewer predicted miRNA-binding sites. All of these novel properties correlated with higher Olfr expression. We also identified striking differences in the post-transcriptional features of the mRNAs from the two major classes of Olfr genes, a finding consistent with their independent evolutionary origin. Together, our results suggest that the Olfr gene family has encountered unusual selective forces in neural cells that have driven them to acquire unique post-transcriptional regulatory features. In support of this possibility, we found that while Olfr mRNAs are degraded by a deadenylation-dependent mechanism, they are largely protected from this decay in neural lineage cells.
INTRODUCTION
Olfactory receptors (OLFRs) are G protein-coupled receptors (GPCRs) essential for odor detection in olfactory sensory neurons (OSNs). These receptors are encoded by the largest gene family in mice, occupying ∼2% of the protein-coding genome (1–3). Olfr genes are divided into 2 classes, each of which has a different evolutionary origin: class-I receptors are thought to be derived from ancestral fish and evidence suggests that class-II receptors evolved from ancestral amphibians (3). These two Olfr gene classes are responsible for generating receptors that detect different odorants; e.g. it has been shown that class-I OLFRs preferentially detect predator-related odorants (4).
Olfr genes are regulated in a unique manner. Only a single Olfr gene allele from ∼1000 Olfr gene choices is selected to be expressed in a given OSN (5–8). The Olfr gene selected by each OSN is not only responsible for detecting odorants in the olfactory epithelium (OE), but it also directs the axons of OSNs that express the same Olfr gene to converge into the same glomerulus in the olfactory bulb (9,10). By controlling both axon guidance and receptor expression, this ‘1-receptor, 1-cell' rule provides the foundation by which the olfactory system distinguishes different odorants (1,3,11). How precisely this 1-receptor, 1-cell rule is implemented at the molecular level remains enigmatic. In principal, it appears to be largely dictated by a selective transcriptional mechanism in which one Olfr gene is transcriptionally activated and all other Olfr genes are transcriptionally repressed in a given OSN. Likely to be involved are transcription factors that regulate Olfr gene expression, including the LHX2 LIM/homeobox transcription factor and members of the OLF-1/EBF (O/E) helix-loop-helix (HLH) family (12–14). The regulation of Olfr gene choice may also be dictated by epigenetic signatures that correlate with transcriptional activity (15,16).
While considerable progress has been defining transcriptional mechanisms acting on Olfr genes, little is known about post-transcriptional mechanisms regulating Olfr mRNAs. This is a large gap in the field given that post-transcriptional regulation has the potential to be critical for regulation of OLFR expression. For example, selective RNA decay mechanisms could contribute to the ‘1-receptor, 1-cell rule’ by degrading non-selected Olfr mRNAs that are expressed from incompletely silenced Olfr genes. Post-transcriptional mechanisms also have the potential to control Olfr mRNA levels during OSN development, as well as in response to acute exposure to odorants.
Post-transcriptional regulation is typically directed toward the 5′ and 3′ untranslated regions (UTRs) of mRNAs, as they house a plethora of cis elements that impact mRNA stability and translation. For example, UTRs harbor sequence motifs and secondary structures that recruit ribosomes and RNA-binding proteins (RBPs) to govern rates of mRNA decay and translation (17–19). Also recruited to UTRs, particularly to 3′ UTRs, are microRNAs (miRNAs), which are short RNAs that elicit translational repression, mRNA destabilization, or both (20). None of these features have been investigated in Olfr mRNAs.
To address post-transcriptional regulatory mechanisms that regulate Olfr transcripts, it is critical to first define Olfr mRNA sequences. Zhang et al. and Shiao et al. detected the expression of ∼800–1200 Olfr mRNAs in the OE using a custom microarray and RNA-seq, respectively, but they did not define the 5′ and 3′ termini of these transcripts or identifying alternative isoforms (21,22). Other studies have used transcription start-site mapping to identify the 5′ UTR and promoter regions of ∼200 Olfr mRNAs (23–26) and one study screened cDNA libraries to identify promoter and 5′ UTR sequences of ∼400 Olfr mRNAs (27). In our study, we employed RNA-seq analysis to analyze Olfr mRNAs with respect to their post-transcriptional features. Our analysis revealed that Olfr mRNAs tend to have several unique features, including a short 3′ UTR, high AU-content, and a high density of AREs and uORFs. After our manuscript describing this work was submitted, another paper was published that characterized Olfr mRNAs using RNA-seq analysis (28). While this Ibarra-Soria et al. paper did not focus on post-transcriptional features, it provided us an opportunity to examine such features using another Olfr data set. As described herein, the data sets from this paper corroborated what we determined with our Olfr data sets. In summary, we have uncovered unusual post-transcriptional features that are unique to the Olfr gene family, thereby shining light into how these genes are potentially regulated in OSNs. In support, we provide in vitro evidence that Olfr mRNAs are post-transcriptionally regulated in a neuronal-specific manner.
MATERIALS AND METHODS
Compiling the Olfr mRNA transcriptome
RNA-seq analysis was performed on two 8 week-old C57BL/6 female OE littermates, who were housed in accordance with UCSD IACUC policies. In brief, dissected OE RNA was isolated using TriZOL (Life Technologies), followed by a secondary purification step using RNeasy columns (Qiagen). RNA-seq was performed at the UCSD Biogem Core. Total RNA was assessed for quality using an Agilent Bioanalyzer, and samples determined to have an RNA Integrity Number (RIN) of at least 8 or greater were used to generate RNA libraries using Illumina's TruSeq RNA Sample Prep Kit following manufacturer specifications, with the RNA fragmentation time adjusted to 5 min. 1 ug of total RNA was used per sample. RNA libraries were multiplexed and sequenced at 10 pM with 50 basepair (bp) single end reads to a depth of approximately 40 million reads per sample on an Illumina HiSeq2000. The OE transcriptome was extracted using the Tuxedo suite, encompassing Bowtie2, Tophat2 and Cufflinks programs (29,30) using the ‘novel gene discovery mode’ as outlined in the Tuxedo suite program manuals (30). Specifically using the CuffCompare program, we extracted information such as the nearest Ensembl reference ID, exon start/end positions, chromosome, gene direction and gene symbol from each gene entry. Next, we fetched the CDS of all entries using the nearest reference IDs from the UCSC Table Browser from the NCBIm37/mm9 genome. Using the exon start/end positions for each reference ID entry, we extracted the exon sequences from the mm9 genome, and ligated them together in silico for each transcript. For each entry, the entire transcript sequence is subtracted from the known CDS sequence obtained above to identify the 5′ and 3′ UTRs. If a 30-nucleotide sequence was matched for the 5′ and 3′ peripheral regions of the nearest reference coding sequence with the corresponding in silico spliced transcript then the transcript was considered complete. For the complete transcripts, the 5′ and 3′ regions flanking the coding sequence were considered 5′ and 3′ UTRs, respectively. Intron sequences were extracted by using the regions between the exon junctions for each transcript. The second set of Olfr transcripts were provided by the supplemental data sets provided by Ibarra-Soria et al. (28). Only transcripts with both a detectable 5′ UTR and 3′UTR were considered in subsequent analysis.
GC-content, ARE and miRNA site search
Once extracted, the lengths and GC-content were calculated for 5′/3′ UTRs, coding sequences and introns. To calculate global GC-content for Olfr, control and GPCR groups we created sets for every position, ranging from −50 to +50, flanking the splice-site of each intron. The GC-content was calculated for each of these positions to construct a general global GC analysis for each group of transcripts. To construct sequence logos, we used all sequences flanking splice-sites, ranging from −15 to +15, in UCB's WebLogo application (http://weblogo.berkeley.edu/) (31). The 3′UTR sequences generated during the annotation were used to scan for AREs. We scanned the 3′UTR sequences for the following n-mers identified by AREsite (http://rna.tbi.univie.ac.at/cgi-bin/AREsite.cgi) (32): AUUUA and WWAUUUAWW. W indicates either an adenine or uracil nucleotide. miRNA sites were discovered using TargetScan 6.0 (33–36). In brief, 9 miRNA mature and whole sequences were scanned along all genes from the three gene groups to find putative miRNA binding sites.
Secondary structure minimum free energy
To select the minimum free energy of the 5′UTR sequences, we used University of Vienna's RNAfold Web Server (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) (37) to calculate the folding energy. The 5′UTR sequences used were generated from the RNA-seq reads, Ensembl reference IDs, and the annotation algorithms described above.
3′UTR cloning, cell culture and luciferase assay
The 3′ UTRs of Olfr212, Olfr1226 and Olfr1242 were amplified by PCR from adult OE cDNA and cloned downstream of the Firefly luciferase ORF (at the SpeI and HindIII restriction enzyme sites) in the pMIR-REPORT vector (Ambion). These reporter constructs were transfected by Lipofectamine 2000 reagent (Life Technologies) into P19 and Neuro2A cells. The P19 cells were cultured in 10% fetal calf serum (Stem Cell Technologies) and MEMα media (Invitrogen); Neuro2A cells were cultured in 10% fetal calf serum (Stem Cell Technologies) and DMEM media (Invitrogen). Also transfected into these cells was the CCR4B expression vector (kindly provided by Jens Lykke-Andersen [UCSD]) and the Ccr4b siRNA and the ONTARGETplus siRNA control (ThermoScientific). The Firefly Renilla luciferase reporter was co-transfected as an internal control. Luciferase activity was assayed as previously described (38) using the Dual Luciferase Assay System (Promega). Cellular RNA was harvested using TriZOL (Invitrogen), followed by iScript Reverse Transcription Kit and SYBR Green qPCR Reagent (BioRad), all according to manufacturer's protocol. The level of Ccr4b mRNA was measured using the following qPCR primers: UCSD8023: CGCGGAGGAGAATGAGACTA and UCSD8024: GTGCAGTGCTGTCAAGTGTG. The endogenous RNA control Rpl19 was used and was amplified using the following qPCR primers: UCSD4667: CTGAAGGTCAAAGGGAATGTG and UCSD4668: GGACAGAGTCTTGATGATCTC.
In situ hybridization
In situ hybridization was performed as previously described previously (38) using 10 μm fresh frozen OE cryosections and LNA probes against miR-741, miR-9 and miR-128 (Exigen).
Identifying uORFs
The 5′ UTR sequences identified through our annotation process (described above when compiling Olfr transcript above) were used to scan for upstream open reading frames that contained particular nucleotides from the Kozak consensus sequence critical for initiating translation (39). In particular, we only considered uORFs ≥ 30 nt long that had a purine at the −3 position or a guanine at the +1 position (relative to the A in the AUG initiation codon [+1]). To reduce the probability of identifying uORFs that can re-initiate translation (and thus escape NMD), one additional criteria is the uORF must not contain the main open reading frame.
Programming language used
Algorithms were generated with the Python programming language (version 2.7.6). Data analysis was performed using both R and GraphPad PRISM.
RESULTS AND DISCUSSION
Genome-wide identification of class I and II Olfr transcripts
Olfr transcripts were defined using RNA-seq data from adult mice OE as outlined in Figure 1A. We detected reads from 1101 Olfr genes, but there were insufficient reads from some of these Olfr genes to accurately determine their 5′ and 3′ termini (data not shown). To exclusively annotate Olfr mRNAs that encode functional proteins, we only analyzed transcripts with RNA-seq reads matching the coding sequence (CDS) of known OLFR proteins, as defined in the Ensembl database. Using this criterion, we identified 805 full-length mRNAs encoded by 554 Olfr genes (Supplementary Table S1). The alternative isoforms included those with different 5′ and 3′ UTR regions and those generated by alternative splicing (Supplementary Table S2). The Olfr transcripts we identified emanate from Olfr genes distributed on the various chromosomes known to have Olfr gene clusters (Figure 1B; 27), suggesting that our analysis was not biased for a particular sector of the genome. Analysis of the reads corresponding to individual Olfr genes showed they exhibited considerable heterogeneity in expression (Figure 1C and D), confirming findings from other studies on Olfr mRNAs (27,28).
Figure 1.
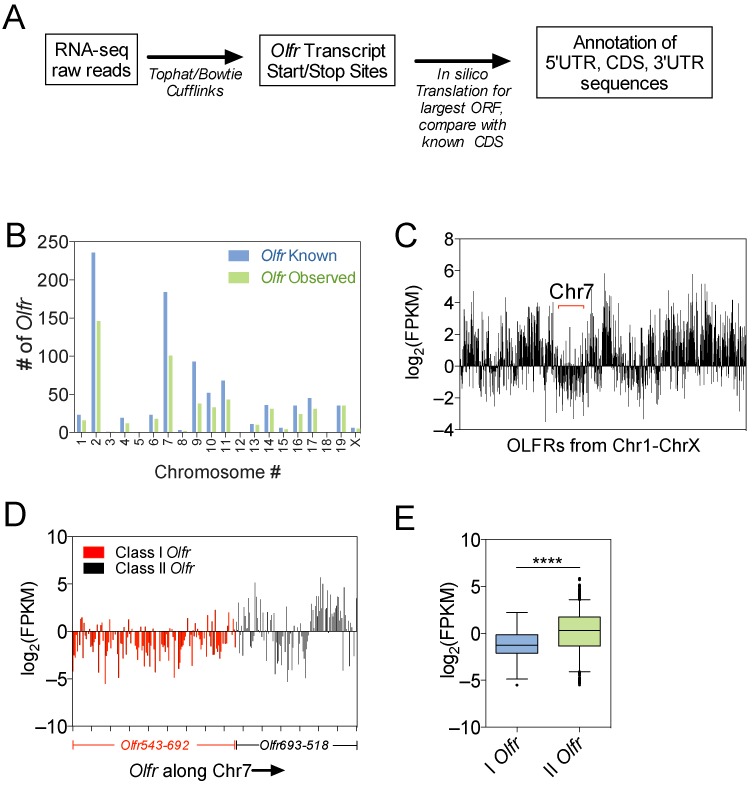
RNA-seq analysis of Olfr mRNAs in the mouse adult OE. (A) Schematic of workflow used to assemble the Olfr transcriptome. (B) Full-length Olfr mRNA transcripts detected by our RNA-seq analysis versus all known Olfr genes in each mouse chromosome. (C) Bar graph of the expression level of individual Olfr genes plotted sequentially along all mouse chromosomes, expressed as Fragments Per Kilobase of transcript per Million mapped reads (FPKM). (D) Bar graph displaying the expression level of individual Olfr genes along chromosome 7. (E) Boxplot of average expression level of class-I versus -II Olfr transcripts. **** P < 0.0001.
We observed that class-I Olfr genes tend to be expressed at strikingly lower level than most class-II Olfr genes (Figure 1C–E). A likely explanation is that class-I Olfr genes are selected to be expressed less often than are class-II Olfr genes in individual OSNs, as class-I Olfr genes comprise only ∼10% of all Olfr genes (40). In further support of this possibility, class-I OLFR-expressing OSNs are only found in the dorsal zone of the OE, whereas class-II OLFR-expressing OSNs are present in virtually all regions of the OE (41,42). A non-mutually exclusive explanation for the lower levels of class-I Olfr mRNAs in the OE is that class-I Olfr genes are transcribed at a lower rate per OSN than are class-II Olfr genes. Since class-I Olfr genes have a distinct evolutionary origin to that of class-II Olfr genes (3), they may have distinct promoters that support lower levels of transcription than do class-II promoters. Finally, it is also possible that class-I Olfr mRNAs are intrinsically less stable than are class-II Olfr mRNAs.
Olfr mRNAs tend to have short 3′ UTRs
To understand how Olfr mRNA characteristics potentially impact their expression, we analyzed Olfr mRNAs we defined by RNA-seq (Figure 2, Supplementary Figure S1A–B, Supplementary Table S3), as Olfr trancripts on publically available databases such as Ensembl and RefSeq have inaccuracies. Unless otherwise noted, all data analysis below was performed in Olfr mRNAs we identified through our RNA-seq analysis. As a basis of comparison, we examined two other gene groups: (i) Gpcr genes encoding 92 non-OLFR GPCR proteins defined by the International Union of Basic and Clinical Pharmacology Database [IUPHAR-DB] (43) that we found were significantly expressed in the OE, based on our RNA-seq analysis, and (ii) Ctrl genes, a randomly selected group of 587 non-Gpcr genes expressed in the OE.
Figure 2.
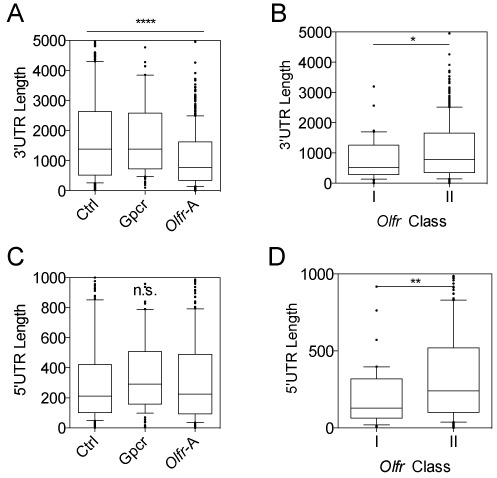
Most Olfr transcripts have short 3′ UTRs. (A, B) Boxplots of 3′ UTR length. (C, D) Boxplots of 5′ UTR length. *P < 0.05, **P < 0.01, ****P < 0.0001; n.s. not statistically significant.
This analysis revealed unique features of Olfr mRNAs as a group. One unusual feature of Olfr mRNAs is 3′ UTR length. The average 3′ UTR length of Olfr mRNAs in our data set (‘Olfr A’, 1094 nt) is dramatically shorter than that of the Ctrl (1895 nt) and Gpcr (1800 nt) group mRNAs (Figure 2A; P < 0.0001). Olfr mRNAs defined by Ibarra-Soria et al. (28) (‘Olfr-B’) also had a shorter 3′ UTR length (1439 nt; P < 0.0001) (Supplementary Figure S1C). Both class-I and -II Olfr mRNAs had shorter 3′ UTRs than control gene groups, but was most evident for the former (786 nt and 1121 nt average 3′ UTR length, respectively; P < 0.05; Figure 2B and Supplementary Table S3). The longer average Olfr mRNA length determined by Ibarra-Soria et al. compared to that found in our study could be due to variability in sampling conditions or transcriptome assembly algorithm differences, or simply because different mice have different repertoires of Olfr mRNAs (22). For example, Ibarra-Soria et al. used both males and female mice for their analysis, while our analysis used only female mice (28). Given that males and females have a vastly different profile of Olfr expression, this experimental difference could explain some transcriptome annotation differences.
The finding that Olfr 3′ UTRs tend to be shorter than control 3′ UTRs (Figure 2A, Supplementary Figure S1C) raised the possibility that there has been strong selection pressure on Olfr genes to evade post-transcriptional regulation pathways that target the 3′ UTR. For example, previous work has suggested that polyadenylation site selection shifts during development and cancer progression to differentially expose recognition sites for RNA-binding proteins (RBPs) or miRNAs for regulatory purposes (44–49). Both RBPs and miRNAs dictate translation efficiency and RNA decay rates, and thus mechanisms that control their ability to be recruited to an mRNA are critical for the fate of an mRNA (50). Below, we consider how the unique characteristics of Olfr 3′ UTRs, including their short length, might shape the post-transcriptional fate of Olfr transcripts.
We also found that there is considerably heterogeneity in the length in Olfr mRNAs encoded by different Olfr genes (Supplementary Figure S1D). This is largely attributable to the heterogeneity of 3′ UTR length, since the coding region (CDS) length of Olfr mRNAs are nearly invariant (Supplementary Figure S1E) and the 5′ UTR region does not contribute much to the variation in Olfr mRNAs (Supplementary Figure S1F). Another contributing factor to Olfr mRNA length heterogeneity is the difference in the average length of class-I and-II Olfr mRNAs; we found that class-I mRNAs are modestly, but significantly shorter than class-II mRNAs (Supplementary Figure S1G).
In contrast to the 3′ UTR, the 5′ UTR was not significantly different in median length in Olfr mRNAs (223 nt in Olfr-A, 241 nt in Olfr-B) as compared to either Ctrl (212 nt) or Gpcr (242 nt) mRNAs (Figure 2C). However, we found that Olfr 5′ UTRs were quite heterogeneous in length, in large part because class-I Olfr 5′ UTRs (129 nt) are, on average, half the length of Class-II Olfr 5′ UTRs (240 nt, Figure 2D; P < 0.01, Supplementary Table S3). Thus, Class-I Olfr 5′ UTRs tend to be much shorter than the average length of 5′ UTRs in mRNAs in general. We suggest that this likely reflects the different evolutionary origins and/or unique selection pressures exerted on class-I Olfr genes (3). 5′ UTR regions are known to be major sites for regulation of translation efficiency and RNA decay rate (19) and thus class-I Olfrs may have been selected to have short 5′ UTR regions over evolutionary time for regulatory purposes.
Olfr mRNAs have few predicted binding sites for OE-expressed miRNAs
We first considered the possibility that Olfr mRNAs have short 3′ UTRs to avoid repression by miRNAs. miRNAs are short (18-22 nucleotides) RNAs that bind by complementarity with their mRNA targets and recruit RBPs that lead to mRNA stabilization and/or translational repression (51). miRNAs typically bind to the 3′ UTR region of their target mRNAs and thus there has been considerable interest in identifying mechanisms that dictate which 3′ UTR regions are accessible to miRNA-mediated regulation (44,47,52). In the case of Olfr mRNAs, we hypothesized that they were selected to harbor short 3′ UTRs to escape repression mediated by miRNAs.
To test this possibility, we used the miRNA target-prediction program, TargetScan, to screen for miRNA-binding seed sequences in the Olfr mRNAs we defined as well as the data set from Ibarra-Soria et al. In particular, we used this in silico screen to examine the following miRNAs: miR-200a/miR-141a, the only miRNA known to have a role in olfactory neurogenesis (53), and eight well-studied neurally expressed miRNAs: miR-7, -9, -124, -128, -132, -134, -138 and let-7 (38,54–61). We found that at least 2 of these miRNAs—miR-9 and -128—are expressed in the OSN layer of the OE (Figure 3A). We functionally tested one of the Olfr 3′ UTRs (Olfr1226) with a predicted miR-9 target site. In support of the accuracy of the TargetScan algorithm, we found that the Olfr1226 3′ UTR conferred repression in response to exogenously expressed miR-9 (Figure 3B).
Figure 3.
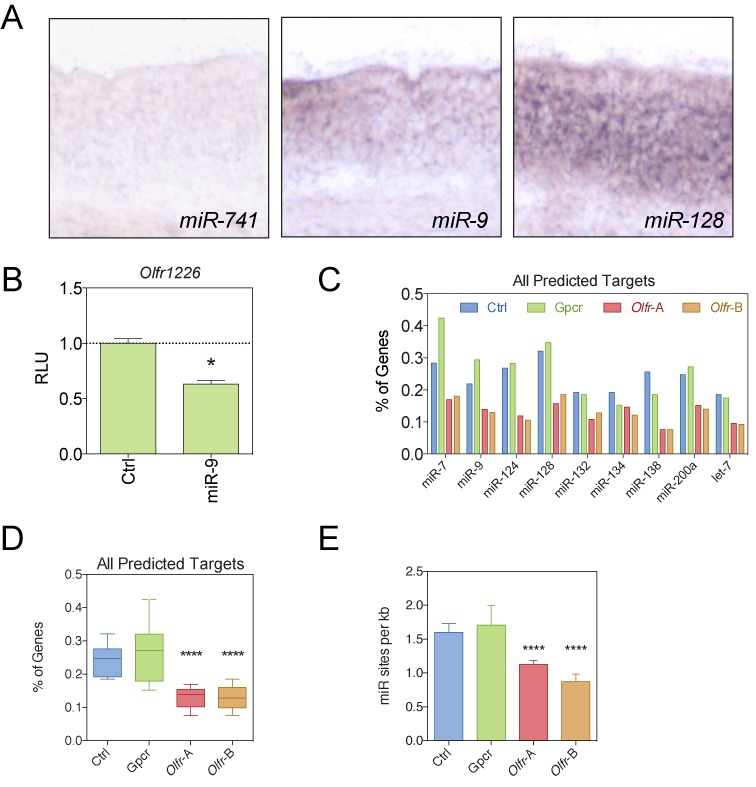
Olfr mRNAs tend to have few predicted target sites for neurally expressed miRNAs. (A) In situ hybridization of neurally expressed miRNAs in the OE. (B) Dual luciferase analysis of the effect of the Olfr1226 3′ UTR in response to ectopic expression of miR-9 in Neuro2A cells. (C) The percentage of mRNAs from the indicated gene groups with predicted miRNA-binding site defined using the TargetScan algorithm. (D) Boxplot representation of the average percentage of miRNA-binding sites determined across all 9 miRNAs, determined from the data in panel C. (E) Average frequency of miRNA sites per kb of 3′UTR in each gene group. *P < 0.05; ****P < 0.0001.
Analysis of predicted target sites revealed less predicted binding sites for 8 out of the 9 miRNAs in Olfr transcripts compared to the Gpcr and Ctrl transcript groups (Figure 3C–D, Supplementary Table S4). As another means to analyze miRNA-regulatory potential, we examined the percentage of mRNAs in a given group predicted to be targeted by the nine neurally expressed miRNAs. We found that a significantly lower percentage of Olfr mRNAs had predicted target sites for these nine miRNAs than did the other mRNA groups (P < 0.0001; Figure 3C).
We considered the possibility that the only reason that Olfr mRNAs have fewer miRNA-target sites than control mRNAs is because their 3′ UTRs are shorter. If this was the case, then the density of miRNA-target sites should be similar between the three groups. Instead, we found that Olfr mRNAs had significantly lower density of predicted binding sites for the nine miRNAs than the control groups (Figure 3E). Thus, the low number of predicted miRNA-target sites in Olfr mRNAs is not only because they tend to have short 3′ UTRs. Together, these data are consistent with a model in which Olfr genes evolved to reduce miRNA-mediated repression by both shortening their 3′ UTRs and reducing the number of miRNA-target sites by mutation. During the course of this analysis, we noted that a relatively large percentage of Gpcr genes (>15%) contain predicted miR-7 and -128 binding sites (Figure 3C), suggesting that miR-7, -200a, -9 and -128, in particular, may be broad regulators of Gpcr mRNAs.
Olfr mRNAs are AU-rich
Previous studies have demonstrated that the average AU-content of mammalian mRNAs is ∼45% (18). In contrast, we found that most Olfr mRNAs have an AU-content of greater than 50%. This characteristic was evident throughout the length of Olfr mRNAs, including both UTRs and the CDS, and even the splice junction region (Figure 4A–B, Supplementary Figure S2A–C). Particularly striking was the Olfr 5′ UTR region, which had ∼60% average AU-content versus the ∼40% average AU-content of the 5′ UTR region in the Ctrl and Gpcr mRNAs (Figure 4B, P < 0.0001, Supplementary Table S5). Of note, both class-I and –II Olfr mRNAs tended to be AU-rich (Supplementary Figure S2D–F), suggesting that this quality was independently selected for during the generation of both classes of Olfr genes.
Figure 4.
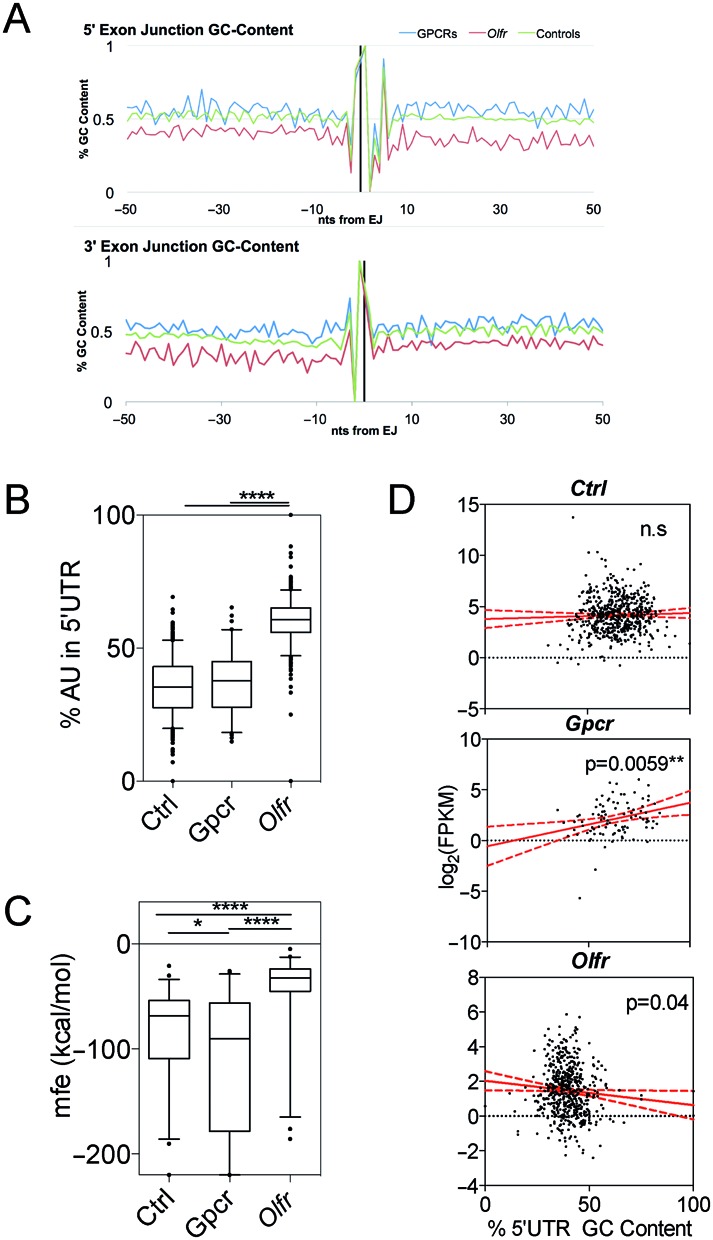
Olfr mRNAs are AU-rich. (A) GC-content along exon-intron junctions (top) and intron-exon junctions (bottom). (B) Boxplot of% of AU-content in the 5′ UTR. (C) Boxplot of minimal free energy (mfe) in the 5′ UTR. (D) Scatterplot and linear regression analysis of expression level versus 5′ UTR% GC-content. * P < 0.05; ** P < 0.01; **** P < 0.0001; n.s., not statistically significant.
The discovery that Olfr mRNAs have a high AU-content raised the possibility that this feature is important for Olfr post-transcriptional regulation. One possibility is that this reduces secondary structure that would otherwise reduce translation efficiency or lead to steric hindrance of interaction with RNA-binding proteins (19). To assess whether Olfr mRNAs have reduced secondary structure relative to control mRNAs, we utilized the RNAFold algorithm (37), which calculates minimum free energy (mfe), a measure of the difficulty to linearize a given sequence. Since 5′ UTR length contributes to mfe calculations, we selected 50 5′ UTRs from each mRNA group that have similar 5′ UTR length. This analysis revealed that Olfr mRNAs have a strikingly higher average mfe compared to Gpcr and Ctrl transcripts (Figure 4C).
mRNAs with low GC-content and secondary structure are thought to be more efficiently translated than those with a high GC-content (19). This is supported by genome-wide studies observing a significant correlation between 5′ UTR secondary structure folding free energy and protein abundance (62). While the molecular mechanism for this is not known, a likely possibility is that less structured 5′ UTRs permit more efficient scanning of the 40S ribosomal subunit as it traverses from the 5′ cap to the start AUG codon of the main ORF (63). This raises the possibility that Olfr genes have been selected to have low GC-content over evolutionary time so that they can translate high levels of Olfr proteins. Because translation can protect mRNAs from decay (64), this also predicts that Olfr mRNAs harboring 5′ UTRs with low GC-content would be more stable than those without. In agreement with this prediction, we found a negative correlation between Olfr expression level and the 5′ UTR GC-content of Olfr mRNAs (Figure 4D). In contrast, we observed a statistically significant positive correlation between the GC-content of Gpcr mRNAs and their expression. While we do not know the underlying mechanism for this, one possibility is higher GC-content alters the spectrum of ribonucleoprotein complexes that form on the 5′ UTR of Gpcr mRNAs in a manner that favors the stability of these mRNAs. No correlation was observed for the Ctrl mRNAs, possibly because they represent a broad group of genes with confounding variables. While there are several explanations for these findings, we suggest that the most parsimonious interpretation is that Olfr mRNAs evolved to have unstructured 5′ UTRs as a means to increase their translation efficiency, which in turn increased the stability of their mRNAs.
Olfr mRNAs tend to be enriched for ARE motifs
Our finding that Olfr mRNAs tend to be AU-rich (Figure 4 and Supplementary Figure S2) raised the possibility that they are enriched for AU-rich binding elements (AREs). AREs are typically permutations of the core sequence AUUUA (or, more rarely, short stretches of U) that serve as cis elements to promote the degradation of mRNAs through a deadenylation-dependent mechanism called ARE-mediated RNA decay (AMD) (65,66).
AREs have been computationally defined in mammalian transcriptomes and deposited in several databases, including ARED and AREsite (32,67). However, these databases do not contain most Olfr mRNAs, including those annotated recently (28). Our annotation of Olfr genes expressed in the mouse OE together with another data set provided an opportunity to fill this gap. We analyzed the average ARE density in Olfr mRNAs and found it is twice as high as in the Gpcr and Ctrl mRNA groups (Figure 5A, Supplementary Table S6). We found this to also be the case when we analyzed the recently defined Olfr mRNA data set from Ibarra-Soria et al. (28) (‘Olfr-B’ in Figure 5A). Given that Olfr mRNAs have much shorter average 3′ UTR length than these other groups (see below), this is consistent with the possibility that there has been strong selection pressure for enrichment of AREs to maintain regulation by ARE-binding proteins. The net result is that AREs are as prevalent in Olfr mRNAs as in the other gene groups (Figure 5B).
Figure 5.
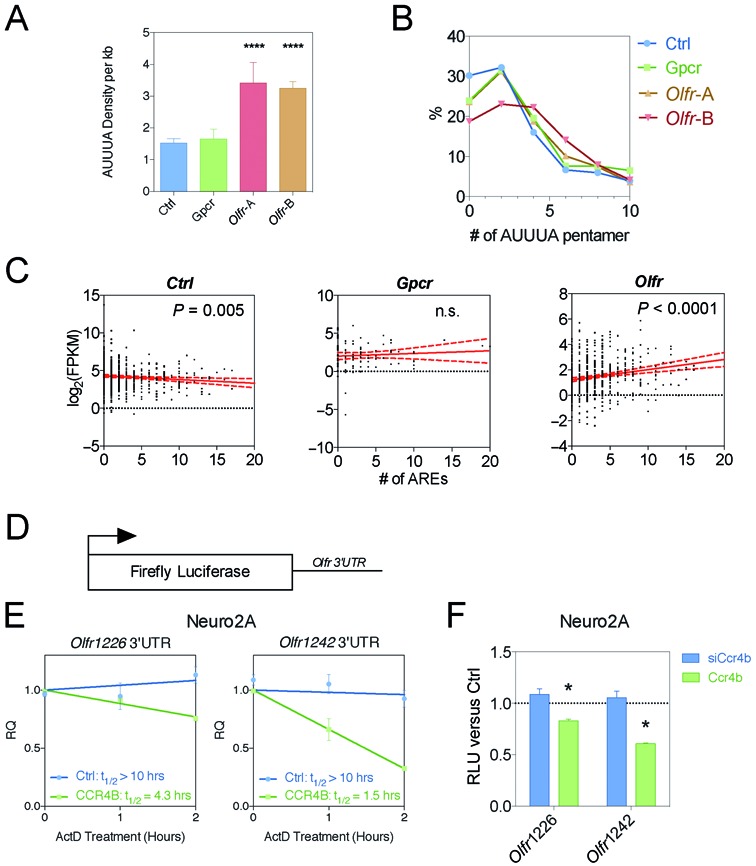
Olfr mRNAs are ARE-rich. (A) ARE pentamer density measured in silico. (B) Density distribution plot of the number of AUUUA ARE pentamers, assessed as in panel (A). (C) Scatterplots and linear regression analysis of expression level versus AUUUA pentamer density, assessed as in panel A. (D) Schematic of luciferase and renilla reporters used. (E,F) Two Olfr 3′ UTRs influence mRNA steady-state level and mRNA half-life in response to CCR4B depletion (using a siRNA targeting CCR4B [siCcr4b]) and CCR4B overexpression (using a CCR4B expression vector). Panel E is measurement of relative mRNA levels (RQ) and panel (F) is measurement of relative luciferase values (RLU). *P < 0.05; n.s. not significant.
To assess the possible impact of AREs on Olfr expression, we asked whether there is an association between ARE density and expression level. We found a strong positive correlation between AUUUA pentamer density and expression level, as assessed by RNA-seq reads (Figure 5C). Linear regression analysis demonstrated that this was highly statistically significant (P < 0.0001). In contrast, there was a negative correlation between ARE density and expression for the Ctrl group and no significant correlation for the Gpcr group (Figure 5C). To extend this analysis, we evaluated the density of another functional ARE motif: the WWAUUUAWW nonamer (32). As with AUUUA pentamer density, WWAUUUAWW density was higher in Olfr mRNAs than in the other groups (Supplementary Figure S3A). The frequency of these nonomers per transcript positively correlated with Olfr mRNA expression level (Supplementary Figure S3B).
Our finding that Olfr 3′ UTRs are both enriched in AREs and expressed in a pattern that positively correlates with ARE density raises the possibility that Olfr mRNAs are a new class of transcripts that are positively regulated by ARE-binding proteins. While AREs are normally associated with decay, they can also promote mRNA stabilization. Some ARE-binding proteins serve as mRNAs stabilizers, including members of the embryonic lethal abnormal vision (ELAV) family member (68). At least one member of this family, HuC, is expressed in the developing OE and other neuronal tissues (69). Another family member, HuD, is neuron-specific in its expression and is responsible for regulating Gap43 mRNA, which encodes a neuron-specific protein critical for neurogenesis (70,71). This raised the possibility that AREs promote the expression of transcripts in a neuronal setting.
Olfr 3′ UTRs confer stability
We examined whether the unique 3′ UTR characteristics of Olfr mRNAs confer stable expression in neural cells. To test this possibility, we cloned the 3′ UTRs of two Olfr genes—Olfr1226 and Olfr1242—downstream of the firefly luciferase ORF in the pMIR-REPORT reporter vector (Figure 5D). We transfected these constructs into a neuronal cell line, Neuro2A, together with an endogenous control containing a Renilla plasmid reporter and measured the stability of the encoded transcripts. We found that both the Olfr1226 and Olfr1242 3′ UTRs confer an extremely long RNA half-life in Neuro2A cells, as measured by treatment with the transcriptional inhibitor, Actinomycin D (>10 h; Figure 5E). This suggests that these 3′ UTRs confer stability to Olfr mRNA in neuronal cells.
A key rate-limiting step of mRNA decay is deadenylation (65). To examine whether Olfr 3′ UTRs confer deadenylation-dependent mRNA downregulation, we measured the effect of depleting or overexpressing the core deadenylase, CCR4B, on reporter mRNAs harboring the two Olfr 3′ UTRs described above. Forced overexpression of CCR4B (Supplementary Figure S3C) downregulated Olfr 3′ UTR-dependent reporter expression and destabilized the reporter mRNA transcript in Neuro2A cells (Figure 5E–F), suggesting that, indeed, Olfr 3′ UTRs are sensitive to deadenylation-dependent decay. In contrast, siRNA-mediated knockdown of CCR4B (Supplementary Figure S3C) did not significantly affect Olfr 3′ UTR-dependent reporter expression (Figure 5F), consistent with our finding that Olfr 3′ UTRs are normally stable in neural cells (Figure 5E).
To assess the specificity of this response, we tested whether Olfr 3′UTR responds to deadenylation-dependent decay in non-neuronal cells. We used P19 embryonal carcinoma cells for this experiment, which are stem-like cells that transfect efficiently (72). As we observed in Neuro2A cells, overexpression of CCR4B from an expression vector in P19 cells (Supplementary Figure S3D) elicited reduced reporter expression from the two Olfr 3′ UTR reporters (Supplementary Figure S3E). In striking contrast to the results obtained with Neuro2A cells, knocking down CCR4B in P19 cells (Supplementary Figure S3D) increased the expression the two Olfr 3′UTR reporters and stabilized its mRNA half-life (Supplementary Figure S3E and F). The difference in Olfr 3′ UTR sensitivity to CCR4B depletion in neuronal versus non-neuronal cells suggests that the Olfr 3′ UTRs have evolved to permit stable mRNA expression specifically in neural cells.
Olfr mRNAs are uORF-rich
Another well-studied deadenylation-dependent RNA degradation mechanism is Nonsense-mediated RNA decay (NMD). This pathway that was originally identified by virtue of its ability to degrade aberrant mRNAs containing premature stop codons as a result of mutations or biosynthetic (e.g. RNA splicing) errors (73,74). More recently, it was discovered that NMD also targets normal mRNAs that have an in-frame stop codon in a context that is recognized as premature (73,75,76). Increasing evidence indicates that the ability of NMD to degrade a subset of normal mRNAs acts as a biological switch to control biological events, including developmental pathways (38,75–80). Thus, we investigated whether Olfr mRNAs might be targeted for decay by NMD.
The best-characterized context that triggers NMD is the presence of an exon-exon junction (intron) at least 55 nt downstream of the stop codon defining the end of the main ORF (81). Most Olfr mRNAs do ‘not’ have this architecture (Supplementary Figure S4). Nevertheless, we identified a small number of Olfr mRNAs (15 of 805) defined by RNA-seq analysis that do possess an ‘NMD-inducing’ architecture; i.e. an exon-exon junction >55 nt downstream of the stop codon (Supplementary Figure S4). Since virtually all mRNAs with this feature are targeted for decay by NMD (73,74), it is likely that this small subset of Olfr transcripts is subject to rapid decay by NMD, perhaps to provide developmental regulation.
Another NMD-inducing feature is the presence of a long 3′ UTR (82,83). While no specific 3′ UTR length has been shown to trigger NMD, our finding that Olfr mRNAs tend to have considerably shorter 3′ UTRs than do control mRNAs (Figure 2A) suggests that it is unlikely that Olfr transcripts are generally degraded by this mechanism.
Additionally, it has been shown that upstream open reading frames (uORFs) can trigger NMD, perhaps by virtue of the ability of the stop codon in the uORF to trigger premature translation termination and thereby recruit mRNA decay enzymes (19,74). We found that majority of the 805 Olfr mRNAs expressed in the OE (67%) have uORFs (Figure 6A, Supplementary Table S7). This is a significantly higher proportion than in the Gpcr and Ctrl groups. Only 38% of the Ctrl mRNA group have uORFs, a value consistent with the average value recently reported for all human mRNAs (84). In addition, Olfr mRNAs from both our data set and Ibarra-Soria et al. have a significantly higher average number of uORFs per transcript than Ctrl and Gpcr group (Figure 6B) despite having a similar average 5′ UTR length as the Ctrl and Gpcr groups (Figure 2C).
Figure 6.
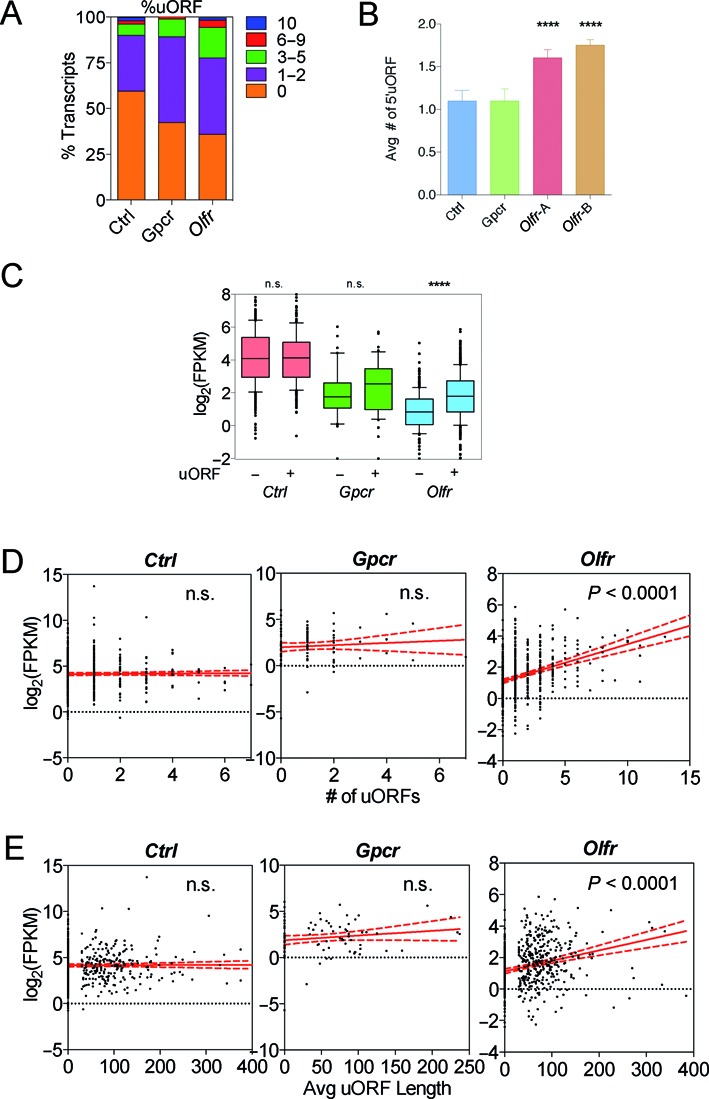
Olfr mRNAs are uORF-rich. (A) The percentage of transcripts from the three groups with the indicated number of uORFs. (B) The average number of uORFs. (C) Box plot comparing the expression level of transcripts harboring or lacking uORFs from the three groups. (D) Scatterplot and linear regression analysis of expression level versus uORFs density. (E) Scatterplot and linear regression analysis of expression level versus average uORF length. **** P < 0.0001; n.s., not statistically significant.
Even though uORFs can trigger NMD, few uORF-containing mRNAs are likely to be subject to NMD regulation (85). This follows from the fact that while ∼40% of human and rodent mRNAs have uORFs (84,86,87) (Figure 6A), at most 5% of mammalian mRNAs are targeted for decay by NMD, based on microarray and RNA-seq analyses of cells depleted of NMD factors (85). Indeed, several mRNAs have been defined that escape uORF-induced NMD (88,89). This predicts that a majority of uORFs found in Olfr mRNAs are not bona fide targets of NMD. Consistent with this prediction, we found that Olfr mRNA harboring uORFs were statistically more highly expressed, when analyzed as a group, than those without (Figure 6C). We also observed a statistically significant positive correlation between the number of uORFs per transcript and Olfr mRNA level (Figure 6D). Finally, we also observed a positive correlation between average uORF length and Olfr mRNA level (Figure 6E). As evidence for specificity, we did not observe a correlation between any of these parameters and the expression of the Gpcr or Ctrl gene groups (Figure 6C–E). Together, these results suggest that uORFs have an overall positive effect on Olfr mRNA expression. It will be interesting in the future to determine the underlying mechanism. One means by which uORF-containing mRNAs may escape uORF-induced NMD is by allowing ribosomes that terminate translation in uORFs to reinitiate downstream. In S. cerevisiae, the 5′ UTR environment surrounding uORFs has been shown to influence this. For example, it has been shown that the AU-rich region downstream of the uORF in gcn4 mRNA allows for efficient ribosome reinitiation, thereby avoiding triggering NMD (90). Given that we found that Olfr 5′ UTRs tend to be AU-rich (Figure 4), this may provide a general mechanism by which they escape uORF-induced NMD. Given that translation is generally regarded as providing a protective role from mRNA degradation (64), one explanation is that the AU-rich environment of Olfr uORFs promotes stabilization of Olfr mRNAs through their ability to promote re-initiation of translation. We note, however, that our results do not rule out that a subset of Olfr mRNAs are targeted for decay through their uORFs by NMD.
SUMMARY
A comprehensive landscape of mRNA features present in Olfr mRNAs revealed several unusual mRNA features in both the 5′ and 3′ UTR of Olfr mRNAs that we posit were selected over evolutionary time to direct the post-transcriptional regulation of Olfr genes. We provide evidence that Olfr mRNAs are extremely stable, which we posit results from one or more of these post-transcriptional features. Mechanisms potentially impacted by the unique features in Olfr mRNAs include ARE-mediated RNA decay, miRNA-mediated regulation and NMD.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
Eleen Y. Shum is a Howard Hughes Medical Institute International Student Fellow. Josh L. Espinoza is a Bridges intern at the California Institute of Regenerative Medicine. We thank the entire Wilkinson Laboratory for their help and support on this manuscript.
FUNDING
National Institutes of Health (NIH) [GM111838 to M.F.W.]. Funding for open access charge: National Institutes of Health [GM111838 to M.F.W.].
Conflict of interest statement. None declared.
REFERENCES
- 1.DeMaria S., Ngai J. The cell biology of smell. J. Cell Biol. 2010;191:443–452. doi: 10.1083/jcb.201008163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoover K.C. Evolution of olfactory receptors. Methods Mol. Biol. 2013;1003:241–249. doi: 10.1007/978-1-62703-377-0_18. [DOI] [PubMed] [Google Scholar]
- 3.Firestein S. How the olfactory system makes sense of scents. Nature. 2001;413:211–218. doi: 10.1038/35093026. [DOI] [PubMed] [Google Scholar]
- 4.Kobayakawa K., Kobayakawa R., Matsumoto H., Oka Y., Imai T., Ikawa M., Okabe M., Ikeda T., Itohara S., Kikusui T., et al. Innate versus learned odour processing in the mouse olfactory bulb. Nature. 2007;450:503–508. doi: 10.1038/nature06281. [DOI] [PubMed] [Google Scholar]
- 5.Ressler K.J., Sullivan S.L., Buck L.B. A zonal organization of odorant receptor gene expression in the olfactory epithelium. Cell. 1993;73:597–609. doi: 10.1016/0092-8674(93)90145-g. [DOI] [PubMed] [Google Scholar]
- 6.Vassar R., Ngai J., Axel R. Spatial segregation of odorant receptor expression in the mammalian olfactory epithelium. Cell. 1993;74:309–318. doi: 10.1016/0092-8674(93)90422-m. [DOI] [PubMed] [Google Scholar]
- 7.Chess A., Simon I., Cedar H., Axel R. Allelic inactivation regulates olfactory receptor gene expression. Cell. 1994;78:823–834. doi: 10.1016/s0092-8674(94)90562-2. [DOI] [PubMed] [Google Scholar]
- 8.Malnic B., Hirono J., Sato T., Buck L.B. Combinatorial receptor codes for odors. Cell. 1999;96:713–723. doi: 10.1016/s0092-8674(00)80581-4. [DOI] [PubMed] [Google Scholar]
- 9.Barnea G., O'Donnell S., Mancia F., Sun X., Nemes A., Mendelsohn M., Axel R. Odorant receptors on axon termini in the brain. Science. 2004;304:1468. doi: 10.1126/science.1096146. [DOI] [PubMed] [Google Scholar]
- 10.Shykind B.M., Rohani S.C., O'Donnell S., Nemes A., Mendelsohn M., Sun Y., Axel R., Barnea G. Gene switching and the stability of odorant receptor gene choice. Cell. 2004;117:801–815. doi: 10.1016/j.cell.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 11.Buck L., Axel R. A novel multigene family may encode odorant receptors: A molecular basis for odor recognition. Cell. 1991;65:175–187. doi: 10.1016/0092-8674(91)90418-x. [DOI] [PubMed] [Google Scholar]
- 12.Wang S.S., Lewcock J.W., Feinstein P., Mombaerts P., Reed R.R. Genetic disruptions of O/E2 and O/E3 genes reveal involvement in olfactory receptor neuron projection. Development. 2004;131:1377–1388. doi: 10.1242/dev.01009. [DOI] [PubMed] [Google Scholar]
- 13.Wang S.S., Tsai R.Y.L., Reed R.R. The characterization of the Olf-1/EBF-like HLH transcription factor family: implications in olfactory gene regulation and neuronal development. J. Neurosci. 1997;17:4149–4158. doi: 10.1523/JNEUROSCI.17-11-04149.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McClintock T.S. Achieving singularity in mammalian odorant receptor gene choice. Chem. Senses. 2010;35:447–457. doi: 10.1093/chemse/bjq041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Magklara A., Yen A., Colquitt B.M., Clowney E.J., Allen W., Markenscoff-Papadimitriou E., Evans Z.A., Kheradpour P., Mountoufaris G., Carey C., et al. An epigenetic signature for monoallelic olfactory receptor expression. Cell. 2011;145:555–570. doi: 10.1016/j.cell.2011.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyons D.B., Allen W.E., Goh T., Tsai L., Barnea G., Lomvardas S. An epigenetic trap stabilizes singular olfactory receptor expression. Cell. 2013;154:325–336. doi: 10.1016/j.cell.2013.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolognani F., Perrone-Bizzozero N.I. RNA-protein interactions and control of mRNA stability in neurons. J. Neurosci. Res. 2008;86:481–489. doi: 10.1002/jnr.21473. [DOI] [PubMed] [Google Scholar]
- 18.Pesole G., Mignone F., Gissi C., Grillo G., Licciulli F., Liuni S. Structural and functional features of eukaryotic mRNA untranslated regions. Gene. 2001;276:73–81. doi: 10.1016/s0378-1119(01)00674-6. [DOI] [PubMed] [Google Scholar]
- 19.Barrett L.W., Fletcher S., Wilton S.D. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell. Mol. Life Sci. 2012;69:3613–3634. doi: 10.1007/s00018-012-0990-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krol J., Loedige I., Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X., Rogers M., Tian H., Zhang X., Zou D.-J., Liu J., Ma M., Shepherd G.M., Firestein S.J. High-throughput microarray detection of olfactory receptor gene expression in the mouse. Proc. Natl. Acad. Sci. U.S.A. 2004;101:14168–14173. doi: 10.1073/pnas.0405350101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shiao M.-S., Chang A.Y.-F., Liao B.-Y., Ching Y.-H., Lu M.-Y.J., Chen S.M., Li W.-H. Transcriptomes of mouse olfactory epithelium reveal sexual differences in odorant detection. Genome Biol. Evol. 2012;4:703–712. doi: 10.1093/gbe/evs039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sosinsky A., Glusman G., Lancet D. The genomic structure of human olfactory receptor genes. Genomics. 2000;70:49–61. doi: 10.1006/geno.2000.6363. [DOI] [PubMed] [Google Scholar]
- 24.Lane R.P., Cutforth T., Young J., Athanasiou M., Friedman C., Rowen L., Evans G., Axel R., Hood L., Trask B.J. Genomic analysis of orthologous mouse and human olfactory receptor loci. Proc. Natl. Acad. Sci. U.S.A. 2001;98:7390–7395. doi: 10.1073/pnas.131215398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michaloski J.S., Galante P.A.F., Malnic B. Identification of potential regulatory motifs in odorant receptor genes by analysis of promoter sequences. Genome Res. 2006;16:1091–1098. doi: 10.1101/gr.5185406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clowney E.J., Magklara A., Colquitt B.M., Pathak N., Lane R.P., Lomvardas S. High-throughput mapping of the promoters of the mouse olfactory receptor genes reveals a new type of mammalian promoter and provides insight into olfactory receptor gene regulation. Genome Res. 2011;21:1249–1259. doi: 10.1101/gr.120162.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young J.M., Shykind B.M., Lane R.P., Tonnes-Priddy L., Ross J.A., Walker M., Williams E.M., Trask B.J. Odorant receptor expressed sequence tags demonstrate olfactory expression of over 400 genes, extensive alternate splicing and unequal expression levels. Genome Biol. 2003;4:R71. doi: 10.1186/gb-2003-4-11-r71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ibarra-Soria X., Levitin M.O., Saraiva L.R., Logan D.W. The olfactory transcriptomes of mice. PLoS Genet. 2014;10:e1004593. doi: 10.1371/journal.pgen.1004593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crooks G.E., Hon G., Chandonia J.-M., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gruber A.R., Fallmann J., Kratochvill F., Kovarik P., Hofacker I.L. AREsite: a database for the comprehensive investigation of AU-rich elements. Nucleic Acids Res. 2011;39:D66–D69. doi: 10.1093/nar/gkq990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewis B.P., Burge C.B., Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 34.Friedman R.C., Farh K.K.-H., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grimson A., Farh K.K.-H., Johnston W.K., Garrett-Engele P., Lim L.P., Bartel D.P. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia D.M., Baek D., Shin C., Bell G.W., Grimson A., Bartel D.P. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol. 2011;18:1139–1146. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hofacker I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruno I.G., Karam R., Huang L., Bhardwaj A., Lou C.H., Shum E.Y., Song H.-W.W., Corbett M.A., Gifford W.D., Gecz J., et al. Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol. Cell. 2011;42:500–510. doi: 10.1016/j.molcel.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 40.Niimura Y., Nei M. Extensive gains and losses of olfactory receptor genes in mammalian evolution. PLoS One. 2007;2:e708. doi: 10.1371/journal.pone.0000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyamichi K., Serizawa S., Kimura H.M., Sakano H. Continuous and overlapping expression domains of odorant receptor genes in the olfactory epithelium determine the dorsal/ventral positioning of glomeruli in the olfactory bulb. J. Neurosci. 2005;25:3586–3592. doi: 10.1523/JNEUROSCI.0324-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsuboi A., Miyazaki T., Imai T., Sakano H. Olfactory sensory neurons expressing class I odorant receptors converge their axons on an antero-dorsal domain of the olfactory bulb in the mouse. Eur. J. Neurosci. 2006;23:1436–1444. doi: 10.1111/j.1460-9568.2006.04675.x. [DOI] [PubMed] [Google Scholar]
- 43.Sharman J.L., Benson H.E., Pawson A.J., Lukito V., Mpamhanga C.P., Bombail V., Davenport A.P., Peters J.A., Spedding M., Harmar A.J. IUPHAR-DB: updated database content and new features. Nucleic Acids Res. 2013;41:D1083–D1088. doi: 10.1093/nar/gks960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spies N., Burge C.B., Bartel D.P. 3′UTR-isoform choice has limited influence on the stability and translational efficiency of most mRNAs in mouse fibroblasts. Genome Res. 2013;23:2078–2090. doi: 10.1101/gr.156919.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mayr C., Bartel D.P. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sandberg R., Neilson J.R., Sarma A., Sharp P.A., Burge C.B. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stark A., Brennecke J., Bushati N., Russell R.B., Cohen S.M. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell. 2005;123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 48.Hogg J.R., Goff S.P. Upf1 senses 3′UTR length to potentiate mRNA decay. Cell. 2010;143:379–389. doi: 10.1016/j.cell.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ji Z., Lee J.Y., Pan Z., Jiang B., Tian B. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc. Natl. Acad. Sci. U.S.A. 2009;106:7028–7033. doi: 10.1073/pnas.0900028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szostak E., Gebauer F. Translational control by 3′-UTR-binding proteins. Brief. Funct. Genomics. 2013;12:58–65. doi: 10.1093/bfgp/els056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bushati N., Cohen S.M. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 52.Bhattacharyya S.N., Habermacher R., Martine U., Closs E.I., Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 53.Choi P.S., Zakhary L., Choi W.-Y., Caron S., Alvarez-Saavedra E., Miska E.A., McManus M., Harfe B., Giraldez A.J., Horvitz H.R., et al. Members of the miRNA-200 family regulate olfactory neurogenesis. Neuron. 2008;57:41–55. doi: 10.1016/j.neuron.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen H., Shalom-Feuerstein R., Riley J., Zhang S.-D., Tucci P., Agostini M., Aberdam D., Knight R.A., Genchi G., Nicotera P., et al. miR-7 and miR-214 are specifically expressed during neuroblastoma differentiation, cortical development and embryonic stem cells differentiation, and control neurite outgrowth in vitro. Biochem. Biophys. Res. Commun. 2010;394:921–927. doi: 10.1016/j.bbrc.2010.03.076. [DOI] [PubMed] [Google Scholar]
- 55.Sempere L.F., Freemantle S., Pitha-Rowe I., Moss E., Dmitrovsky E., Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao C., Sun G., Li S., Shi Y. A feedback regulatory loop involving microRNA-9 and nuclear receptor TLX in neural stem cell fate determination. Nat. Struct. Mol. Biol. 2009;16:365–371. doi: 10.1038/nsmb.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Makeyev E. V, Zhang J., Carrasco M.A., Maniatis T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell. 2007;27:435–448. doi: 10.1016/j.molcel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edbauer D., Neilson J.R., Foster K.A., Wang C.-F., Seeburg D.P., Batterton M.N., Tada T., Dolan B.M., Sharp P.A., Sheng M. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron. 2010;65:373–384. doi: 10.1016/j.neuron.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lagos D., Pollara G., Henderson S., Gratrix F., Fabani M., Milne R.S.B., Gotch F., Boshoff C. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat. Cell Biol. 2010;12:513–519. doi: 10.1038/ncb2054. [DOI] [PubMed] [Google Scholar]
- 60.Siegel G., Obernosterer G., Fiore R., Oehmen M., Bicker S., Christensen M., Khudayberdiev S., Leuschner P.F., Busch C.J.L., Kane C., et al. A functional screen implicates microRNA-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat. Cell Biol. 2009;11:705–716. doi: 10.1038/ncb1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wulczyn F.G., Smirnova L., Rybak A., Brandt C., Kwidzinski E., Ninnemann O., Strehle M., Seiler A., Schumacher S., Nitsch R. Post-transcriptional regulation of the let-7 microRNA during neural cell specification. FASEB J. 2007;21:415–426. doi: 10.1096/fj.06-6130com. [DOI] [PubMed] [Google Scholar]
- 62.Ringnér M., Krogh M. Folding free energies of 5′-UTRs impact post-transcriptional regulation on a genomic scale in yeast. PLoS Comput. Biol. 2005;1:e72. doi: 10.1371/journal.pcbi.0010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kozak M. The scanning model for translation: an update. J. Cell Biol. 1989;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roy B., Jacobson A. The intimate relationships of mRNA decay and translation. Trends Genet. 2013;29:691–699. doi: 10.1016/j.tig.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen C.-Y.A., Shyu A.-B. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 66.Barreau C., Paillard L., Osborne H.B. AU-rich elements and associated factors: are there unifying principles. Nucleic Acids Res. 2005;33:7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bakheet T., Frevel M., Williams B.R., Greer W., Khabar K.S. ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 2001;29:246–254. doi: 10.1093/nar/29.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ford L.P., Watson J., Keene J.D., Wilusz J. ELAV proteins stabilize deadenylated intermediates in a novel in vitro mRNA deadenylation/degradation system. Genes Dev. 1999;13:188–201. doi: 10.1101/gad.13.2.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Okano H.J., Darnell R.B. A Hierarchy of Hu RNA Binding Proteins in Developing and Adult Neurons. J. Neurosci. 1997;17:3024–3037. doi: 10.1523/JNEUROSCI.17-09-03024.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anderson K.D., Morin M.A., Beckel-Mitchener A., Mobarak C.D., Neve R.L., Furneaux H.M., Burry R., Perrone-Bizzozero N.I. Overexpression of HuD, but not of its truncated form HuD I+II, promotes GAP-43 gene expression and neurite outgrowth in PC12 cells in the absence of nerve growth factor. J. Neurochem. 2002;75:1103–1114. doi: 10.1046/j.1471-4159.2000.0751103.x. [DOI] [PubMed] [Google Scholar]
- 71.Mobarak C.D., Anderson K.D., Morin M., Beckel-Mitchener A., Rogers S.L., Furneaux H., King P., Perrone-Bizzozero N.I. The RNA-binding protein HuD is required for GAP-43 mRNA stability, GAP-43 gene expression, and PKC-dependent neurite outgrowth in PC12 cells. Mol. Biol. Cell. 2000;11:3191–3203. doi: 10.1091/mbc.11.9.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McBurney M.W. P19 embryonal carcinoma cells. Int. J. Dev. Biol. 1993;37:135–140. [PubMed] [Google Scholar]
- 73.Popp M.W.-L., Maquat L.E. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu. Rev. Genet. 2013;47:139–165. doi: 10.1146/annurev-genet-111212-133424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang Y.-F., Imam J.S., Wilkinson M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 75.Huang L., Wilkinson M.F. Regulation of nonsense-mediated mRNA decay. Wiley Interdiscip. Rev. RNA. 2012;3:807–828. doi: 10.1002/wrna.1137. [DOI] [PubMed] [Google Scholar]
- 76.Karam R., Wilkinson M. A conserved microRNA/NMD regulatory circuit controls gene expression. RNA Biol. 2012;9:22–26. doi: 10.4161/rna.9.1.18010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lareau L.F., Inada M., Green R.E., Wengrod J.C., Brenner S.E. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature. 2007;446:926–929. doi: 10.1038/nature05676. [DOI] [PubMed] [Google Scholar]
- 78.Gong C., Kim Y.K., Woeller C.F., Tang Y., Maquat L.E. SMD and NMD are competitive pathways that contribute to myogenesis: effects on PAX3 and myogenin mRNAs. Genes Dev. 2009;23:54–66. doi: 10.1101/gad.1717309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Colak D., Ji S.-J., Porse B.T., Jaffrey S.R. Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell. 2013;153:1252–1265. doi: 10.1016/j.cell.2013.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lou C.H., Shao A., Shum E.Y., Espinoza J.L., Huang L., Karam R., Wilkinson M.F. Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Rep. 2014;6:748–764. doi: 10.1016/j.celrep.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nagy E., Maquat L.E. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci. 1998;23:198–199. doi: 10.1016/s0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- 82.Nicholson P., Yepiskoposyan H., Metze S., Zamudio Orozco R., Kleinschmidt N., Mühlemann O., Muhlemann O. Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell. Mol. life Sci. 2010;67:677–700. doi: 10.1007/s00018-009-0177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rebbapragada I., Lykke-Andersen J. Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr. Opin. Cell Biol. 2009;21:394–402. doi: 10.1016/j.ceb.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 84.Calvo S.E., Pagliarini D.J., Mootha V.K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. U.S.A. 2009;106:7507–7512. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hurt J.A., Robertson A.D., Burge C.B. Global analyses of UPF1 binding and function reveal expanded scope of nonsense-mediated mRNA decay. Genome Res. 2013;23:1636–1650. doi: 10.1101/gr.157354.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Iacono M., Mignone F., Pesole G. uAUG and uORFs in human and rodent 5′untranslated mRNAs. Gene. 2005;349:97–105. doi: 10.1016/j.gene.2004.11.041. [DOI] [PubMed] [Google Scholar]
- 87.Matsui M., Yachie N., Okada Y., Saito R., Tomita M. Bioinformatic analysis of post-transcriptional regulation by uORF in human and mouse. FEBS Lett. 2007;581:4184–4188. doi: 10.1016/j.febslet.2007.07.057. [DOI] [PubMed] [Google Scholar]
- 88.Stockklausner C., Breit S., Neu-Yilik G., Echner N., Hentze M.W., Kulozik A.E., Gehring N.H. The uORF-containing thrombopoietin mRNA escapes nonsense-mediated decay (NMD) Nucleic Acids Res. 2006;34:2355–2363. doi: 10.1093/nar/gkl277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Silva A.L., Pereira F.J.C., Morgado A., Kong J., Martins R., Faustino P., Liebhaber S.A., Romão L. The canonical UPF1-dependent nonsense-mediated mRNA decay is inhibited in transcripts carrying a short open reading frame independent of sequence context. RNA. 2006;12:2160–2170. doi: 10.1261/rna.201406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hinnebusch A.G. Translational regulation of yeast GCN4. A window on factors that control initiator-trna binding to the ribosome. J. Biol. Chem. 1997;272:21661–21664. doi: 10.1074/jbc.272.35.21661. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.