


Abstract
The ω gene is encoded in broad-host range and low-copy plasmids. It is genetically linked to antibiotic resistance genes of the major human pathogens of phylum Firmicutes. The homodimeric forms of ω (ω2) coordinate the plasmid copy number control, faithful partition (ω2 and δ2) and better-than-random segregation (ζϵ2ζ) systems. The promoter (P) of the ωϵζ operon (Pω) transiently interacts with ω2. Adding δ2 facilitates the formation of stable ω2·Pω complexes. Here we show that limiting ω2 interacts with the N-terminal domain of the β’ subunit of the Bacillus subtilis RNA polymerase (RNAP-σA) vegetative holoenzyme. In this way ω2 recruits RNAP-σA onto Pω DNA. Partial Pω occupancy by ω2 increases the rate at which RNAP-σA complex shifts from its closed (RPC) to open (RPO) form. This shift increases transcription activation. Adding δ2 further increases the rate of Pω transcription initiation, perhaps by stabilizing the ω2·Pω complex. In contrast, full operator occupancy by ω2 facilitates RPC formation, but it blocks RPO isomerization and represses Pω utilization. The stimulation and inhibition of RPO formation is the mechanism whereby ω2 mediates copy number fluctuation and stable plasmid segregation. By this mechanism, ω2 also indirectly influences the acquisition of antibiotic resistance genes.
INTRODUCTION
Resistance to glycopeptides, macrolides, pleuromutilins, phenicols, linezolid and other antibiotics among Gram-positive cocci is generally linked to plasmids whose copy number control, partitioning and post-segregational killing is regulated by the ω cassette (1,2). This cassette contains sequences that encode the ω or ω2 gene products (1). It is thus important to understand how homodimeric ω (ω2) (or ω2 [ω22]) functions, not only because of its intrinsic biological interest, but also because of its relevance to antibiotic resistance transmission (1,2). Plasmids of the inc18 family are commonly found in Enterococcus and Streptococcus. These plasmids have a broad host range in Firmicutes. Here, ω forms an operon with ϵ and ζ. Meanwhile, ω2 forms an operon with the ermB gene (1).
A transcriptional analysis of few inc18 plasmids (e.g. pSM19035, pIP501 and pAMβ1) revealed that ω2 controlled the expression of the copy control gene copS. Also mediated by ω2 was the expression of plasmid partition genes, such as δ and ω, and toxin-antitoxin systems, such as the ωϵζ operon (Figure 1A) (1). The inc18 plasmids persist in the population through a variety of mechanisms controlled by ω2. The ω2 protein is a ParB homologue and binds to parS centromers. In concert with δ2 (a ParA ATPase), ω2 is involved in accurate plasmid partitioning and coupling plasmid replication to faithful segregation (1,3–5). All three parS sites (Figure 1A) can cause partition-mediated incompatibility (5). Furthermore, toxin ζ stabilizes bacterial plasmids by programming the death of any host cell that fails to inherit a plasmid copy during cell division (1,6). Toxins ζ and RelE are the most ubiquitous toxins in nature. In contrast, much less is known about the purpose of the ω2 gene product, which is truncated in some members of the family (e.g. pSM19035) (Figure 1A) (1).
Figure 1.
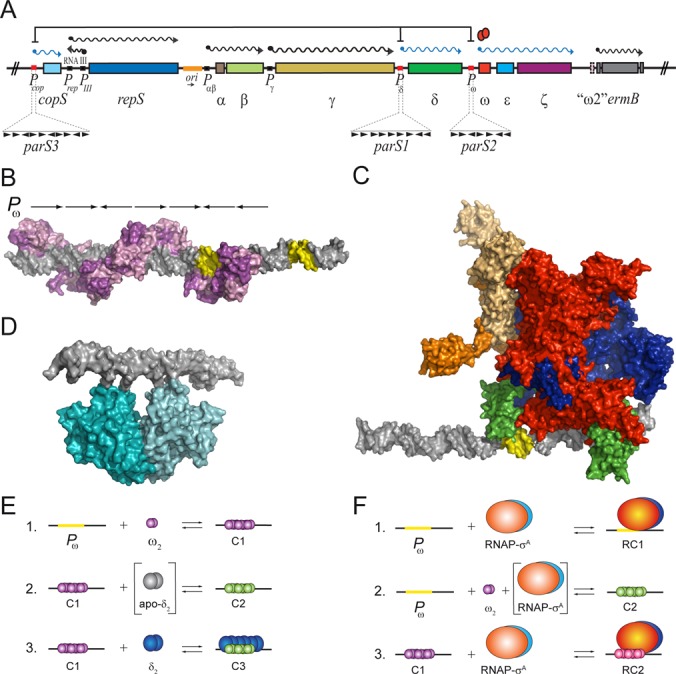
Interaction of ω2, δ2 or RNAP-σA with the Pω operator sites. (A) Genome organization of the relevant region of plasmid pSM19035. The promoters (P), the mRNAs and the genes are symbolized by boxes, wavy lanes and rectangles, respectively. The plasmid replication origin (ori) is labelled in orange. The direction of replication is denoted by a black arrows below. Protein ω2-mediated transcriptional repression is indicated (ω2, red ovals). The upstream regions of PcopS, Pδ and Pω (red box) which constitute the cis-acting parS centromeric sites magnified. The ω2 cognate sites consist of a variable number of contiguous 7-bp heptad repeats (iterons) symbolized by ▸ (in the direct orientation) or ◂ (in the inverted orientation). The number of repeats and their relative orientations are indicated. The genes involved in replication (copS, repS, RNAIII and γ), dimer resolution (β), faithful partition (δ and ω), stable segregation (ϵ and ζ) are indicated. The antibiotic resistance gene ermB and the truncated version of ω2 (‘ω2’) are also indicated. (B) A structural model of ω2-bound to Pω DNA which derived from the crystal structure of the complex of the minimal operator site and ω2Δ19 (PDB ID 1IRQ, 2BNW and 2BNZ). Pink/purple ω2 molecules form a left-handed protein-matrix winding around the nearly linear operator DNA. The DNA is represented in grey with the −35 and −10 elements in yellow. (C) Model of RNAP-σA which is derived from crystal structures of the homologous protein from T. aquaticus and T. thermophilus (PDB ID 1IW7, 2A6H) together with the Pω DNA from PDB ID 2CAX. Colour coding: brown and light brown refer to α2; blue, β; red, β’ and green, σ subunit. (D) Model of δ2 binding to DNA. The atomic coordinates of (δ·ATPγS·Mg2+)2 were taken from the 2OZE PDB entry. The modelled structures were prepared and visualized with PyMOL version 1.5.0.4. (E) Interactions of ω2 and δ2 with Pω DNA and each other: 1. ω2 (in purple) transiently interacts with Pω DNA forming complex C1; 2. The interaction of ω2 with δ2-apo (in grey), leads to functional transition of ω2 (in green) and formation of the durable C2 complex; 3. In the presence of ATP, δ2 (in blue) binds to C1 to generate C3. (F) Complexes formed by ω2 and RNAP-σA upon binding to Pω DNA: 1. RNAP-σA bound to Pω DNA forms complex RC1; 2. the interaction of RNAP-σA with limiting concentrations of ω2 leads to a functional transition of ω2 and formation of C2; and 3. RNAP-σA bound to C1 makes RC2.
Streptococcus pyogenes monomeric ω (71-residue long, 7.9 kDa) has an unstructured N-terminal domain (NTD, residues 1–24) followed by a ribbon-helix-helix (RHH) fold (residues 25–71). The latter facilitates the formation, in solution, of a dimer that has a pseudo-2-fold symmetry (7–9). The RHH domain recognizes the parS centromers embedded in the promoter regions of the cop, δ, ω and ω2 genes (Figure 1A and Supplementary Figure S1) (4). The operator binding sites are comprised of a series of 6–10 unspaced heptad repeats (5′-T/AATCACT/A-3′) in a forward orientation. Alternatively, they consist of two or three repeats of the following: two heptads in a forward orientation followed by one in an inverse orientation (→→←) (Supplementary Figure S1). Both the ω2 and the NTD lacking ω2ΔN19 mutant transiently bind promoters and repress promoter utilization both in vivo and in vitro (4,9–13).
The minimal ω2 binding site is comprised of two contiguous heptads in a forward (→→) or inverted (→←) orientation. It has higher affinity for the latter (see 10). The structure of the complex of ω2 bound to →→ DNA is very similar to the one of ω2 bound to →← DNA. In neither case does ω2 distort the DNA when binding to it (9,14). These structures show that a pair of positively charged antiparallel β strands from ω2 insert into the major groove of DNA. The β strands make specific and sequence-dependent contacts with symmetric or asymmetric repetitive sequences that deviate 0.3 Å with respect to the central C-G pair of each repetition (8,9,14). In a full cognate site, ω2 is displaced ∼7-bp and rotated 252º with respect to its neighbouring dimer. The negatively charged sugar-phosphate DNA backbone faces the positively charged surface of the protein (Figure 1B) (9).
Protein ω2 transiently binds with high affinity (apparent dissociation constant [KDapp] = 5 ± 1 nM) and cooperativity to Pω DNA (Figure 1E, condition 1 [C1]) (13). The physical interaction of the apo form of δ2 with ω2 bound to Pω DNA facilitates a structural transition in ω2 that might involve folding of its unstructured NTD (Figure 1E, condition 2) (see 13,15). ω2 stably binds Pω DNA with high affinity (KDapp = 0.7 ± 0.1 nM), forming the C2 complex (ω2·Pω DNA) (Figure 1E, condition 2). The C2 complex is stable, with a half-life of >30 min, whereas the C1 complex is transient, with half-life of <1 min (10,13,15). However, despite the difference in stability, C1 and C2 have a similar mobility in a PAGE at low protein concentrations.
The δ2 protein is a U-shaped ATPase that in its ATP-bound form, binds non-specifically to DNA (Figure 1D) (16). In the presence of ATP, δ2 interacts with C1 to form the C3 complex (Figure 1E, condition 3) (13). Since it lacks the unfolded NTD, ω2ΔN19 cannot facilitate C2 and C3 formation (13). Given that δ2 (a ParA ATPase) works together with ω2 (a ParB centromeric binding protein) bound to parS (e.g. Pω DNA) to promote faithful plasmid segregation (12,13), it is likely that δ2 also contributes to ω2-mediated transcription regulation.
Transcription initiation by the multisubunit RNA polymerase (RNAP) is an intricate multistep process (17–20). Bacterial RNAP exists in two forms: i) the ubiquitous core enzyme, which consists of the dimeric form of α (α2), the monomeric form of β, β’, and one or more of small non-essential subunits; this carries out processive transcription elongation followed by termination; and ii) the RNAP-σ holoenzyme, in which a dissociable σ subunit, essential for promoter recognition, has joined the core enzyme (21–23). The Bacillus subtilis vegetative RNAP-σA holoenzyme binds to specific −10 and −35 promoter (P) elements to form an unstable closed binary complex (RPC) (21,24–27). A RNAP-σA-assisted isomerization step then occurs. This is mediated by kinetically unstable intermediates (RPI). This, in turn, leads to P melting of ∼14-bp (−12 to +2) in the DNA surrounding the transcription start site. This process yields the catalytically active, open RNAP-σA·P DNA complex (RPO) (17,28). The structures responsible for the functions associated with RPo formation are predominantly located in the σ, β and β’ subunits of the RNAP-σ (18,22,23,25,28). In the presence of nucleotide triphosphates, an initiation complex (RPINIT) is formed. This complex is a prerequisite for displacement of RNAP-σA from the promoter through an elongation complex (RPE) (24,25,28). RNAP subunits δ, ϵ and ω are not essential for this process and their roles are therefore poorly understood.
As a result of the association of regulatory elements to promoter-embedded operator sequences, gene regulation is often achieved at the level of transcription initiation (29). The ω2 protein interacts with its cognate sites as a left-handed protein helix wrapped around a nearly linear Pω DNA (9). In this structure the −35 and −10 elements are free to interact with RNAP-σA (Figure 1B, yellow regions). A model of RNAP-σ bound to Pω DNA suggests that ω2 might repress transcription by steric hindrance (Figure 1C). However, preliminary results indicate that ω2 forms a ternary complex with RNAP-σA and PcopS DNA (4). These data suggest that ω2 regulates transcription through a mechanism that does not exclude the RNAP-σA from the RPC. It is assumed that this mechanism also applies to Pω and Pδ. In this study, we aimed to unravel the mechanism of ω2-mediated transcriptional regulation of Pω DNA, in vitro, and Pδ utilization, in vivo. We first characterized the effect that ω2 binding to Pω DNA had on RNAP-σA promoter recognition. We also tested whether or not modifying the stoichiometry of ω2, δ2 and RNAP-σA resulted in variations in their affinity for Pω DNA. Also investigated was whether or not transcription activation or repression by ω2 required direct contacts with RNAP-σA and δ2. Another important question was whether or not this binding was cooperative. Based on the results of this study, we present a model that explains how ω2-mediated transcriptional regulation functions.
MATERIALS AND METHODS
Bacterial strains and plasmids
The E. coli strains DH5α (Invitrogen) and ER2566 (New England Biolabs) and the B. subtilis strains BG214, BG508 (4) and NIG2001 (30) were used. The BG508 strain carries Pδ fused to a promoter-less lacZ gene. This construct was integrated as a unique copy into the amyE locus of the B. subtilis chromosome (4). In the NIG2001 strain, the wild-type (wt) rpoC gene was substituted in the B. subtilis genome with a version that had a His-tag coding sequence fused to the 3′-end (30). The Pω bearing pCB30 plasmid was used for promoter analysis, and pHP14 was used for cloning purposes (4). The plasmids used for gene over-expression were pT712ω bearing ω, pCB746 bearing δ (4,11,16), and pT712ωD56A bearing the ωD56A gene (this work). The single mutations in the ω gene were obtained by gene synthesis (Genscript). BG508 cells bearing pHP14 carrying either the ω, δ, ωδ, ωΔN19, ω2, ωK52A, ωE53A, ωD56A, ωR64A or ωK70A genes were used for the β-galactosidase assays. The native promoters of these genes were also incorporated into the constructs.
DNA, RNA, proteins and reagents
Plasmid DNA was purified as described (4). The multiple mutations in heptads 1, 1 plus 2, 7 and 7 plus 6 were obtained by in vitro synthesis (Genscript). DNA restriction and modification enzymes and RNaseA were purchased from Boehringer Mannheim and the nucleotides were purchased from Sigma. Gel-purified DNA fragments were end-labelled as described (4). The amount of DNA was quantified using molar extinction coefficient of 6500 M−1 cm−1 at 260 nm and was expressed in moles of DNA molecules.
The RNAP-σA was purified using Ni-NTA and Q-sepharose columns as described (30). The ω2 and δ2 proteins were purified as described (4,11,16). Protein ωD56A was purified as wt ω2. Protein concentrations were calculated using molar extinction coefficients at 280 nm of 2980, 2980, 38 850 and 236 000 M−1 cm−1 for ω2, ωD56A, δ2, and RNAP-σA, respectively. Concentrations were expressed in molarity of protein monomers for RNAP-σA and of dimers for δ2, ω2 and ω2 derivatives. Note that unless otherwise stated, δ2 is in the ATP·Mg2+-bound form.
B. subtilis BG508 harbouring different plasmids was grown to OD600 = ∼0.5 and aliquots were used for β-galactosidase assays (4). The cultures were pelleted and resuspended in buffer B (10 mM Na2HPO4/NaH2PO4 pH 7.2, 50 mM β-mercaptoethanol, 1 mM MgCl2, 10 mM KCl) containing 0.4 μg/ml lysozyme (4). After a 5 min incubation at 37ºC, the lysates were clarified by centrifugation for 5 min at 12,000 g and assayed for β-galactosidase activity, as described (31).
Protein cross-linking was used to study potential protein–protein interactions. For this, bisdisuccinimidyl suberate (DSS) was employed as the crosslinking agent and SDS-PAGE was used to visualize the result (13). Two-dimensional gel electrophoresis (2D) was performed essentially as described (32). The resolved proteins were transferred onto a 0.45 μm polyvinylidene fluoride membrane (PVDF, Millipore). Rabbit polyclonal anti-ω2 and anti-RNAP-σA antibodies were obtained using standard techniques (4).
Far-western blotting was used to probe the direct interaction between ω2 and RNAP-σA. The prey used were ω2 (1 μg), RNAP-σA (1 μg) and bovine serum albumin (BSA, 5 μg used as a control); these were resolved by SDS-PAGE and transferred to a PVDF membrane. The protein was renatured by incubation of the membrane in TBS containing 0.05% Tween, 10% glycerol and 5 mM β-mercaptoethanol followed by a blocking step with 5% skim milk, as described (4). The efficiency of the protein transfer was checked by Ponceau staining. The membrane was then incubated with 2 μg/ml of bait protein. To detect interactions between the bait and prey, rabbit polyclonal antibodies against the bait were employed as described (33).
Tryptic digestion of gel-purified protein bands, spotting onto the MALDI-targets, and MALDI-TOF-TOF of the spotted peptides were carried out as previously described (34).
In vitro transcription experiments
A 423-bp Pω DNA sequence (5 nM) was used as a template for in vitro transcription run-off assays. 20 μl reaction mixtures containing 20 nM B. subtilis RNAP-σA, variable concentrations of ω2, δ2 or both, 0.5 mM each of ATP, CTP, GTP and UTP plus 3000 Ci/mmol [α-32P]-UTP in buffer C (25 mM Tris-HCl, pH 8, 6 mM MgOAc, 5 mM DTT), and 20 U RNasin (Promega) were prepared. After 6 min of incubation at 37°C, the reactions were stopped by adding 10 μl of formamide. RNAs were analysed by 8% denaturing (d) polyacrylamide gel electrophoresis (PAGE), and autoradiographed. Chemical sequencing reactions of the purines were run in parallel to determine the sizes of the cDNAs.
Protein-DNA complexes
For electrophoretic mobility shift assays (EMSA), the 423-bp [α32P]-Pω DNA (0.1 nM) was incubated either with a variety of concentrations of ω2, δ2 or RNAP-σA or with a constant concentration of one component and a range of concentrations of the others. Incubations were performed in buffer D (50 mM Tris-HCl pH 7.5, 50 mM NaCl, 10 mM MgCl2) for 15 min at 37°C in a 20 μl reaction. Mixtures were subjected to 6% PAGE in 1xTAE at 4ºC. Gels were dried prior to autoradiographical analysis.
In order to obtain KDapp values from the EMSA experiments, the relative concentrations of free DNA and protein·DNA complexes were densitometrically determined under non-saturating conditions using differently exposed autoradiographs of the EMSA gels. The protein concentration needed to trap 50% of the free, labelled DNA containing the same molar concentration of heptads, in complexes is approximately equal to the KDapp under conditions where the DNA concentration is much lower than the KDapp.
Reaction conditions similar to those used for EMSA were employed in footprinting experiments. The 423-bp [α32P]-Pω DNA (1 nM) was incubated with variable protein concentrations and treated with DNaseI, as previously described (4). The samples were resolved by 6% dPAGE and the gel was dried prior to autoradiographical analysis. For KMnO4 footprinting, the samples were treated with 1 mM KMnO4 for 0.5 min at 37º C, after which the DNA was cleaved with piperidine (35).
RESULTS
RNAP-σA facilitates ω2·Pω DNA complex formation
In order to unravel the mechanism of ω2-mediated regulation of Pω utilization, the interaction of ω2 and/or RNAP-σA with Pω DNA was assayed by EMSA (Figure 2A and 2B). Pω DNA has 7 discrete ω2 cognate sites. A 423-bp DNA segment containing Pω bound ω2 with high affinity to form ω2·Pω DNA also known as the C1 complex (KDapp of 6 ± 1.7 nM) (Figure 2C filled circles). When Pω DNA was replaced with a non-specific DNA, the affinity of ω2 was low, with a KDapp > 500 nM (data not shown) (4). This confirmed previously reported data indicating a high affinity of ω2 for PcopS, Pδ and Pω (10). In addition, binding was found to be cooperative (Figure 2C). Pω also bound RNAP-σA with high affinity to form RNAP-σA·Pω DNA, also known as the RC1 complex (KDapp of 29.4 ± 9 nM, Figure 2C filled rombs).
Figure 2.
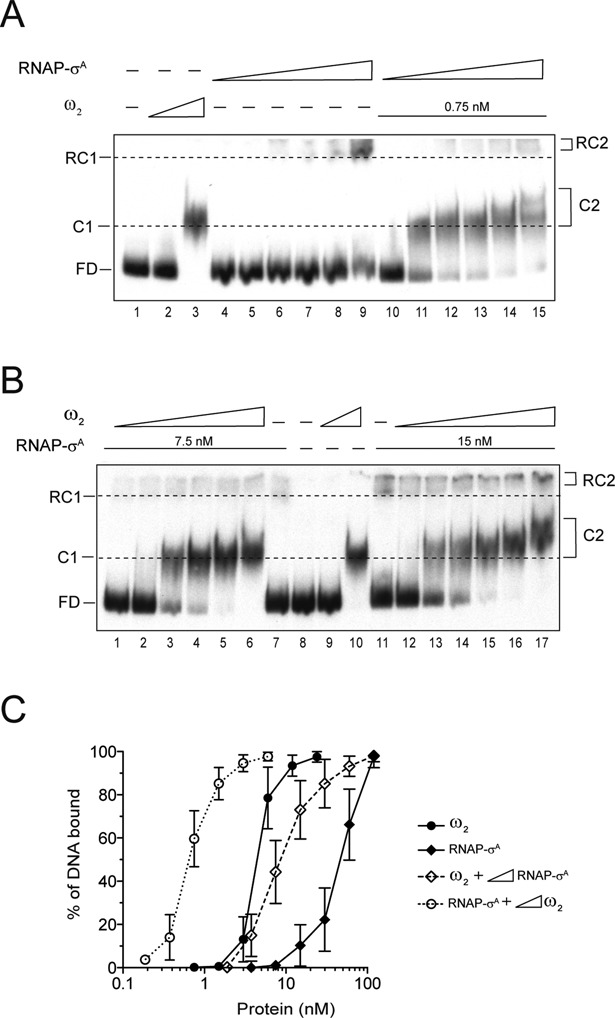
Cooperative binding of ω2 and RNAP-σA to Pω DNA. (A) EMSA of (0.1 nM) 423-bp [α32P]-Pω DNA incubated with 0.75 nM and 6 nM ω2 (lanes 2 and 3), increasing concentrations of RNAP-σA (1.9, 3.7, 7.5, 15, 30 and 60 nM, lanes 4–9), or fix ω2 (0.75 nM) and increasing concentrations of RNAP-σA (1.9–60 nM) (lanes 10–15) in buffer D. (B) EMSA of [α32P]-Pω DNA with of 0.75 nM and 6 nM ω2 (lanes 9 and 10, respectively) or with 7.5 nM (lanes 1–7) or 15 nM (lanes 11–17) RNAP-σA and increasing concentrations of ω2 (0.19, 0.37, 0.75, 1.5, 3 and 6 nM) in buffer D. (C) Graph showing the percentage of Pω DNA bound to the proteins based on densitometric data of bands from the above gels. The signals present in the protein-DNA complex and in the free-DNA (FD) were determined by densitometry. The data presented here are averages and standard deviations of the results of at least four independent experiments.
In the presence of limiting ω2 (0.75 nM) concentrations, ω2·Pω DNA complex formation was not observed (Figure 2A, lane 2 and 2C filled circles). Increased concentrations of RNAP-σA enhanced recruitment of ω2 to the promoter region by at least 8-fold. This was observed as an increase in the formation of high affinity C2 complexes. This effect was detected using limiting RNAP-σA concentrations (3.7 nM) (Figure 2A, lanes 11–15). The ‘cooperative’ binding leading to C2 complex formation could not be attributed to molecular crowding, because the BSA control did not function as a substitute for RNAP-σA (data not shown). The stable C2 and the transient C1 had a similar mobility. However, at higher concentrations of ω2, the mobility of C2 was even further diminished (Figure 2A, lanes 3 versus 11). The C1 complex had a half-life of <1 min (10). The formation of the ω2·Pω DNA complex was also enhanced at least 8-fold (KDapp of 0.6 ± 0.2 nM) when a fixed limiting concentration of RNAP-σA (∼4-fold below KDapp) and increasing concentrations of ω2 were incubated with Pω DNA (Figure 2B, lanes 1–6 and 2C open circles).
To distinguish cooperative binding from a mechanism whereby a protein–protein interaction preceded binding to Pω DNA, the experiment was modified by doubling the concentration of RNAP (∼2-fold below KDapp). In the presence of sub-stoichiometric concentrations of RNAP-σA (15 nM), the ω2·Pω DNA complex formation became enhanced by at least 10-fold. This resulted in a stoichiometry of ∼2 ω2/Pω DNA (Figure 2B, lanes 12–17 and 2C, open circles). This is consistent with the known characteristics of ω2 binding to Pω DNA which had a stoichiometry of ∼1 ± 0.2 ω2 /heptad. The minimal ω2 binding site consisted of two contiguous heptads (9–12). It is therefore likely that ω2 interacts with RNAP-σA, and that such an interaction induces a conformational change in the former that increases its apparent affinity for Pω DNA. This favours the formation of the C2 complex. Meanwhile, in the absence of RNAP-σA, ω2-bound to Pω forms the C1 complex with an 8-fold lower apparent affinity (KDapp of 6 ± 1.7 nM).
A low concentration of ω2 facilitates the formation of the RNAP-σA·Pω DNA complex
Limiting concentrations of ω2 enhanced the recruitment of RNAP-σA to Pω DNA (Figure 2B, lanes 1–3 and 12–14). 7.5 nM RNAP-σA was the limiting concentration of RNAP-σA necessary to detect the RC2 complex (RNAP-σA·Pω DNA·ω2). This was ∼4-fold less than the KDapp (Figure 2C open versus filled rombs). It is therefore likely that ω2 interacts with RNAP-σA and facilitates a functional transition of RNAP-σA. To test whether or not ω2 and RNAP-σA co-localize in a RC2 complex, ω2 and RNAP-σA were incubated with Pω DNA and subjected to DNase I footprinting analysis. The ω2 protein protected nucleotides −22 to −75 (with a numbering relative to the +1 transcription start site) (Supplementary Figure S2A, lanes 4–5). Meanwhile, RNAP-σA made a weak but significant contact with a segment located between positions −53 to +18. In parallel, a clearly hypersensitive site appeared at position −37. This is denoted by a dotted line square in Supplementary Figure S2A, lanes 6–9. Addition of a limiting concentration of ω2 of ∼4-fold below the KDapp resulted in the disappearance of the hypersensitive site. This effect was reversed upon increasing the concentration of RNAP-σA (Supplementary Figure S2A, lanes 10–13). It was also reversed by increasing the concentration of ω2 to ∼2-fold below the KDapp (Supplementary Figure S2B, lanes 13–16). Judging by the fading out of the hypersensitive site at position −37, it is likely that limiting concentrations of ω2 or RNAP-σA reposition RNAP-σA on the Pω DNA.
Addition of sub- to stoichiometric concentrations of ω2 led to an increase in the formation of RC2 (RNAP-σA·Pω DNA·ω2) complex formation by at least 3-fold (KDapp of 9.5 ± 3.4 nM) (Figure 2B, lane 17 and 2C, open rombs). Equilibrium was thus reached at about ∼4 ω2/RNAP-σA/Pω DNA. These results suggested that there was not a sufficient number of ω2 molecules to occupy the seven Pω heptads. Since ω2 binds with a slightly higher affinity and cooperativity to heptad pairs in the →← than in the →→ orientation (10), we favour the hypothesis that the →← heptads at positions −41 to −27, which overlap the −35 element and its neighbours, might be the ones recruited by ω2 to interact with RNAP-σA. Indeed, sub- to stoichiometric concentrations of ω2 bound to Pω DNA protected this region from DNase I attack (Supplementary Figure S2B, lanes 3–6). Meanwhile at stoichiometric concentrations RNAP-σA made weak but extensive contacts with the upstream region, which had the same exposed hypersensitive site at position −37 (Supplementary Figure S2B, lanes 8–11). At sub-stoichiometric concentrations of ω2, RNAP-σA made extensive contacts with the upstream −35 region. The hypersensitive site at position −37 remained exposed (Supplementary Figure S2B, lanes 13–16). This hypersensitive site was lost in the presence of stoichiometric concentrations of ω2. Meanwhile, protection from DNase I was only observed in a stretch of DNA between positions −72 to −21 (Supplementary Figure S2B, lanes 18–21). In light of these results, it is likely that: (i) ω2 physically interacts with RNAP-σA; and (ii) depending on the experimental conditions, ω2 either displaces RNAP-σA from or re-localises with it on Pω DNA.
The interaction between ω2 and δ2 does not affect RNAP-σA binding to Pω DNA
Protein δ2 bound non-specific DNA (KDapp 130 ± 20 nM) in the presence of ATP. This lead to the formation of the DC complex (Supplementary Figure S3A, lanes 6–8) (13). Binding of δ2 to non-specific DNA increased 3 to 4-fold in the presence of ω2·Pω DNA (Supplementary Figure S3A, lanes 10–12, and S3C, empty squares). In the absence of ATP, δ2 only augmented the affinity of ω2 for Pω DNA by 6- to 10-fold (C2 formation) (Supplementary Figure S3B, lanes 10–15 and Figure 1E, condition 2). This was consistent with observations that: (i) the presence of apo-δ2 decreased the off rate of ω2 from ω2·Pω DNA complexes; (ii) the presence of ω2 significantly increased the half-life of the δ2·Pω DNA complexes in the presence of ATP (13); and (iii) upon interacting with δ2, ω2 that is bound to Pω DNA (parS) undergoes a structural transition that might involve the formation of an α-helix in the normally unstructured NTD (see 15).
To test whether the interaction of ω2 with δ2 or RNAP-σA are mutually exclusive or if the interaction between δ2 and ω2 affects ω2-mediated recruitment onto the Pω DNA of RNAP-σA, limiting concentrations of ω2 (∼8-fold below KDapp) and/or RNAP-σA (∼4-fold below KDapp) were incubated with increasing δ2 concentrations and subjected to an EMSA (Supplementary Figure S3A and S3B, lanes 10–15). In the presence of ATP, assembly of the ternary C3 complex (δ2·ω2·Pω DNA) occurred (Supplementary Figure S3A, lanes 10–15). However, these results were not observed when ATP was omitted (Supplementary Figure S3B, lanes 10–15). Meanwhile, assembly of the ternary RC2 complex (ω2·Pω·RNAP-σA) was observed without ATP (Supplementary Figure S3A and S3B, lanes 13–15). Finally, in the presence of ATP, δ2 did not significantly affect the affinity of RNAP-σA for Pω in the presence of ATP (Supplementary Figure S3D, lanes 8–16).
Whether ω2 functions as an activator or repressor is dependent on its concentration
Protein ω2 binds PcopS, Pδ and Pω DNA with a stoichiometry of ∼1 ± 0.2 ω2 /heptad (9,12). We previously mapped the pSM19035 transcription start sites of PcopS, Pδ and Pω (see Supplementary Figure S1). In that study, we showed that 7.5 – 15 ω2/PcopS, Pδ or Pω DNA represses promoter utilization (4). To gain insight into the mechanism by which ω2 regulates promoter utilization, we performed transcription run-off experiments using RNAP-σA (at KDapp) in the presence of increasing concentrations of ω2. Linear Pω DNA containing seven heptads was used as the template (Figure 3A). As expected, 282-nt mRNA transcripts were produced (Figure 3B, lane 1). This result is consistent with the location of the initial nucleotides of Pω, which had been previously mapped in vivo (4).
Figure 3.
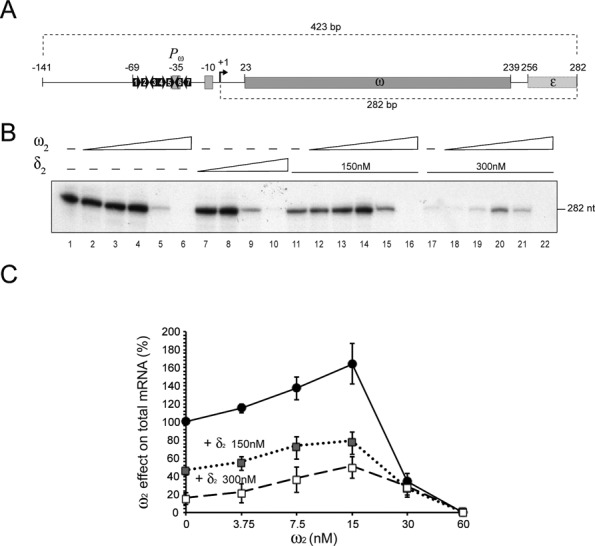
RNAP-σA mediated transcription as a function of the presence or absence of ω2 and δ2. (A) A 423-bp DNA segment containing Pω is depicted. The heptads are labelled and their relative orientations are represented by arrows. The positions of the −35 and −10 elements are indicated with filled rectangles. The transcription start site is represented with a solid arrow bent 90º. The ω and part of the ϵ gene are indicated as well. (B) Run-off experiments: the 423-bp Pω DNA (5 nM) was the template in an in vitro transcription experiment using [α32P]-UTP in buffer C. RNAP-σA (20 nM) was present in all cases. Results shown for transcription in the absence (lane 1) or presence of increasing concentrations of either ω2 (3.7, 7.5, 15, 30 and 60 nM, lanes 2–6) or δ2 (37, 75, 150, 300 nM, lanes 7–10). Also shown are results of assays in the presence of either 150 or 300 nM δ2 (lanes 11–16 or 17–22 respectively) with increasing concentrations of ω2. (C) Quantification of mRNA synthesis in the presence of increasing concentrations ω2 alone or in the presence of a fix concentration of δ2 (150 or 300 nM). Shown here are the analysed results from five independent experiments.
In the presence of limiting concentrations of ω2 (0.9 and 1.8 ω2/Pω DNA), transcriptional activation was modest (1.4- to 1.7-fold), but reproducible (Figure 3B, lanes 2–4, and 3C). Protein ω2 might preferentially bind the heptads of the Pω DNA overlapping the −35 element. To test this hypothesis, the heptad 7, heptads 6 plus 7 and, as controls, heptads 1 or 1 plus 2 were inactivated (Figure 3A). As documented in Supplemental material Annex 1, the selective occupancy of heptads 6 and 7 versus 1 and 2 plays a minor role, if at all, in ω2-mediated activation of Pω utilization.
Stoichiometric concentrations of 7.5 ω2/Pω DNA inhibited Pω expression by 4- to 8-fold. At slightly saturating conditions (15 ω2/Pω DNA or ∼2 ω2/heptad) mRNA synthesis halted completely (>50-fold) (Figure 3B, lanes 5 and 6). Concentrations of ω2 equal to or higher than those required to repress Pω did not affect the expression of the unrelated promoter (Pcro of phage A2) (data not shown). We could hence rule out RNase contamination or any other nonspecific effect as the reason for the lack of RNA synthesis. It is therefore likely that ω2 has a dual activity: at limiting concentrations it facilitates the Pω-RNAP-σA interaction, but at stoichiometric concentrations and higher, transcriptional repression results.
Limiting concentrations of ω2 facilitate the transition from RPC to RPO and stoichiometric concentrations of ω2 block this shift
To discern the mechanism of ω2-mediated repression of Pω, we investigated RPO complex formation and abortive initiation (RPINIT) effects in the presence of variable concentrations of ω2. For this, we carried out KMnO4 footprinting assays in the presence or absence of GTP and ATP. Up to 9-nt long transcripts were synthesized in these assays (see Supplementary Figure S1).
No oxidized thymines were detected on the template strand after 50 s of KMnO4 exposure (Figure 4, lanes 1, 7, 13 and 19). In the absence of nucleotide precursors, the position in the Pω DNA of non-base-paired thymines preferentially attacked by KMnO4 revealed that RNAP-σA promoted spontaneous formation of a RPO complex centred at position −11T and −10T of the template strand, rather than an extended P melting of ∼14-bp (from −12 to +2) (Figure 4, lanes 2 and 8). KMnO4-promoted cleavage of RNAP-σA bound template increased in the presence of sub-stoichiometric concentrations of ω2 (Figure 4, Supplementary Figure S4A, lanes 3–4). Cleavage was inhibited at saturating concentrations of ω2 (Figure 4, Supplementary Figure S4A, lanes 5–6). However, increased cleavage was not observed when sub-stoichiometric ω2 concentrations were added to reactions containing RNAP-σA, i.e. with a pre-formed RPO (Figure 4, lanes 9–10).
Figure 4.
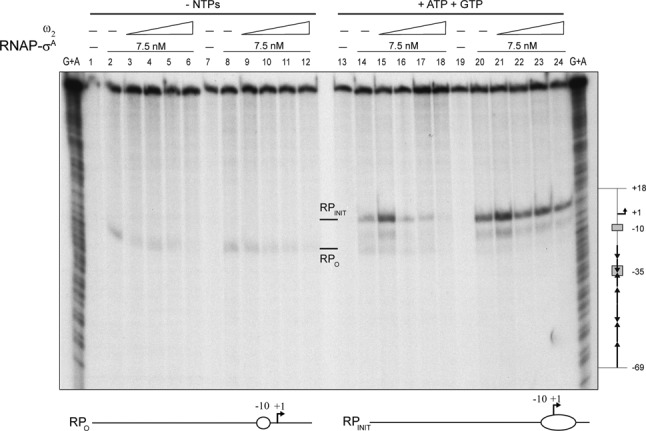
Effect of ω2 on the formation of RPO at Pω. The 423-bp [α32P]-Pω DNA (1 nM) was pre-incubated with increasing concentrations of ω2 (7.5, 15, 30 and 60 nM; lanes 2–6 and 14–18) or with 7.5 nM RNAP-σA (lanes 8–12 and 20–24) in buffer C. A second protein was added along with the initiating nucleotides, GTP and ATP (as indicated). DNA melting was probed by KMnO4 footprinting as a way of observing the open complex. The positions hypersensitive to KMnO4 are labelled (RPO and RPINIT) and depicted at the bottom of the figure. The coordinates are relative to the transcription start point. Chemical sequencing reactions for purines (G +A) are shown and the relevant regions of Pω depicted to the right of the figure.
In the presence of ATP and GTP, RNAP-σA promoted abortive initiation and synthesis of up to 9-nt oligonucleotides (RPINIT) (data not shown). KMnO4 attack revealed the formation of an extended single-stranded bubble that cleaved at positions −10T, −6T, −5T and +7T (Figure 4, lanes 14 and 20). When ω2 and RNAP-σA were left out, no cleavage was observed (Figure 4, lanes 13 and 19). Pre-incubation of Pω DNA with sub-saturating concentrations of ω2 followed by addition of RNAP-σA resulted in a significant increase (∼2.5-fold) in KMnO4 cleavage (Figure 4, lane 15 and 21 and Supplementary Figure S4B). At higher ω2 concentrations, the reaction became inhibited (Figure 4, lanes 17–18, Supplementary Figure S4A, lanes 15–16 and S4B). However, RNAP-σA-mediated RPO formation was hardly, if at all, affected by addition of ω2 to the preformed RNAP-σA·Pω complexes (Figure 4, lanes 21–24).
Altogether, these data suggest that: (i) ω2 does not repress transcription by sterically hindering the interaction between RNAP-σA and Pω DNA; (ii) in the absence of the nucleotides cofactors, RNAP-σA forms a short RPO complex on Pω centred at position −11T and −10T; (iii) limiting concentrations of ω2 push RPO to begin RNA synthesis (RPINIT); (iv) stoichiometric concentrations of ω2 inhibit RPO formation, with ω2 blocking the isomerization of RPC to RPO; and (v) ω2 has no apparent effect on pre-formed RPO. We cannot rule out that saturating concentrations of ω2 may inhibit transcription by steric occlusion or relocation of RNAP-σA on preformed ω2·Pω complexes (see Supplementary Figure S2B, lanes 18–21).
Protein δ2 represses Pω expression
In the presence of ATP, δ2 binds non-specific DNA with a KDapp = 130 ± 20 nM) (Supplementary Figure S3A, lanes 6–8) (13). As shown in Figure 3B (lane 1 versus 7–10), limiting concentrations of δ2 did not affect transcription of Pω. Meanwhile, stoichiometric and saturating concentrations of δ2 inhibited Pω utilization (2.8- to 3.5-fold). Ultimately utilization was blocked entirely.
To unravel the mechanism of δ2-mediated Pω repression, KMnO4 cleavage experiments were performed. Pre-incubation of Pω DNA with sub-stoichiometric to stoichiometric concentrations of δ2 followed by addition of RNAP-σA did not alter the pattern of KMnO4 cleavage obtained in the absence of δ2 (Supplementary Figure S5, lanes 1–4 versus 5). This result suggested that δ2 represses Pω (Figure 3, lanes 9–10) through gene silencing, i.e. halting RNAP elongation (Figure 3, lanes 9–10). The same model has been as proposed for other ParAB systems to which δ2 and ω2 belong (36,37). This model is also consistent with the observation that non-specific binding of δ2 to DNA might occlude RNAP-σA clearance or affect RNAP mediated elongation.
Protein δ2 acts as a co-activator of Pω expression
Upon coming into contact with ω2·Pω complexes, δ2 protein bound to non-specific DNA relocates onto ω2·Pω to form ternary C3 complexes (Figure 1E, condition 3) (12). The C3 complex is characterized by a longer half-life than is C1 (ω2·Pω DNA). To test whether C3 might affect transcription, Pω run-off experiments were performed (Figure 3B, lanes 11–22). At 150 nM, a concentration of δ2 equivalent to its KDapp, addition of limiting concentrations of ω2 (1.8 to 3.7 ω2/Pω DNA) significantly stimulated mRNA synthesis by >3-fold (Figure 3B, lanes 12–14 and 3C). Nevertheless, synthesis was attenuated and ultimately blocked at higher concentrations of ω2 (Figure 3B, lanes 15–16).
At 300 nM, δ2 significantly reduced Pω utilization by ∼14-fold (Figure 3B, lanes 1 versus 17). Addition of limiting concentrations of ω2 (1.8–3.7 ω2/Pω DNA or 0.2–0.5 ω2/heptad) significantly stimulated Pω dependent mRNA synthesis by >6-fold (Figure 3B, lanes 18–20, and 3C). As expected, ω2 blocked Pω utilization when used at the slightly saturating concentrations of 15 ω2/Pω DNA (Figure 3B, lane 22). It is likely, therefore, that whether ω2 acts as a transcriptional activator or a repressor hinges on its concentration (Figure 3C). Moreover, while by itself repressing Pω utilization, δ2 apparently behaves as a transcriptional co-activator. This is consistent with the observations that: i) the half-life of ω2·Pω complex increased ∼30-fold in the presence of δ2 (10,13); and ii) upon interacting with δ2, ω2 bound to Pω DNA, a rearrangement of its unstructured NTD occurs (see 15).
Protein ω2 interacts with the NH2-terminal half of the RNAP-σA β’ subunit
RNAP-σA and ω2 cooperatively bind Pω DNA cooperatively and create higher-order nucleoprotein complexes that reflect the combinatorial control of gene expression (Figures 2–4). This effect is likely attributed to direct protein–protein interactions between adjacent DNA-binding factors that promote the assembly of higher-order complexes. To determine whether or not RNAP-σA and ω2 physically interacted, RNAP-σA was bound to a Ni2+ agarose column through coordination with the C-terminal histidine tag of its β’ subunit (30). The RNAP-σA-bound matrix retained ω2, even in the absence of Pω DNA. These proteins also co-eluted from the matrix during an elution step (Supplementary Figure S6).
Initial identification of the RNAP-σA subunit(s) responsible for association to ω2 was achieved by carrying out far-western blots of RNAP-σA, ω2 and BSA as control. These proteins were resolved by SDS-PAGE under conditions under which the small ω protein (7.9 kDa) migrated with the front. After renaturation of prey proteins and a membrane blocking step, the bait, ω2, was added. Protein–protein interactions were detected using anti-ω2 polyclonal antibodies. As expected, ω2 interacted with itself and also with the co-migrating β and/or β’ subunits of RNAP-σA. No other subunits gave a signal (Figure 5A). Due to a similarity in mass, β (133.6 kDa) and β’ (134.2 kDa) subunits could not be distinguished. We therefore took advantage of the fact that all B. subtilis RNAP-σA subunits, except for β’ (pI 8.8), have acidic pIs and repeated the far-western experiments, this time using two-dimensional (2D)-PAGE. Protein ω2 interacted with β’ and several proteins in unexpected spots (termed 1–2, 3–4 and 5). These had masses of ∼34 kDa and were located in the basic region of the gel (Figure 5B, Ab-anti ω2 condition). Corresponding polypeptides were extracted from the gel, subjected to mass spectrometry analysis, and identified as RNAP-σA β’ subunit NTDs with slight variations in the C-termini (Figure 5C). Taking into account the sizes observed, it was assumed that the Sw2 structural module (residues 316–342) was missing from these NTD variants of the β’ subunit.
Figure 5.
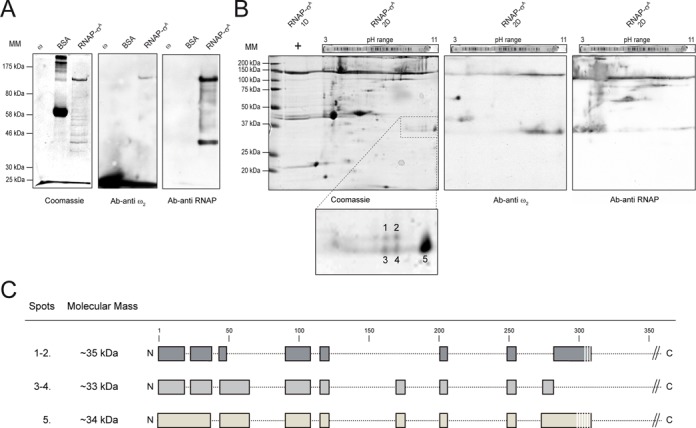
Far-western blotting of ω2 and RNAP-σA. (A) 1 μg ω2, 1 μg RNAP-σA or 5 μg BSA (as control) were resolved by SDS-PAGE and either stained with Coomasie blue or transferred onto nitrocellulose membranes. Membranes underwent a renaturing step and incubated with the specific ω2 and RNAP-σA antibodies (Ab). (B) 1 μg RNAP-σA was resolved by iso-electric focusing in a pH 3–11 gradient followed by an SDS-PAGE. Protein was then transferred to a membrane, renatured, incubated with the ω2 bait and highlighted with either Ab-anti ω2 or Ab-anti RNAP-σA. (C) Basic, ∼34 kDa polypeptides (1–5) that had reacted with Ab-anti ω2 were gel purified and identified by mass spectrometry. The regions identified are shown in this figure. The sequence coverage was > 40%.
The central and C-terminal regions of ω2 appears not to interact with RNAP-σA
The ω2 protein has three functional regions: (i) the unstructured NTD (residues 1–24), which is essential for the ω2·δ2 interaction (11,15); (ii) the β-sheet domain (residues 28–32), which is required for ω2 recognition of its cognate DNA site (9,14); and (iii) the α-helix α1 (residues 34–46) which, in concert with the α2 helix (residues 51–64) contributes to monomer-monomer and dimer-dimer interfaces (8,9). The function, if any, of the C-terminal region (residues 65–71) is unknown (7,8).
Significantly, the 79-residue long ω2 (9.0 kDa) shares a 98% identity with the 71-residue ω for the first 55 residues (Supplemental material Annex 2). However, they only share an 18% identity in the last 24 residues (Supplementary Figure S7A). The ω2 protein repressed Pδ utilization in vivo nearly as efficiently as wt ω2 (Supplemental material Annex 2, Supplementary Figure S7B). Similar results were observed when the Pδ-lacZ fusion was replaced by the Pω-lacZ fusion (data not shown). Furthermore, plasmid-borne ω2 and δ genes which were transcribed from Pω (parS2) and Pδ (parS1) (Figure 1A), were necessary and sufficient for stabilizing of an otherwise unstable plasmid in B. subtilis cells (38, our unpublished results). It is likely that: (i) the dimer is the functional unit of ω2; (ii) ω22 interacts with δ2 just like ω2; and (iii) the different C-terminal domains of ω and ω2 are not involved in gene repression.
Protein docking experiments predicted that the unstructured ω NTD folded into an α-helical structure that interacted with the β’ NTD of RNAP-σA (Supplementary Figure S8). In contrast, several charged amino acids located in the coiled region between the α-helices α1 and α2 (residues 47–52) and in the α2 helix itself (residues 51–64) could make contacts with the oppositely charged residues in the β’ subunit of RNAP-σA. To test this hypothesis (Supplemental material Annex 3), the charged residues that were accessible in these domains, K52, E53, D56, R64 and K70, were replaced with alanine. Subsequently, the in vivo behaviour of these mutants was investigated (Supplemental material Annex 3). With the exception of ωD56A, the ω mutant variants repressed Pδ transcription in vivo as efficiently as wt ω2 (Supplementary Figure S7B) (Supplemental material Annex 3). The D56A mutant only reduced Pδ transcription ∼6-fold. As described in Supplementary Annex 3, purified ωD56A also formed dimers, albeit in a far smaller proportion than wt ω2 (Supplementary Figure S7C). Since in the dimeric form of ω, the β-sheet domain adopts an antiparallel configuration before binding Pω, the primary defect of ωD56A might be a poor ability to dimerize. Therefore, it was not further analysed. It would be very interesting to determine whether or not the ω2 NTD can, by itself recruit RNAP-σA to the ω2·Pω DNA complex.
DISCUSSION
Direct contacts between ω2 and RNAP-σA stimulate RPC complex formation and its subsequent isomerization to RPO. This appears to be the mechanism by which limiting concentrations of ω2 activate Pω transcription. This process is further enhanced by δ2 (Figure 3B). However, stoichiometric concentrations of ω2 have the opposite effect: here the recruited RNAP-σA inefficiently isomerizes into RPO and represses Pω or Pδ transcription both in vivo and in vitro. It would be highly interesting to determine whether or not ω2 functions as an activator to repressor switch of PcopS. However, preliminary results indicate that ω2·PcopS DNA forms a ternary complex with RNAP-σA and that ω2 regulates transcription through a mechanism that does not exclude the RNAP-σA from the RPC (4).
The dual activity of the ω2 regulator
Plasmid-encoded ω2, from Gram-positive cocci, is the only one out of the more than 2000 RHH2 proteins that can either activate or repress the utilization of a single promoter (Pω) in a concentration-dependent manner. This is true at least in a simplified in vitro system. The majority of RHH2 proteins are predicted to be transcriptional repressors (39). However, four of them act both as activators and as repressors. These are: a P22-Arc variant, Mer, AmrZ [AlgZ] and NikR. They bind to a variety of promoters, functioning as repressors for some while behaving as activators for others (40–43). In the case of other putative regulators, various metal cofactors and different stoichiometries might also influence the effect they have on promoter functioning (40–43).
The activator to repressor switch function of ω2 appears to be managed by the diverse modes of ω2 binding to the operator region of Pω. These modes of binding have distinct effects on the initial activity of RNAP-σA. Under limiting concentrations, ω2 promotes RNAP-σA binding to operator sequences that overlap the −35 sequence. This results in the stimulation of RPC formation and in an increase in the rate of isomerization from RPC to RPO. It is thus likely that: (i) ω2 increases the local concentration of both proteins, leading to a ternary ω2·Pω·RNAP-σA complex; (ii) this ternary complex facilitates the rate of isomerization from RPC to RPO and increases Pω dependent mRNA synthesis; and (iii) δ2 may act as a co-activator by increasing the half-life of ω2·Pω DNA complexes (see 13).
With stoichiometric concentrations of ω2 full operator occupancy was achieved, and a different outcome was observed. Under these conditions, ω2 assembles into a left-handed matrix that wraps around right-handed, straight Pω DNA (see Figure 1B). This assembly makes Pω DNA accessible to RNAP-σA (RPC formation), while simultaneously inhibiting isomerization to RPO (transcriptional repression) (Figure 4, lanes 17–18). In vitro, we observed that δ2 contributed to Pδ repression (Figure 3B), and the presence of both ω2 and δ2 transcriptional repression of Pδ and Pω was elevated in vivo with respect to wt ω2 alone (Supplementary Figure S7B, data not shown). When stoichiometric concentrations of ω2 were added to preformed RPO complexes, a moderate effect on Pω utilization was observed (see Figures 3 and 4). This suggested that RNAP-σA transcription was influenced by ω2 that was bound to its cognate promoters. In sum, we describe here a previously uncharacterized mechanism of transcription regulation in bacteria belonging to the phylum Firmicutes.
Several different models can be considered in order to explain the specific transcriptional repression resulting from the full occupancy of the operator sequences by ω2: (i) relocation of RNAP-σA to a position unfavourable for efficient RPO formation; (ii) ‘locking’ RNAP-σA into a conformation unfavourable for RPO formation; (iii) blocking the interaction between the β’ NTD and the DNA, which may be an essential step for RPO formation; and (iv) inhibition of RNAP-σA mediated transcription by hindering any putative upstream element. We favour the first model because RPC formation is stimulated by the interaction between ω2 and RNAP-σA, while the subsequent isomerization step producing stable RPO (through the unstable intermediate, RPI) becomes inhibited. Also consistent with this model is the fact that ω2 establishes interactions with operator sites when RNAP-σA is already bound to Pω DNA. Meanwhile, ω2 fails to inhibit pre-formed RPO, Also in line with this model is the observation that, in the presence of limiting amounts of ω2 bound to Pω DNA, RNAP-σA moves onto Pω DNA (Supplementary Figure S2A), and that while under saturating concentrations of ω2, RNAP-σA moves off of Pω DNA (Supplementary Figure S2B).
The interplay between the ω2 regulator and the β’ subunit of RNAP holoenzyme
The work presented here establishes that ω2 is a global regulator of plasmid biology through its effect on replication, faithful partitioning and better-than random segregation (see Introduction). Importantly, ω2 represents an exception to the accepted prokaryotic transcription regulation paradigm, which asserts that, there are proteins that can act as either activators or repressors, but that the same protein cannot act as both. Since ω2 regulates the expression of plasmid encoded genes that are harboured in different Firmicutes bacteria, it presumably recognizes the RNAP β’ NTD of all of them. This recognition might be limited to the first 316 amino acids, the length of the shortest polypeptide of the β’ subunit that binds ω2 (Figure 5B). This would not be surprising because the β’ subunits of all Firmicutes share a high degree of sequence identity. Consistent with this, ω2 does not regulate transcription of a genetically distant bacterium (e.g. E. coli) (16). These facts imply that the regions of the Firmicutes β’ subunits that show a significant degree of sequence divergence are not involved in ω2 binding, such as residues 124–165 and 178–208 (22% and 13%, respectively). Presumably, residues 260–271, which match almost perfectly between the β’ subunits of E. coli and B. subtilis, is a region also not involved in ω2 binding.
Few proteins have been observed to interact with the RNAP β’ subunit. Most of these are encoded by proteobacterial phages. The phage Xp10-p7 factor interacts with the first 10 residues of the NTD of β’ (44). The Mu-C protein binds to part of region F (b7) (45). T7-Gp2 recognizes part of the jaw (b9-b10) and σ 1.1 domains (46,47). And lastly, N4-SSB interacts with part of region H (b11) at the CTD (48). These regulators do not share a specific target domain and have different modes of action: N4-SSB and Mu-C specifically act as transcription activators (45,48), while T7-Gp2 and Xp10-p7 are repressors (44,46). In contrast, ω2 has a dual function (Figure 3B). Of these regulators, only T7-Gp2, Xp10-p7, and ω2 act during the early stages of RNAP-σ isomerization. Meanwhile, T7-Gp2 and Xp10-p7 directly interact with RNAP-σ rather than binding to P DNA as does ω2 (44,46,47).
Biological implication of ω2-mediated transcription regulation
A growing number of plasmid-encoded genetic determinants for resistance to diverse antimicrobials among streptococci, enterococci and staphylococci has been shown to be regulated by ω-like cofactors. They act either as part of the ωϵζ operon or on their own as part of the ω cassette (ω or ω2 genes) (1). In conjunction with δ2 and RNAP-σ, the biological role of ω2 as a dual regulator is to control vital plasmid functions in Firmicutes. It corrects the downward fluctuations in plasmid copy number through regulation of the synthesis of CopS (also termed CopF, CopR). CopS is a repressor of the initiator RepS (also termed RepE, RepR) protein. ω2 also controls the synthesis of the toxin-antitoxin module, which in turn restricts the survival of plasmid-free segregants. The ω2 protein mediates the synthesis of the partition system by regulating the expression of δ2 and ω2. Protein ω22 manages the expression of the ermB gene (4,16,49). With the help of δ2 (the ParA ATPase), the ω2 (the ParB centromere binding) protein also safeguards plasmid faithful segregation via the ParAB system. The regulation of Firmicutes RNAP-σA by the ω cassette is a newly characterized mechanism through which bacterial transcription of a large number of antibiotic resistance genes is regulated.
Supplementary Material
Acknowledgments
We thank Margarita Salas and José M Lazaro for their generous gift of rabbit polyclonal anti-RNAP-σA. This work was supported in part by grant BFU2012–39879-C02-01 awarded to J.C.A by the Dirección General de Investigación-Ministerio de Economía y Competitividad (DGI-MINECO). A.V. thanks the Consejería de Educación de la Comunidad de Madrid for its fellowship (CPI/0266/2008) and the European Social Fund (ESF). M.T. is a PhD fellow supported by the La Caixa Foundation International Fellowship Programme of La Caixa/CNB.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Dirección General de Investigación-Ministerio de Economía y Competitividad [BFU2012-39879-C02-01 to J.C.A.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Volante A., Soberón N.E., Ayora S., Alonso J.C. The interplay between different stability systems contributes to faithful segregation: Streptococcus pyogenes pSM19035 as a model. Microbiol. Spectrum. 2014;2 doi: 10.1128/microbiolspec.PLAS-0007-2013. PLAS-0007. [DOI] [PubMed] [Google Scholar]
- 2.Croucher N.J., Harris S.R., Fraser C., Quail M.A., Burton J., van der Linden M., McGee L., von Gottberg A., Song J.H., Ko K.S., et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;331:430–434. doi: 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Camacho A.G., Misselwitz R., Behlke J., Ayora S., Welfle K., Meinhart A., Lara B., Saenger W., Welfle H., Alonso J.C. In vitro and in vivo stability of the ϵ2ζ2 protein complex of the broad host-range Streptococcus pyogenes pSM19035 addiction system. Biol. Chem. 2002;383:1701–1713. doi: 10.1515/BC.2002.191. [DOI] [PubMed] [Google Scholar]
- 4.de la Hoz A.B., Ayora S., Sitkiewicz I., Fernandez S., Pankiewicz R., Alonso J.C., Ceglowski P. Plasmid copy-number control and better-than-random segregation genes of pSM19035 share a common regulator. Proc. Natl. Acad. Sci. U.S.A. 2000;97:728–733. doi: 10.1073/pnas.97.2.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dmowski M., Sitkiewicz I., Ceglowski P. Characterization of a novel partition system encoded by the δ and ω genes from the streptococcal plasmid pSM19035. J. Bacteriol. 2006;188:4362–4372. doi: 10.1128/JB.01922-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lioy V.S., Pratto F., de la Hoz A.B., Ayora S., Alonso J.C. Plasmid pSM19035, a model to study stable maintenance in Firmicutes. Plasmid. 2010;64:1–17. doi: 10.1016/j.plasmid.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Murayama K., de la Hoz A.B., Alings C., Lopez G., Orth P., Alonso J.C., Saenger W. Crystallization and preliminary X-ray diffraction studies of Streptococcus pyogenes plasmid pSM19035-encoded ω transcriptional repressor. Acta Crystallogr. D Biol. Crystallogr. 1999;55:2041–2042. doi: 10.1107/s0907444999012275. [DOI] [PubMed] [Google Scholar]
- 8.Murayama K., Orth P., de la Hoz A.B., Alonso J.C., Saenger W. Crystal structure of ω transcriptional repressor encoded by Streptococcus pyogenes plasmid pSM19035 at 1.5 A resolution. J. Mol. Biol. 2001;314:789–796. doi: 10.1006/jmbi.2001.5157. [DOI] [PubMed] [Google Scholar]
- 9.Weihofen W.A., Cicek A., Pratto F., Alonso J.C., Saenger W. Structures of ω repressors bound to direct and inverted DNA repeats explain modulation of transcription. Nucleic Acids Res. 2006;34:1450–1458. doi: 10.1093/nar/gkl015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de la Hoz A.B., Pratto F., Misselwitz R., Speck C., Weihofen W., Welfle K., Saenger W., Welfle H., Alonso J.C. Recognition of DNA by ω protein from the broad-host range Streptococcus pyogenes plasmid pSM19035: analysis of binding to operator DNA with one to four heptad repeats. Nucleic Acids Res. 2004;32:3136–3147. doi: 10.1093/nar/gkh633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Welfle K., Pratto F., Misselwitz R., Behlke J., Alonso J.C., Welfle H. Role of the N-terminal region and of β-sheet residue Thr29 on the activity of the ω2 global regulator from the broad-host range Streptococcus pyogenes plasmid pSM19035. Biol. Chem. 2005;386:881–894. doi: 10.1515/BC.2005.103. [DOI] [PubMed] [Google Scholar]
- 12.Pratto F., Suzuki Y., Takeyasu K., Alonso J.C. Single-molecule analysis of protein·DNA complexes formed during partition of newly replicated plasmid molecules in Streptococcus pyogenes. J. Biol. Chem. 2009;284:30298–30306. doi: 10.1074/jbc.M109.035410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soberón N.E., Lioy V.S., Pratto F., Volante A., Alonso J.C. Molecular anatomy of the Streptococcus pyogenes pSM19035 partition and segrosome complexes. Nucleic Acids Res. 2011;39:2624–2637. doi: 10.1093/nar/gkq1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dostál L., Pratto F., Alonso J.C., Welfle H. Binding of regulatory protein ω from Streptococcus pyogenes plasmid pSM19035 to direct and inverted 7-Base pair repeats of operator DNA. J. Raman Spectrosc. 2007;38:166–175. [Google Scholar]
- 15.Volante A., Alonso J.C. Molecular anatomy of ParA-ParA and ParA-ParB interactions during plasmid partitioning. J. Biol. Chem. 2015;290:18782–18795. doi: 10.1074/jbc.M115.649632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pratto F., Cicek A., Weihofen W.A., Lurz R., Saenger W., Alonso J.C. Streptococcus pyogenes pSM19035 requires dynamic assembly of ATP-bound ParA and ParB on parS DNA during plasmid segregation. Nucleic Acids Res. 2008;36:3676–3689. doi: 10.1093/nar/gkn170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Record M. Jr, Reznikoff W., Craig M., McQuade K., Schlax P. In: In Escherichia coli and Salmonella: Cellular and Molecular Biology. 2nd Edn. Neidhardt F, Curtiss RI, Ingraham J, Lin E, Low K, Magasanik B, Reznikoff W, Riley M, Schaechter M, Umbarger H, editors. Washington, DC: ASM Press; 1996. pp. 792–821. [Google Scholar]
- 18.Vassylyev D.G., Vassylyeva M.N., Perederina A., Tahirov T.H., Artsimovitch I. Structural basis for transcription elongation by bacterial RNA polymerase. Nature. 2007;448:157–162. doi: 10.1038/nature05932. [DOI] [PubMed] [Google Scholar]
- 19.Lane W.J., Darst S.A. Molecular evolution of multisubunit RNA polymerases: structural analysis. J. Mol. Biol. 2010;395:686–704. doi: 10.1016/j.jmb.2009.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y., Feng Y., Chatterjee S., Tuske S., Ho M.X., Arnold E., Ebright R.H. Structural basis of transcription initiation. Science. 2012;338:1076–1080. doi: 10.1126/science.1227786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Darst S.A. Bacterial RNA polymerase. Curr. Opin. Struct. Biol. 2001;11:155–162. doi: 10.1016/s0959-440x(00)00185-8. [DOI] [PubMed] [Google Scholar]
- 22.Murakami K.S., Darst S.A. Bacterial RNA polymerases: the wholo story. Curr. Opin. Struct. Biol. 2003;13:31–39. doi: 10.1016/s0959-440x(02)00005-2. [DOI] [PubMed] [Google Scholar]
- 23.Gruber T.M., Gross C.A. Multiple σ subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol. 2003;57:441–466. doi: 10.1146/annurev.micro.57.030502.090913. [DOI] [PubMed] [Google Scholar]
- 24.Ebright R.H. RNA polymerase: structural similarities between bacterial RNA polymerase and eukaryotic RNA polymerase II. J. Mol. Biol. 2000;304:687–698. doi: 10.1006/jmbi.2000.4309. [DOI] [PubMed] [Google Scholar]
- 25.Haugen S.P., Ross W., Gourse R.L. Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat. Rev. Microbiol. 2008;6:507–519. doi: 10.1038/nrmicro1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murakami K.S., Masuda S., Campbell E.A., Muzzin O., Darst S.A. Structural basis of transcription initiation: an RNA polymerase holoenzyme-DNA complex. Science. 2002;296:1285–1290. doi: 10.1126/science.1069595. [DOI] [PubMed] [Google Scholar]
- 27.Vassylyev D.G., Sekine S., Laptenko O., Lee J., Vassylyeva M.N., Borukhov S., Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 Å resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- 28.Saecker R.M., Record M.T. Jr, Dehaseth P.L. Mechanism of bacterial transcription initiation: RNA polymerase - promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J. Mol. Biol. 2011;412:754–771. doi: 10.1016/j.jmb.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Browning D.F., Busby S.J. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2004;2:57–65. doi: 10.1038/nrmicro787. [DOI] [PubMed] [Google Scholar]
- 30.Fujita M., Sadaie Y. Rapid isolation of RNA polymerase from sporulating cells of Bacillus subtilis. Gene. 1998;221:185–190. doi: 10.1016/s0378-1119(98)00452-1. [DOI] [PubMed] [Google Scholar]
- 31.Miller J.H. Experiments in Molecular Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1972. [Google Scholar]
- 32.Zechel K. On the resolution of polypeptides by isoelectric focusing in polyacrylamide gels in the presence of urea and nonidet-p40. Anal. Biochem. 1977;83:240–251. doi: 10.1016/0003-2697(77)90532-2. [DOI] [PubMed] [Google Scholar]
- 33.Wu Y., Li Q., Chen X.Z. Detecting protein-protein interactions by Far western blotting. Nat. Protoc. 2007;2:3278–3284. doi: 10.1038/nprot.2007.459. [DOI] [PubMed] [Google Scholar]
- 34.Lioy V.S., Martin M.T., Camacho A.G., Lurz R., Antelmann H., Hecker M., Hitchin E., Ridge Y., Wells J.M., Alonso J.C. pSM19035-encoded ζ toxin induces stasis followed by death in a subpopulation of cells. Microbiology. 2006;152:2365–2379. doi: 10.1099/mic.0.28950-0. [DOI] [PubMed] [Google Scholar]
- 35.Tapias A., Fernandez S., Alonso J.C., Barbe J. Rhodobacter sphaeroides LexA has dual activity: optimising and repressing recA gene transcription. Nucleic Acids Res. 2002;30:1539–1546. doi: 10.1093/nar/30.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodionov O., Lobocka M., Yarmolinsky M. Silencing of genes flanking the P1 plasmid centromere. Science. 1999;283:546–549. doi: 10.1126/science.283.5401.546. [DOI] [PubMed] [Google Scholar]
- 37.Lynch A.S., Wang J.C. SopB protein-mediated silencing of genes linked to the sopC locus of Escherichia coli F plasmid. Proc. Natl. Acad. Sci. U.S.A. 1995;92:1896–1900. doi: 10.1073/pnas.92.6.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lioy V.S., Volante A., Soberón N.E., Lurz R., Ayora S., Alonso J.C. ParAB partition dynamics in Firmicutes: nucleoid bound ParA captures and tethers ParB-plasmid complexes. PLoS One. 2015;10:e0131943. doi: 10.1371/journal.pone.0131943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schreiter E.R., Drennan C.L. Ribbon-helix-helix transcription factors: variations on a theme. Nat. Rev. Microbiol. 2007;5:710–720. doi: 10.1038/nrmicro1717. [DOI] [PubMed] [Google Scholar]
- 40.Smith T.L., Sauer R.T. Dual regulation of open-complex formation and promoter clearance by Arc explains a novel repressor to activator switch. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8868–8872. doi: 10.1073/pnas.93.17.8868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schreiter E.R., Sintchak M.D., Guo Y., Chivers P.T., Sauer R.T., Drennan C.L. Crystal structure of the nickel-responsive transcription factor NikR. Nat. Struct. Biol. 2003;10:794–799. doi: 10.1038/nsb985. [DOI] [PubMed] [Google Scholar]
- 42.Muller C., Bahlawane C., Aubert S., Delay C.M., Schauer K., Michaud-Soret I., De Reuse H. Hierarchical regulation of the NikR-mediated nickel response in Helicobacter pylori. Nucleic Acids Res. 2011;39:7564–7575. doi: 10.1093/nar/gkr460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pryor E.E. Jr, Waligora E.A., Xu B., Dellos-Nolan S., Wozniak D.J., Hollis T. The transcription factor AmrZ utilizes multiple DNA binding modes to recognize activator and repressor sequences of Pseudomonas aeruginosa virulence genes. PLoS Pathog. 2012;8:e1002648. doi: 10.1371/journal.ppat.1002648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuzenkova Y., Zenkin N., Severinov K. Mapping of RNA polymerase residues that interact with bacteriophage Xp10 transcription antitermination factor p7. J. Mol. Biol. 2008;375:29–35. doi: 10.1016/j.jmb.2007.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swapna G., Chakraborty A., Kumari V., Sen R., Nagaraja V. Mutations in beta’ subunit of Escherichia coli RNA polymerase perturb the activator polymerase functional interaction required for promoter clearance. Mol. Microbiol. 2011;80:1169–1185. doi: 10.1111/j.1365-2958.2011.07636.x. [DOI] [PubMed] [Google Scholar]
- 46.James E., Liu M., Sheppard C., Mekler V., Camara B., Liu B., Simpson P., Cota E., Severinov K., Matthews S., et al. Structural and mechanistic basis for the inhibition of Escherichia coli RNA polymerase by T7 Gp2. Mol. Cell. 2012;47:755–766. doi: 10.1016/j.molcel.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bae B., Davis E., Brown D., Campbell E.A., Wigneshweraraj S., Darst S.A. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of σ70 domain 1.1. Proc. Natl. Acad. Sci. U.S.A. 2013;110:19772–19777. doi: 10.1073/pnas.1314576110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller A., Wood D., Ebright R.H., Rothman-Denes L.B. RNA polymerase beta’ subunit: a target of DNA binding-independent activation. Science. 1997;275:1655–1657. doi: 10.1126/science.275.5306.1655. [DOI] [PubMed] [Google Scholar]
- 49.Lioy V.S., Machon C., Tabone M., Gonzalez-Pastor J.E., Daugelavicius R., Ayora S., Alonso J.C. The ζ toxin induces a set of protective responses and dormancy. PLoS One. 2012;7:e30282. doi: 10.1371/journal.pone.0030282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.