


Abstract
We here use our site-specific base analog mapping approach to study the interactions and binding equilibria of cooperatively-bound clusters of the single-stranded DNA binding protein (gp32) of the T4 DNA replication complex with longer ssDNA (and dsDNA) lattices. We show that in cooperatively bound clusters the binding free energy appears to be equi-partitioned between the gp32 monomers of the cluster, so that all bind to the ssDNA lattice with comparable affinity, but also that the outer domains of the gp32 monomers at the ends of the cluster can fluctuate on and off the lattice and that the clusters of gp32 monomers can slide along the ssDNA. We also show that at very low binding densities gp32 monomers bind to the ssDNA lattice at random, but that cooperatively bound gp32 clusters bind preferentially at the 5′-end of the ssDNA lattice. We use these results and the gp32 monomer-binding results of the companion paper to propose a detailed model for how gp32 might bind to and interact with ssDNA lattices in its various binding modes, and also consider how these clusters might interact with other components of the T4 DNA replication complex.
INTRODUCTION
In the companion paper (1) we described the interactions of the single-stranded DNA binding protein of bacteriophage T4 (gp32) with short (8-mer) ssDNA lattices containing site-specifically placed 2-aminopurine (2-AP) base analog as spectroscopic probes. These lattices were just long enough to span the binding surface of a single gp32 monomer, and by monitoring the changes in the fluorescence and circular dichroism (CD) spectra of the 2-AP dimer probes as a function of probe position we were able to ‘map’ the binding surface of an individual (isolated) gp32 molecule from the nucleic acid perspective. Our results suggested that the primary source of binding free energy for the monomer gp32–ssDNA interaction involves a strong interaction of the protein with the sugar-phosphate backbone of a 2–3 nucleotide (nt) sequence located near the 3′-end of these 8-mer ssDNA lattices, accompanied by local base unstacking and extension of the ssDNA backbone. The remainder of the binding was attributed to electrostatic interactions of DNA phosphates of the middle and 5′-end of the 8-mer ssDNA oligomers with a sequence of basic amino acid residues present in the gp32 DNA binding cleft. These latter electrostatic interactions do not appear to add much net binding free energy to the complex, presumably because this binding requires the concomitant displacement of the negatively charged C-terminal domain of the protein from the binding cleft. These results also provided a structural basis and molecular interpretation of the binding polarity of the ssDNA lattices, and comparison with the structural details of the crystal structure of the gp32 core binding domain (2) suggested how this ssDNA binding polarity could be interpreted in terms of specific ssDNA interactions with structures on the protein surface as well. (For models and details of the monomer binding interaction see the ‘Discussion’ section and Figure 6 of the companion paper (1).
Figure 6.
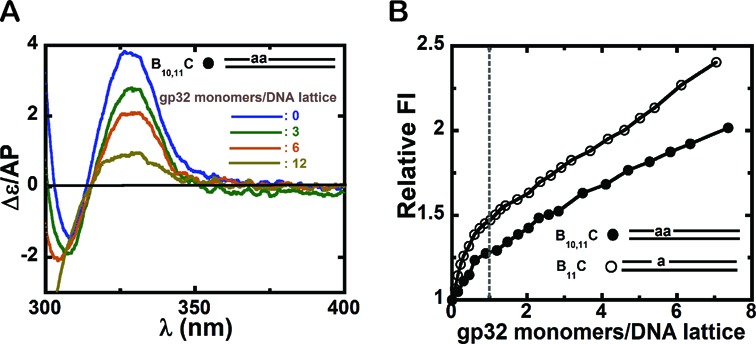
Circular dichroism and fluorescence intensity changes observed for dsDNA constructs with 2-AP dimer or monomer probes site-specifically positioned within duplex DNA constructs. (A) CD spectra for a 25B10,11–25C dsDNA construct consisting of a 25B10,11 ssDNA lattice complexed to a fully complementary 25C ssDNA lattice in the presence of increasing concentrations of gp32 monomers. blue: free DNA construct; green: 3; brown: 6; and olive green: 12 gp32 monomers present per dsDNA lattice. (B) Normalized (to unity) fluorescence intensity changes at 370 nm for 1 μM 25B10,11–25C and 25B11–25C dsDNA constructs plotted as a function of gp32 monomers per dsDNA construct. Construct 25B11–25C (open circles) and 25B10,11–25C (closed circles). The dashed vertical line marks the point at which one gp32 monomer has been added per dsDNA construct.
In this paper we extend these approaches and observations to the ‘cooperative’ binding of ‘blocks’ of gp32 monomers to longer ssDNA lattices. Such binding cooperativity is, in principle, essential to the function of this protein (although this has been shown directly only for in vitro model systems), because in the absence of such cooperativity gp32 protein binding to the transient single-stranded sequences formed within the DNA replication complex would be both weak and incomplete. And this, in turn, would undermine the primary roles of this protein in protecting the ssDNA templates from nuclease degradation and secondary structure formation, as well as its auto-regulatory interactions with sequences devoid of secondary structure in the gp32 mRNA molecule (see (3,4)). In this paper we use our base analogue approaches to define structural and thermodynamic aspects of gp32 binding cooperativity that are central both to the function of this single stranded DNA binding protein with the T4 replication complex and to the interactions of this protein with the transiently single-stranded DNA sequences exposed by the primosome helicase and utilized as templates by the DNA polymerases in T4 DNA replication.
MATERIALS AND METHODS
Most of the Materials and Methods used in this study have been described in the companion paper (1), and the information presented in this section supplements those descriptions for experiments specific to this paper.
DNA constructs, protein purification and buffers
Unlabeled and 2-AP-labeled DNA oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA, USA) and 6-MI labeled DNA oligonucleotides from Fidelity Systems (Gaithersburg, MD, USA). Oligonucleotide concentrations were determined using extinction coefficients furnished by the manufacturer. Duplex constructs were made by heating equimolar concentrations of complementary strands to 90°C, followed by gradual cooling. The sequences and nomenclature of the DNA constructs used in this study are shown and described in Table 1. Proteins were purified and characterized as described in the companion paper (1) and unless stated otherwise all experiments were performed at 20°C in buffer containing 10 mM HEPES (pH 7.5) and 30 mM potassium acetate.
Table 1. Nomenclature and sequences of the DNA constructs.
| Construct | Sequence |
|---|---|
| 25B11 | 5′GCA CCA TAT AaT CGC TCG CAT ATT A |
| 25B2, 3 | 5′Taa TCA CCA TAT TCG CTC GCA TAT T |
| 25B10,11 | 5′GCA CCA TAT aaT CGC TCG CAT ATT A |
| 25B23,24 | 5′GCA CCA TAT TCG CTC GCA CTA Taa T |
| 25G10,11 | 5′GCA CCA TAT ggT CGC TCG CAT ATT A |
| 56F4 | 5′CAG aCA TAA TAT GCG TCG CTA ATA TAC CAC TTT TCA CAC TTT CAC TCA CGT CTT AC |
| 56F21 | 5′CAG TCA TAA TAT GCG TCG CTa ATA TAC CAC TTT TCA CAC TTT CAC TCA CGT CTT AC |
| 56F53 | 5′CAG TCA TAA TAT GCG TCG CTA ATA TAC CAC TTT TCA CAC TTT CAC TCA CGT CaT AC |
| 25B11–25C | 5′GCA CCA TAT AaT CGC TCG CAT ATT A |
| 3′CGT GGT ATA TTA GCG AGC GTA TAA T | |
| 25B10,11 - 25C | 5′GCA CCA TAT aaT CGC TCG CAT ATT A |
| 3′CGT GGT ATA TTA GCG AGC GTA TAA T |
* ‘T’, ‘A’, ‘G’ and ‘C’ represent the canonical DNA nucleotide residues, ‘a’ represents the 2-AP probe(s) and ‘g’ the 6-MI probe(s). The subscripts before the construct names indicate the total length of the ssDNA lattice and the subscripts that follow the names denote the positions of the probes within each construct.
Spectroscopic procedures
CD spectra and fluorescence spectra were measured as described previously (5–7) for both the 2-AP and 6-MI labeled oligonucleotide constructs used in this study. DNA constructs were manually titrated with successive aliquots of gp32 at 20°C. The samples were gently mixed for 2 min at each gp32 concentration, and then fluorescence emission values from 330 to 450 nm or 355 to 500 nm were collected at excitation wavelengths of 315 or 350 nm for 2-AP- or 6MI-labeled oligonucleotide constructs, respectively. Raw data were corrected for dark counts and for Raman scattering. Corrections for protein based or background fluorescence were made by subtracting the fluorescence intensities obtained with an unlabeled ssDNA construct of the same length following titration with the same amounts of gp32 as used in the experiment. The total concentrations of ssDNA oligomer and gp32 were calculated from the added volumes and the input concentrations of gp32 monomers per binding site assuming a binding site size of 7 nts for ssDNA (8–10), and 5 bps for dsDNA (8,10). The fluorescence intensities were either plotted as raw values or as normalized fluorescence intensity changes by using the intensity measured for ssDNA divided by the plateau value for the titration profile. The error bars shown for the fluorescence measurements represent average deviations obtained from two to four independent experiments.
RESULTS
Changes in the fluorescence and CD spectra of base analog probes site-specifically positioned in DNA during biologically relevant protein binding and interaction events can be used to elucidate structural and mechanistic understanding of macromolecular machines. By monitoring these spectral changes we have been able to track local conformational changes in nucleic acids, as well as to pinpoint physiologically relevant base and base pair stacking and unstacking reactions within a variety of protein–nucleic acid complexes (6,11–14). In the companion paper (1) we have used these techniques to map the details of the binding of isolated gp32 molecules to short ssDNA oligonucleotide lattices, establishing that the binding has a defined polarity with respect to both the ssDNA and the protein and that strong gp32 interactions with the sugar-phosphate backbone occur at a 2–3 nts sequence near the 3′ end of 8-mer ssDNA lattices that fully span the protein surface (see Figure 6 and ‘Discussion’ section of companion paper (1)).
Building on these earlier results, we here extend our studies to the cooperative binding of short clusters of gp32 molecules to longer ssDNA lattices. This work has permitted us to use our base analog approaches to map the lattice positions and nucleic acid binding surfaces of cooperatively bound gp32 molecules, leading to a better definition of the molecular origins of the centrally important property of this paradigmatic single-stranded DNA binding protein. And this approach, in turn, now provides a background for understanding how these cooperatively bound ‘blocks’ or ‘clusters’ of gp32 molecules might interact with, move on, and be manipulated by, the other proteins of the T4 DNA replication complex.
DNA constructs and nomenclature
The occlusion binding site-size of gp32 molecules that are cooperatively bound to long ssDNA lattices is ∼7 nts per gp32 monomer (8,15). As substrates to study the cooperative binding of gp32 to ssDNA lattices, we designed the series of defined polynucleotide ssDNA constructs shown in Table 1. The constructs included ssDNA polynucleotide sequences ranging from 25 to 56 nts in length (corresponding to ∼3 to ∼8 contiguous gp32 monomer binding sites) and with isolated or pairs of fluorescent base analog probes (either 2-AP or 6-MI) positioned at specific sites to monitor the local changes in DNA conformation resulting from gp32 binding. The total length of each construct is indicated by a subscript integer preceding the construct name, and the locations of the fluorescent base(s) are indicated following the construct name by a second subscript integer, with one number indicating the position of a single base-analog probe (for example, 25B11 represents a construct that is 25 nts in length, and carries an analog base located at 11th position from 5′-end) and two numbers for a construct containing a dimer probe (for example, 25B2,3 where the analog is at second and third positions from 5′-end). The fluorescent base analog 2-AP is designated by ‘a’ and 6-MI by ‘g’. The base sequences of all the constructs used were designed to be as closely identical as we could make them and still have the monomer or dimer base analog probes flanked by the same bases in most cases, since the immediately flanking bases seem to perturb the spectroscopic properties of the probes significantly more than sequence changes further away (N. Johnson, D. Jose and W. Lee, unpublished results). In earlier work (9,10,15) we have shown that the binding affinity of gp32 to ssDNA lattices is relatively independent of base sequence or composition, except that binding is approximately three-fold tighter to runs of dT residues.
The fluorescence intensities of 2-AP dimer probes increase with gp32 binding to ssDNA polynucleotide strands
As shown in earlier titration studies in which binding was monitored by tracking changes in the intrinsic protein fluorescence of the protein, gp32 binds cooperatively to ssDNA lattices and the binding is expected to be close to stoichiometric under the solvent conditions used in our experiments (30 mM KOAc and 10 mM HEPES concentrations at 20°C) (10,15–17). Based on these earlier results, we expected the association binding constant of an isolated gp32 monomer (K) at these salt concentrations to be ∼105 M−1, and the cooperativity parameter (ω) to be ∼103. Thus, the net equilibrium association constant for a ‘cluster’ of three gp32 molecules contiguously and cooperatively bound to a ssDNA lattice containing 3 gp32 binding sites (assuming no partial binding of an additional gp32 molecule to any ‘extra’ nts present) should be ∼K3ω2 M−3, or Kω2/3 M−1 per bound gp32 monomer, where K is the association constant for an isolated gp32 monomer and ω is the (unitless) cooperativity parameter that multiplies K for each contiguous gp32–gp32 subunit interface formed.
In keeping with our findings for short (8-mer) ssDNA lattices containing 2-AP probes [Figure 3A–C, companion paper (1)], the fluorescence spectra obtained for 2-AP probes embedded in longer ssDNA lattices also consisted of a single peak centered at 370 nm, with the binding of gp32 again increasing the fluorescence intensity without a significant shift of the peak position. Thus in presenting gp32 titrations in this paper we have generally plotted only the intensity (or amplitude), or changes in intensity, of the fluorescence peak.
Figure 3.
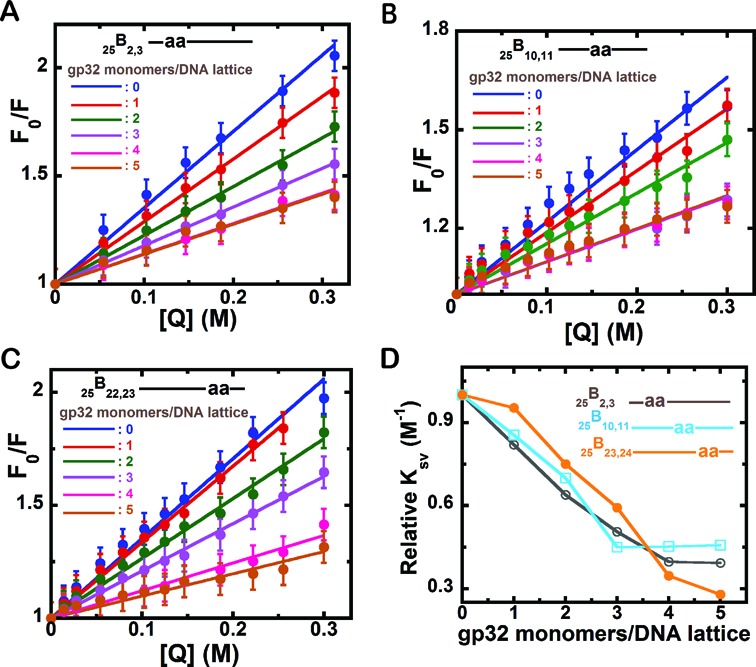
Acrylamide quenching Stern–Volmer plots of 2-AP dimer-labeled 25-mer-ssDNA constructs with increasing concentrations of gp32 monomer added per 25-mer ssDNA lattice. (A) 25B2,3 construct; (B) 25B10,11; and (C) 25B23,24. The individual plots within each panel correspond to the various input concentrations of gp32 expressed as gp32 monomers added per ssDNA lattice, and are (for each spectrum): blue: 0; red: 1; green: 2; purple: 3; pink: 4; and brown: 5 gp32 monomers added per 25-mer lattice. Fo/F (y-axis) corresponds to the fluorescence intensity in the absence of quencher divided by the fluorescence at the given quencher concentration and [Q] (x-axis) is the molar concentration of acrylamide monomers. The initial concentrations of the ssDNA lattice constructs in each titration were 1 μM. (D) Relative changes in the slope of the Stern–Volmer plots for each construct with increasing concentrations of gp32. 25B2,3 construct (grey, open circles); 25B10,11 (cyan, open squares) and 25B23,24 (orange, closed circles). The x-axis is expressed in units of gp32 monomers added per ssDNA lattice present.
The normalized fluorescence intensity changes accompanying the titration of 1 μM solutions of the three 25-mer ssDNA constructs containing 2-AP dimer probes that are listed in Table 1 (25B2,3, 25B10,11 and 25B23,24) are plotted in Figure 1A as a function of the number of gp32 monomers added per ssDNA lattice. (This mode of parameterizing the x-axis has the advantage of taking into account the dilution of the DNA as titration proceeds.) We note that these 25-mer ssDNA substrates were designed to be somewhat longer than 3 (times 7 = 21 nts) gp32 binding ‘site-sizes’ to avoid possible complications from ‘end-effects’ (presumably due to decreased cation condensation at the end residues, see (17), that might result in weaker gp32 subunit binding to the 3′- and 5′- end residues of the ssDNA lattice. This issue is further explored in this paper—see subsequent ‘Results’ section.) All three constructs (representing individually the binding interactions with probes that separately sense binding to each of the three potential gp32 binding sites of the 25-mer ssDNA lattice) showed comparable (and close to linear) increases in fluorescence intensity up to a sharp break at close to 3 gp32 monomers per 25-mer ssDNA lattice, followed by a saturation plateau, regardless of the position of the dimer probes within the ssDNA constructs. This presumably reflects the fact that gp32 binding at sub-stoichiometric concentrations of gp32 is expected to take advantage of its large cooperativity parameter to largely saturate one ssDNA lattice after another, after initially binding at random as isolated monomers on all the lattice sites available. Based on a binding site size of ∼7 nts per gp32 monomer bound, all three constructs in Figure 1A show the expected binding saturation at close to three gp32 monomers bound per 25-mer ssDNA lattice.
Figure 1.
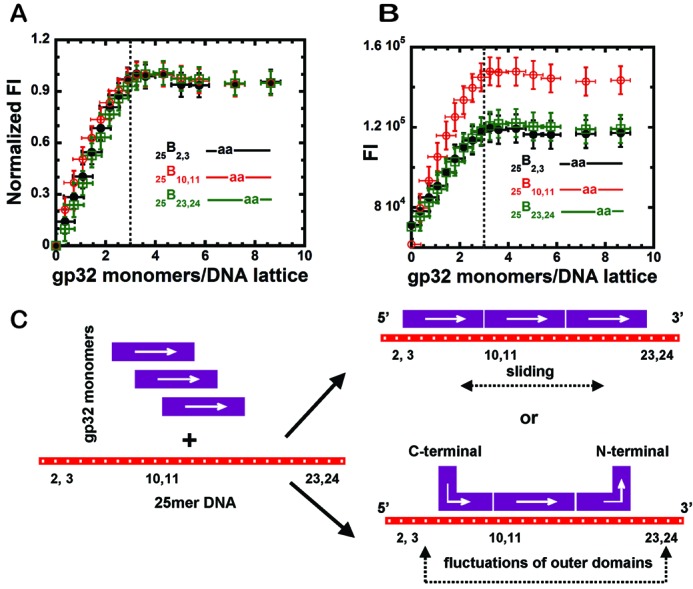
Fluorescence intensity changes at 370 nm observed for gp32 binding to 25-mer ssDNA constructs with 2-AP dimer probes located at different positions, and a schematic model for the binding. (A) Normalized fluorescence intensity changes of the dimer probes for ssDNA constructs at 1 μM concentration as a function of gp32 monomers added per 25-mer ssDNA lattice. 25B2,3 construct (black, closed circles); 25B10,11 construct (red, open circles) and 25B23,24 construct (green, open squares). The dashed vertical line in (B) represents the point at which 3.0 gp32 monomers have been added per 25-mer ssDNA lattice. (B) Raw fluorescence intensity changes for the titration curves of the same ssDNA constructs as in Figure 1A (same color-coding). (C) Schematic model of possible binding modes of trimeric gp32 monomer clusters to these 25mer ssDNA lattices.
This observation of essentially fully stoichiometric binding at all three lattice positions tested suggests that the binding affinity for the three gp32 molecules that comprise the cooperatively binding cluster is approximately equi-partitioned across all three gp32 subunits of the cluster, resulting in a net association constant per gp32 monomer of Kω2/3, or ∼107 M−1 under the solvent and temperature conditions used here. In contrast, if the binding free energy were not equi-partitioned across all three gp32 subunits of the cluster, then one of the ‘end’ subunits might bind more weakly (with K ≈105 M−1) and we would expect a significantly rounded (non-saturated) binding profile in Figure 1A for one of the ‘end’ probe positions, and this is not seen. We have checked this assumption by carrying out theoretical calculations for binding expectations for these ssDNA lattices, based on the above binding parameters and using the finite lattice techniques of Epstein (18), and the results (see Supplementary Data and Supplementary Figure S1) confirm our expectation that all three subunits bind reasonably tightly and with comparable binding affinities.
On the other hand, if we plot the raw (not normalized) fluorescence data for these ssDNA substrates for each of the three probes separately (Figure 1B), we note that the ‘amplitudes’ of the fluorescence intensity changes observed at the three probe positions are very different, with the fluorescence increase seen at the ‘middle’ probe being significantly larger than the amplitude of the increases observed at the two ‘end’ probe positions. Given that the binding affinity at all three probe positions are approximately equal, and that the binding as three subunit clusters is close to stoichiometric, this suggests that the outer portions of the two ‘external’ gp32 subunits of the cluster can fluctuate ‘on and off’ their respective binding probes at equilibrium while the gp32 molecules themselves remain bound. This notion is illustrated schematically in Figure 1C. An alternative interpretation at this point (see also Figure 1C) is that the 3-subunit gp32 cluster can slide back and forth on the 25-mer ssDNA lattice as a ‘block’, resulting in a partial equilibrium exposure of both pairs of probes located near the ends of the ssDNA lattice, relative to the middle probe (see Table 1). Experiments described below (with ssDNA lattices that are closer to an exact integral number of gp32 ‘occlusion site sizes’), as well as single molecule studies of gp32 binding dynamics (W. Lee, J. Gillies, D. Jose, C. Phelps and B. Israels, unpublished data), suggest that both binding domain fluctuations at the ends of the clusters and the sliding of the clusters themselves are likely to be responsible for reducing the observed fluorescence changes seen at these flanking probe positions.
Circular dichroism (CD) spectra of site-specifically labeled ssDNA constructs also change with added gp32
As shown in the companion paper (1) for the binding of single gp32 monomers to 8-mer ssDNA lattices, changes in the CD spectra of 2-AP dimer probes within ssDNA lattices can also serve as a sensitive measure of local DNA conformation changes induced by gp32 binding. Here we have used this approach with longer lattices to look for and characterize position-specific effects within the three gp32 binding sites on the ssDNA lattices for 25-mer constructs with 2-AP dimer probes located respectively near the 5′-end (25B2,3), near the middle (25B10,11) and near the 3′-end (25B23,24) of the lattice. Based on the fluorescence binding studies summarized in Figure 1, we expected each of these 25-mer constructs to bind essentially stoichiometrically and cooperatively to clusters of three gp32 monomers. The CD spectra obtained at each of the three probe positions as a function of number of gp32 monomers present per ssDNA lattice binding site are shown in Figure 2A–C. The concentration of ssDNA construct used in each experiment was 3 μM (see blue spectra—ssDNA alone), and then gp32 subunits were added to each sample to concentrations of ∼3 μM gp32 (red spectra), ∼6 μM gp32 (green spectra) and ∼9 μM gp32 (purple spectra), with saturation of the 3 full gp32 binding sites expected at the 9 μM gp32 concentration. The titration results obtained appear to be largely consistent with our interpretations of the fluorescence intensity changes presented in Figure 1.
Figure 2.
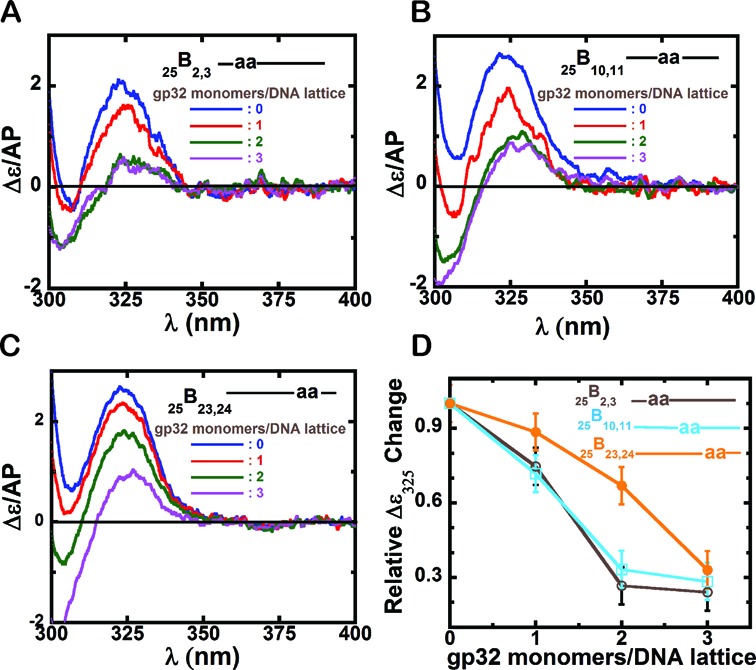
Circular Dichroism spectral changes for 25-mer ssDNA constructs with 2-AP dimer probes at various positions within the ssDNA lattice. Panels A–C show CD spectra of the various ssDNA constructs upon addition of increasing concentrations of gene 32 protein, expressed as gp32 monomers added per ssDNA lattice present. (A) 25B2,3 construct; (B) 25B10,11 construct; and (C) 25B23,24 construct. In each panel the color code for the CD spectra are as follows: blue: ssDNA construct only; red: 1 gp32 monomers per 25-mer ssDNA lattice; green: 2 gp32 monomers per lattice; and purple: 3 gp32 monomers per lattice. (D) Relative Δϵ/AP change at 325 nm for each construct with increasing concentrations of gp32. The x-axis is expressed in units of gp32 monomers added per ssDNA lattice. 25B2,3 construct (grey, open circles); 25B10,11 (cyan, open squares) and 25B23,24 (orange, closed circles).
Figure 2A shows the CD spectral changes observed on protein binding to the 25B2,3 construct, which has a 2-AP dimer probe near the 5′ end. The blue spectrum shows the CD signal for the construct alone, which is typical for a 2-AP dimer-probe-labeled ssDNA lattice (9), with the CD peak at ∼325 nm reflecting strong exciton coupling within the dimer probe. The observed weakening of the intensity of the 325 nm peak with gp32 addition is consistent with the notion that exciton coupling decreases as the bases are pulled apart (unstacked) as a consequence of backbone extension caused by gp32 binding to the ssDNA construct. At an input concentration of ∼1 gp32 monomer per 25-mer ssDNA lattice (red spectrum, Figure 2A), the height of the 325 nm peak is significantly decreased, and at ∼2 gp32 monomers per lattice (green spectrum, Figure 2A) the peak height drops significantly further, suggesting additional extension of the sugar-phosphate backbone at these probe positions (green curve, Figure 2A). This backbone extension is clearly significant, because at this level of gp32 addition ∼70% of the exciton coupling has already been lost. The purple spectrum in Figure 2A shows the CD spectrum of 3 μM DNA with an input concentration of ∼3 gp32 monomers per ssDNA lattice, which should come close to saturating the three full gp32 binding sites of the ssDNA construct. We note that the 325 nm peak in Figure 2A at this concentration of gp32 did not decrease significantly beyond the value observed with ∼2 gp32 monomers per 25-mer ssDNA lattice, suggesting that the dimer probe at the 2,3 positions along the ssDNA lattice was already fully covered at an approximately two-thirds saturation with gp32.
The CD changes of the 25B10,11 construct, with the 2-AP probes located near the middle of the DNA strand, are plotted in Figure 2B. The changes observed were similar to those seen for the 25B2,3 construct, again showing a decrease in 325 nm peak intensity upon gp32 addition, with no significant change beyond approximately the two-thirds saturation level, suggesting that binding to the central dimer probe position was also essentially saturated with the addition of approximately two gp32 monomers per 25-mer ssDNA lattice.
The CD changes of the 25B23,24 construct (Figure 2C), with the 2-AP probes located at lattice positions near the 3′-end showed somewhat different behavior. Here the red curve, corresponding to an approximately one gp32 monomer per 25-mer ssDNA lattice, showed a smaller CD intensity change in the 325 nm peak height than seen with the other constructs for this input gp32 concentration. Furthermore, binding at these probe positions also did not reach the full plateau level at the approximately two-thirds saturation level (green curve), suggesting that three (or more) gp32 monomers per ssDNA lattice were required to ‘cover’ the probes located at the 3′ end of the lattice.
These position-specific probe titrations are summarized in Figure 2D, which plots (y-axis) the normalized (to 1.0 for ssDNA in the absence of added gp32) change at the 325 nm peak for each probe position as a function of lattice saturation. The results are consistent with the notion that backbone extension and unstacking of the probe bases at the 5′-end and the center of the lattice go essentially to completion when ∼2 gp32 monomers have been added per 25-mer ssDNA lattice, while unstacking of the probes at the 3′-end of the lattice requiring significantly higher input gp32 concentrations. These differences could reflect incomplete coverage at the end probes due either to fluctuations at the extreme 3′-end of the gp32 unit at the 23,24 lattice positions or to ‘whole block sliding’ at the 3′ probes (see schematics in Figure 1C).
Gp32 monomers bind at random on a 25-mer ssDNA lattice at low concentrations of added protein, but show a preference for 5′-end binding upon the onset of contiguous (cooperative) cluster formation
The CD spectra observed for the 25-mer ssDNA constructs with probes at different lattice positions at input concentrations of gp32 sufficient to cover ∼30% of the potential gp32 binding sites on the ssDNA constructs showed a decrease in exciton coupling at all three probe positions, indicating that backbone extension is being manifested at all three probe positions. This suggests that gp32 binding is initially random over all lattice positions, because if preferential binding at one of the lattice ends were occurring at low binding density we would have observed differences in the CD signals from the 25B2,3 and 25B23,24 constructs from the onset of the titration. However, as the input concentration of gp32 was increased, a plateau in the CD signal change is observed for the constructs with 2-AP dimer probes located near the 5′ end and near the middle of the lattice, whereas such plateauing was not observed for the construct with probes near the 3′ end (Figure 2D). This suggests that when the titration reaches a point at which contiguous binding is initiated and cooperative gp32 cluster formation becomes significant, the resulting gp32 clusters do exhibit preferential binding at the 5′-end of the ssDNA polynucleotide lattice. Possible molecular mechanisms for this apparent switch in binding preference with cluster formation are considered in the ‘Discussion’ section.
Acrylamide quenching data provide an independent measure of gp32 ‘coverage’ at the various probe positions
We have previously used the collisional quenching of the fluorescence of our base analog probes by acrylamide monomers to measure solvent accessibility of these probes under various conditions (6,11). The extent of quenching of the probes in a free ssDNA construct is expected to differ from that of a protein-bound construct if a conformational change occurs at the probe position upon protein binding or if the bound protein partially ‘covers’ the probe bases, thereby reducing the accessibility of these bases to solvent.
To examine the accessibility of the dimer probes in our 25-mer ssDNA constructs as a function of gp32 binding, increasing concentrations of acrylamide quencher were added to DNA in the presence of various concentrations of gp32, and the changes in probe fluorescence intensities were monitored at 370 nm. The results are presented in the form of Stern–Volmer plots for each 25-mer, with F0/F (the initial fluorescence in the absence of quencher, F0, divided by the observed fluorescence in the presence of each concentration of quencher, F) plotted as a function of quencher concentration ([Q]). Straight lines in such plots confirm that quenching is ‘dynamic’ (or ‘collisional’), and thus a good measure of solvent exposure, while curved lines would suggest that the quenching might have some ‘static’ character.
Figure 3A–C show the results of our acrylamide quenching experiments for the 25B2,3, 25B10,11 and 25B23,24 ssDNA constructs at various DNA:gp32 monomer ratios. Here the concentration of ssDNA molecules was held constant at 1 μM, progressive addition of quencher to all three ssDNA constructs resulted in collisional quenching at all three probe positions (blue lines), and quenching was reduced at all three probe positions with increasing gp32 binding. For the probes at the 10,11 positions, the reduction in quenching was complete with the addition of 3 μM gp32 (purple line), as expected at these central lattice positions for cooperative and stoichiometric binding of gp32 to the three full gp32 monomer binding sites present on these 25-mer ssDNA constructs. As also expected from our fluorescence and CD titrations (Figures 1 and 2), accessibility to the acrylamide quencher continued to decrease at the 2,3 and 23,24 lattice positions on addition of gp32 protein, even at gp32 protein concentrations beyond the saturation level, again suggesting that probe coverage at these positions near the ends of the lattice is incomplete at stoichiometric gp32 binding. This conclusion is consistent either with fluctuations of the ‘outer’ domains of the gp32 molecules bound at the end positions of the lattice or with block gp32 ‘sliding’ on the lattice—see Figure 1C. Again, both effects could contribute.
These acrylamide quenching results are roughly consistent with the CD spectral changes observed at the different probe positions (Figure 2) as a function of gp32 addition. The fact that probe quenching decreased linearly with increasing (sub-stoichiometric) gp32 concentrations at all three probe positions reflects expectations—after initial random binding—for essentially cooperative gp32 binding, with all three gp32 binding sites (and thus all three pairs of probe positions) being largely ‘filled’ together on one ssDNA lattice after another. However, we note again that the change in solvent accessibility at the central probe positions goes to completion at added gp32 concentrations close to binding saturation (purple line), while excess gp32 seemed to be required to saturate the quenching effect at the 5′ probe positions 2,3. The probes close to the 3′-end (positions 23,24) were not fully saturated even upon the addition of significant excess gp32.
Figure 3D show the relative changes in the slope of the Stern–Volmer plots for all three constructs plotted as a function of gp32 monomers/binding site. At initial concentrations of gp32 a steep decrease in the slope is observed for the 25B2,3 and 25B10,11 constructs, whereas the 25B23,24 construct shows a lag initially followed by a steep decrease. The decrease in the steepness of the slope for all constructs, coupled with the lag observed for the 25B23,24 construct, is in agreement with the CD result that suggested that gp32 monomers initially bound randomly to long ssDNA at low levels of binding density and then, as the binding density was increased, the binding became contiguous and cooperative. Furthermore, the preferential plateauing observed for the 25B10,11 and 25B2,3 constructs at lower concentrations of gp32 than seen with the 25B23,24 construct is consistent with the preference for 5′-end binding of gp32 upon cooperative cluster formation.
Similar gp32 binding effects are seen with 6-MI base analog probes
To test whether these spectral consequences of gp32 binding might somehow reflect specific interactions with the 2-AP dimer probes, we also performed gp32 titrations on the 25-mer 25G10,11 ssDNA construct (see Table 1). This construct has the same nucleotide sequence as the 25B10,11 construct, but contains a pair of 6-methyl isoxanthopterin [6-MI—a fluorescent base analog of guanine (13,19)] probes at the 10,11 positions instead of the 2-AP dimer probes used previously. CD and fluorescence titrations with gp32 of ssDNA construct 25G10,11 are presented in Figure 4A and B, respectively, and show CD and fluorescence intensity changes with increasing gp32 concentrations that closely resemble those seen with the 25B10,11 construct (Figures 1B and 2B), including cooperative and stoichiometric binding as manifested by the fluorescence intensity change (here at 425 nm) as a function of gp32 monomers added per ssDNA lattice (Figure 4C).
Figure 4.
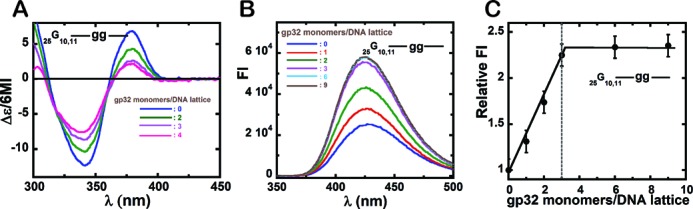
Circular dichroism and fluorescence spectral changes observed for DNA construct 25G10,11 containing a pair of 6-MI dimer probes at positions 10,11. (A) CD spectra of the DNA construct upon addition of various amounts of gene 32 protein. Blue: free ssDNA construct; green: 2; purple: 3; and pink: 4 gp32 monomers added per 25-mer ssDNA lattice. (B) Fluorescence spectra for the ssDNA construct with 6-MI dimer probes at the 10,11 lattice positions. Blue: free DNA construct; red: 1; green: 2; purple: 3; cyan: 6; and grey: 9 gp32 monomers added per 25-mer lattice. (C) Normalized fluorescence intensity changes at 425 nm as a function of gp32 monomers added per ssDNA lattice. Binding is stoichiometric and saturates at ∼3 gp32 monomers bound per 25-mer ssDNA lattice (dashed vertical line).
Figure 4A shows the CD spectral changes observed for the 25G10,11 construct at various gp32 concentrations. Increasing concentrations of gp32 were accompanied by strong decreases in exciton coupling, as observed with the 2-AP labeled constructs and again are consistent with backbone extension of the ssDNA lattice upon gp32 binding. This result suggests that the changes observed with the 2-AP dimer probes do not reflect unique features of that particular base analog, but instead general properties of the ssDNA-gp32 interaction. Figure 4B shows the fluorescence intensity changes (at 425 nm) of the 6-MI dimer probe on gp32 binding, again showing increasing fluorescent amplitudes with gp32 addition and that these amplitude changes reach a plateau at stoichiometric protein concentrations. The position of the fluorescence peak did not shift with increasing gp32 concentration, which is again consistent with the interpretation that the observed fluorescence intensity changes arise solely from the effect of gp32 on the base analogue probes, and also that the aromatic amino acid residues of the protein did not contribute to, or otherwise interfere with, the fluorescence changes observed with either set of probes.
The emission maximum for 6-MI dimer probes in ssDNA occurs at ∼425 nm (13), and the peak intensity is plotted as a function of gp32 monomers added per gp32 binding site in Figure 4C, which shows that the fluorescence intensity changes reach a maximum at close to a stoichiometric concentration ratio of DNA to gp32 protein, again consistent with a gp32 occlusion binding site size of ∼7 nts and with highly cooperative gp32 binding to this 25-mer ssDNA lattice containing 6-MI probes.
Monitoring the fluorescence intensity changes of base analog probes at various positions in ssDNA lattices containing (at saturation) eight gp32 binding sites as a function of input gp32 concentration confirms that gp32 monomer binding is initially random, but that higher gp32 binding densities favor binding at the 5′-end of the ssDNA lattice
As polynucleotide lattices become longer and thus more potential binding sites are available at low (well below stoichiometric) gp32 concentrations, entropy considerations dictate that even large values of the cooperativity parameter (ω) will not suffice to bring about fully sequential loading of one individual ssDNA lattice after another, and small differences in the binding affinity (K) of gp32 monomers as a function of base sequence or lattice position can lead to preferential (initial) binding at some lattice positions relative to others. To learn more about this interplay between cooperativity and site-specific preferential binding of gp32 to longer polynucleotide lattices three additional ssDNA constructs were synthesized, each containing 56 nts with a single 2-AP probe located at position 4 (the 56F4 construct), position 21 (56F21) and position 53 (56F53)—see Table 1. Since the binding site size of gp32 is ∼7 nts, a saturated 56 nts ssDNA lattice should contain exactly eight gp32 monomer binding sites.
The raw and normalized (to an initial value of unity) fluorescence intensity profiles at 370 nm, showing gp32 titrations of 1 μM concentrations of these 56-mer ssDNA constructs with single 2-AP probes located at various lattice positions, are plotted in Figure 5A and B, respectively. The first additions of gp32 to the 56F4 construct (black titrations) resulted in a sharp jump in the initial fluorescence intensity, with a rough plateau being reached at a gp32 concentration of ∼5 gp32 monomers per 56-mer ssDNA lattice (Figure 5A and B, closed circles). Clearly the binding plateau for construct 56F4, in which the 2-AP probe is located near to 5′-end of the ssDNA lattice, is reached well before the expected stoichiometric saturation level, suggesting that at equilibrium cooperative clusters of gp32 molecules again appeared to bind preferentially at the 5′-end of the lattice.
Figure 5.
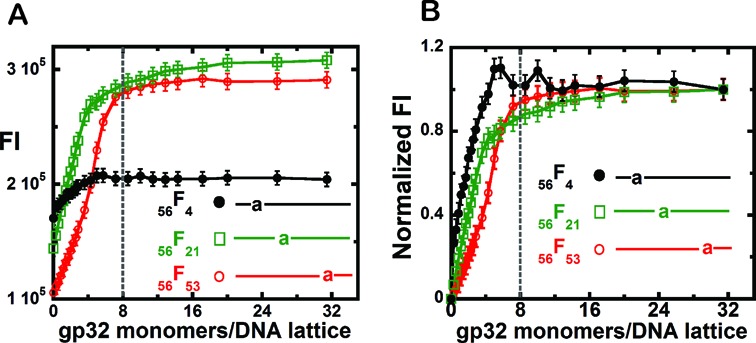
Fluorescence intensity changes at 370 nm observed for the gp32 titration of 56-mer-ssDNA constructs with 2-AP monomer probes at various positions. (A) Fluorescence intensity changes of the DNA constructs (1 μM) upon the addition of various concentrations of gp32 per 56-mer ssDNA lattice. 56F4 construct (black curve, closed circles); 56F21 construct (green curve, open squares); and 56F53 construct (red curve, open circles). (B) Normalized fluorescence intensity changes of these DNA constructs (1 μM) upon addition of increasing concentrations of gp32 per ssDNA lattice. Same constructs and color-coding as in (A). The dashed vertical lines represent the expected saturation point at 8 gp32 monomers added per 56-mer ssDNA lattice.
This interpretation is borne out when we examine the titration profiles of the 56F21 (green – open squares) and 56F53 (red—open circles) constructs, containing 2-AP probes located near the center or near the 3′-end of the lattice, respectively. We note the titration of the probe in position 21 (open squares) is shifted somewhat to the right (relative to the titration of the 56F4 construct), but still appears to reach effective saturation before gp32 stoichiometry has been attained, while the 56F53 construct shows a major lag and requires somewhat more than stoichiometric concentrations of gp32 to reach a final binding plateau. (These latter features are more clearly seen in Figure 5B, in which the titration profiles have been normalized to a constant amplitude.) However, we note (see Figure 5A) that the ‘actual’ amplitude of the titration profile for the 56F4 construct (with the 2-AP probe close to the 5′-end) is much smaller than the amplitude of the titration profiles for the other constructs, suggesting that this probe—either because of its exact position ‘under’ the first bound gp32 monomer or because of its base sequence environment—might be less exposed to solvent after gp32 binding than the probes in the other constructs. In any case these results suggest that at equilibrium gp32 binding sites located near the 5′-end of the ssDNA lattice are favored for the binding of gp32 clusters, perhaps either because cluster dissociation proceeds preferentially from the 3′ ends of cooperatively bound blocks of gp32 monomers, or because cluster binding is favored at the 5′-end of the lattice. These points are considered further in the ‘Discussion’ section.
Binding of gp32 protein to dsDNA is not cooperative, but does result in some base unstacking and presumably extension of the DNA backbone
In addition to the binding to ssDNA (and ssRNA) lattices, it is known that gp32 monomers can also bind to duplex DNA sequences and also that this binding affinity for dsDNA sequences can be altered by changing the salt concentration of the solution (8). It was shown previously that gp32 binds more tightly to ssDNA than to dsDNA lattices, as expected given that the primary functional role of this protein in DNA replication is to coat the ssDNA sequences transiently formed by the primosome helicase and position them for template-dependent synthesis of the DNA ‘daughter’ strands by the replication polymerases (15). The preference of gp32 for binding to ssDNA (or ssRNA) over dsDNA reflects in part the decreased binding affinities of gp32 monomers for the dsDNA form, but is primarily a consequence of the fact that gp32 cannot bind to dsDNA cooperatively, and thus cannot utilize the free energy contributed by the cooperative binding interaction (ω) to strengthen the binding reaction (8).
We show in Figure 6 that the binding of gp32 to dsDNA, and the conformational changes that result in the DNA duplex, can also be monitored by following the low energy CD spectral and fluorescence intensity changes of 2-AP monomer or dimer probes that occur as a consequence of gp32 binding. dsDNA constructs with a 2-AP dimer or monomer probes positioned near the middle of the duplex were formed by annealing the 25B10,11 or the 25B11 ssDNA constructs with their unlabeled 25C ssDNA complement (see Table 1) to form the 25B10,11–25C or 25B11–25C duplex constructs.
Figure 6A shows the changes in the CD spectra that result from gp32 addition to an initial 3 μM concentration of the duplex 25B10,11–25C construct containing a 2-AP dimer probe. Clearly the spectral changes with increasing gp32 concentrations resemble those seen with our (initially partially stacked) ssDNA constructs containing dimer 2-AP probes (see Figure 2). These results are consistent with binding mechanisms involving partial opening of the duplex and a partial separation of the DNA strands to permit local binding of gp32 in the ssDNA binding mode, resulting in unstacking of the dimer probe as manifested by reduced exciton coupling. We use an estimated binding site size of ∼5 bps per gp32 monomer for the initial binding of gp32 to the dsDNA constructs (8), and the color-coded spectra correspond to 0 (blue), 3 (green), 6 (brown) and 12 (olive green) gp32 monomers added per 25 bp dsDNA lattice present. This interpretation of the dsDNA binding interaction of gp32 with initially duplex DNA is also consistent with the observed reduction in binding affinity (relative to ssDNA), since some of the binding free energy of gp32-ssDNA complex formation must be expended in partially opening the DNA duplex to provide access to a binding site that is at least partially single-stranded.
Figure 6B tracks the fluorescence intensity change at 370 nm of a 2-AP dimer or monomer probe within an initially 1 μM concentration of 25B10,11–25C or 25B11–25C dsDNA constructs as a function of gp32 added per initial dsDNA gp32 binding site, and shows that after an initially steep jump the fluorescence continues to increase linearly up to at least a 1.5-fold molar excess of added gp32 monomers. This result is consistent with the interpretation that, while gp32 can bind to duplex DNA and partially unstack the bases of the dimer probe as a consequence, this binding does not bring about complete dsDNA melting or strand separation and is not cooperative at the gp32 concentrations tested. The observed progressive increase in fluorescence with gp32 addition is also consistent with previous findings (8,15) that gp32 binding to dsDNA is relative weak and non-cooperative.
DISCUSSION
Mapping the interaction surfaces of DNA regulatory proteins from the nucleic acid perspective
Generally the nonspecific binding of DNA regulatory proteins to their ssDNA and dsDNA targets in solution has been measured via relatively undefined (in structural terms) assays from the perspective of the protein; for example, by monitoring the quenching or enhancement of the fluorescence of one or more aromatic amino acid residue located in or near the binding surface of the protein—(8), or the nucleic acid (by using changes in the overall absorbance or hypochromicity at 260 nm—(8,20)), or by tracking parameters reflecting large-scale changes in the frictional properties of the complex on protein binding (such as changes in fluorescence depolarization anisotropy). The results of such binding assays are generally couched in terms of three thermodynamic parameters: K, the equilibrium constant for binding an isolated protein monomer; ω, the cooperativity constant that describes the added binding free energy resulting from contiguous protein monomer binding; and n, the occlusion site size of the protein monomer bound to the DNA lattice (21). Furthermore, for reasonably long ssDNA lattices (10 or more gp32 binding sites per lattice) the binding distributions of protein monomers on the DNA lattice as a function of degree of lattice saturation can be described by simple conditional probability models couched in terms of these equilibrium binding parameters (for example, see (22)), although binding to shorter lattices requires the use of the finite binding models of Epstein (3,15,18).
These models have been useful in providing thermodynamic explanations of certain aspects of biological function, including how positive binding cooperativity competes in these proteins with the consequences of overlap binding (each bound protein covers several adjacent DNA positions—see (22)) to make it possible to saturate ssDNA lattices at finite protein concentrations, why long single-stranded binding sites on mRNA are saturated before short ones (leading to a mechanistic explanation of the auto-regulation of gp32 synthesis during DNA replication (3,4,23)), and other related thermodynamic issues. However, they provide no mechanistic information about the actual interactions between these single-stranded DNA binding (ssb) proteins and their target lattice sites, the molecular origin(s) of binding polarity, the interactions between the individual ssb proteins (either directly or through the DNA) that are responsible for binding cooperativity, and how the binding mechanisms of the individual ssb proteins can be exploited by competing proteins that cycle on and off the DNA and must interact with, or displace, ssb proteins during the various phases of DNA replication (and recombination and repair).
Mechanistic information of this sort can be obtained by ‘mapping’ the binding surfaces of the proteins, thus providing detailed positional information to determine how these interfaces interact with the complementary surfaces of the underlying DNA. In this paper and its companion (1) we have begun to develop a systematic way of doing this, in the context of understanding the isolated and cooperative binding of gp32 protein to ssDNA, by site-specifically inserting base analog probes that display fluorescence and CD spectra at wavelengths above 300 nm (where the DNA canonical bases—and the amino acid residues of the binding proteins—are transparent) and interpreting the resulting spectral data in structural terms.
Gp32 monomers bind contiguously and cooperatively to long ssDNA lattices at high binding densities
In the companion paper (1) we proposed a model for the binding of gp32 monomers to short (one gp32 site-size) ssDNA lattices in which the strong component of the binding involved direct interaction with the ssDNA backbone at a pair of ssDNA nucleotide residues located near the 3′-end of the lattice, with the 5′-end of the protein fluctuating on and off the lattice depending on the position of C-terminal ‘interaction arm’ domain. We proposed that this relatively weak binding might serve as a model for the initial isolated binding of gp32 monomers at random ssDNA polynucleotide sites at low protein binding densities.
In Figure 1 of this paper we summarized the fluorescence changes observed with 2-AP dimer probes positioned at the two ends, and near the middle, of 25-mer ssDNA constructs containing three potential full gp32 binding sites at saturation, plus a couple of extra residues at each end to minimize end-effects, to monitor the progression of the binding of multiple gp32 to these longer lattices. The results showed that binding of gp32 to these longer ssDNA constructs was essentially fully cooperative, with all three monomers bound with essentially the same net affinities, meaning that the total binding free energy of each ‘block’ of three cooperatively bound monomers per lattice was essentially equi-partitioned between the three monomers, corresponding to a binding constant per gp32 monomer of ∼Kω2/3. Figure 1B, however, showed that the ‘amplitude’ of the fluorescence change was very different for the probes at the three lattice positions tested, with the amplitude at the central position being significantly larger than the amplitudes for the pairs of probes located at the two ends of the lattice. These results led us to the two possible binding models schematized in Figure 1C, suggesting that the amplitudes of the fluorescent signals at the two ends might be less than that in the middle either because sliding of the contiguously bound ‘block’ of three gp32 monomers partially exposed the two end positions, or because the outer ends of the two flanking gp32 molecules might fluctuate on and off the ssDNA lattice without dissociation of the bound gp32 monomers, with cooperative binding preventing such fluctuations for the central gp32 monomer.
Gp32 monomers bind randomly to ssDNA lattices containing multiple gp32 binding sites at low binding density, and then shift to bind preferentially at the 5′-end as the binding density increases and binding becomes contiguous and cooperative
The CD experiments performed on ssDNA constructs containing multiple gp32 binding sites with 2-AP dimer probes placed at different positions within the lattice showed a decrease in the CD signal at sub-stoichiometric concentrations of gp32 protein at all lattice positions (Figure 2 A–C). This suggests that, unlike for gp32 binding to 8-mer oligomers for which the binding register was fixed by the fact that the binding site size of the protein was essentially equivalent to the length of the lattice, the initial binding of gp32 protein monomers to a longer (polynucleotide) lattice is essentially random. As the input concentration of gp32 was increased, the CD signal for these longer lattices showed a further decrease in intensity, but the general shape of the curve remained the same. This suggests that the isolated protein interacts with backbone sugar-phosphate sites, resulting in the bases in this interaction region being pulled apart (unstacked) with a concomitant loss of exciton coupling between the adjacent bases. Figure 2D showed the relative change in the amplitude of the CD signal at 325 nm for all the constructs as a function of input protein concentration. These data confirmed that initial gp32 binding was random, because if binding at sub-stoichiometric concentrations of gp32 occurred only at one end of the lattice, then the CD spectra for probes located at the other end of the lattice should have remained unperturbed.
However, Figure 2 showed that, while all probe positions of the ssDNA constructs showed an initial CD change at low concentrations of gp32, these changes—like those in the fluorescence amplitudes measured in Figure 1—differed in amplitude for the different constructs. Thus at gp32 input levels representing ∼30% of lattice saturation the amplitude of CD signal showed a decrease of ∼10% for the 25B23,24 construct, whereas for 25B2,3 and 25B10,11 constructs the decrease in amplitude of the CD peaks was ∼30%. This leads us to suggest that, after initial random binding, increasing binding density (and thus binding cooperativity) appears to induce the bound protein blocks to ‘slide’ or ‘hop’ toward the 5′-end of the lattice. This suggestion is further supported by the fact that the CD signal changes at 325 nm for both the 25B2,3 and 25B10,11 constructs reached a plateau at ∼60% of binding saturation, while the CD changes for the 25B23,24 construct had not reached a plateau even at binding saturation.
The observed changes in fluorescence as a consequence of acrylamide quenching at the various probe positions with increasing gp32 concentration shown in Figure 3 are also consistent with the above interpretations of the CD titration data. Thus the observation that 25B2,3 and 25B10,11 constructs reached quenching minima at stoichiometric gp32 concentrations or less also implies that after initial binding the protein again preferentially covered the probes at the 5′-end and the middle of the lattice, while binding at the probe positions of the 25B23,24 lattice did not saturate even at input gp32 concentrations well above saturation. The observation (Figure 3D) that the slopes of the Stern–Volmer plots at all the probe positions initially decreased at about the same rate as a function of added gp32 (though with a slight initial lag for the probes at the 23,24 positions) is consist with initial binding being close to random, followed by differential changes in probe occlusion as a consequence of shifts of the bound ‘blocks’ of gp32 molecules toward the 5′-end of the lattices as cooperative binding ‘kicks in’.
Such initial random binding, followed by migration to the 5′-end as cooperative binding takes over, was also observed for the titration of polynucleotide constructs 56 residues in length (Figure 5), which should bind ∼8 gp32 monomers per ssDNA lattice at binding saturation. Figure 5A and B show that a steep rise in fluorescence intensity was observed at lower concentrations of gp32 for the 2-AP probes located near the 5′-end of the lattice, (construct 56F4) whereas a clear lag was observed for constructs with the 2-AP probe at the 3′ end (construct 56F53). The construct (56F21) with the 2-AP probe near the middle of the lattice showed an intermediate lag.
These differences in binding behavior of gp32 to short oligomers that are essentially only one gp32 binding site in length, and longer ssDNA lattices where a number of gp32 molecules can bind contiguously and cooperatively, are consistent with the model put forward in the companion paper (1), where we suggested the presence of the C-terminal arm prevents the movement (‘shuffling’) of the protein across the short (8-mer) oligomer, whereas movement is possible for blocks of gp32 molecules on longer lattices because cooperative binding forces the C-terminal arm into a displaced position on ‘top’ of the gp32 monomer (see also (1,10)). These issues are further developed in the model figure (Figure 7) and the related text below.
Figure 7.
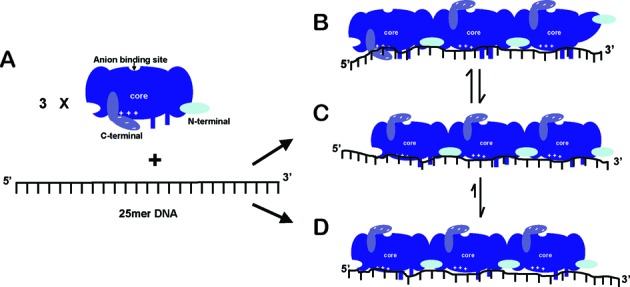
A schematic and mechanistic model for the binding of a ‘block’ of three cooperatively bound gp32 monomers to a 25-mer ssDNA lattice. (A) A 25mer ssDNA construct and a gp32 monomer are drawn showing the three distinct domains of the intact gp32 molecule: the core DNA binding domain (blue); the C-terminal interaction domain (violet blue); and the N-terminal cooperativity domain (light blue). The electropositive cleft in the core domain and the acidic C-terminal are depicted by the ‘+’ and ‘−’ symbols, and the anion binding site is indicated. Panels B–D show three possible cooperative binding modes for a ‘block’ of three gp32 monomers on a 25mer polynucleotide lattice (such as the ssDNA constructs used in Figures 1–3). (B) Binding mode 1—a breathing or fluctuating mode. The C-terminal acidic domain and the 5′-end of the DNA construct compete for the electropositive cleft of the core domain, perhaps helping to make the 5′-end of the gp32 cluster available for interaction with other proteins. The absence of a contiguously bound protein at the 3′-end of the DNA construct may also permit the N-terminus of the protein at the 3′ end to fluctuate and assume positions that provide interaction possibilities for other T4 DNA replication proteins. (C) Binding mode 2—a sliding mode. The C-terminal domain flips up and the 5′ end of the DNA is bound to the electropositive cleft of the core domain and the N-terminal of the core domain is fully bound to the ssDNA construct, perhaps enabling the protein to slide along the polynucleotide lattice in a thermally-driven one-dimensional random walk, and also further favoring gp32-induced ssDNA lattice extension. (D) Binding mode 3—the preferred binding mode at the 5′-end of the ssDNA lattice. The gp32 cluster slides to and preferentially binds at the 5′ end of the ssDNA lattice (or perhaps at the junction of a DNA replication fork on the longer DNA structures present in vivo), further extending and stabilizing that end of the lattice and perhaps optimizing this position for interactions with other replication proteins.
A mechanistic model for the cooperative binding of gp32 to ssDNA lattices and functional implications for the bacteriophage T4-coded DNA replication complex
In the preceding paper (1) we presented a complete study of the isolated binding of individual gp32 monomers to 8-mer ssDNA lattices that were just long enough to span the complete protein binding site, using site-specifically positioned 2-AP spectral probes to map the protein binding surface. We showed that the protein binds relatively weakly to these oligomers, with essentially the same binding affinity to all, that it does not ‘shuffle’ on these oligomers and that binding is polar relative to the 5′→3′ backbone of the ssDNA lattice. This polarity was manifested by the differential responses of the 2-AP dimer probes to gp32 addition as a function of probe position, with bases located near the 3′-end of the lattice showing significantly more unstacking than those near the 5′-end.
Based on these results and the known x-ray structure of the DNA binding domain of gp32 (2), we were able to use our results to infer the orientational polarity of the gp32 monomer protein bound to ssDNA, and also to suggest how the N-terminal and C-terminal interaction domains of the protein might be involved in the binding interactions. Our present structural model for the binding of the monomer is presented in Figure 6E and F of the companion paper (1), and shows that the protein binds tightly to the backbone of the ssDNA at a di-nucleotide position near the 3′-end of the lattice, resulting in an extension of the backbone and unstacking of the vicinal bases. We suggested (see also earlier models—see (10,24) that this binding is accompanied by a fluctuation of the C-terminal arm of the gp32 monomer between a position in which it impedes the full interaction of the ssDNA with the binding cleft of the gp32 monomer and a position in which the C-terminal arm ‘swings out-of-the-way’ and full electrostatically-driven binding of the 5′ end of the lattice to the gp32 binding cleft is achieved. Under monomer binding conditions we suggested that the protein fluctuates between a partially bound state with the C-terminal arm swung down (see Figure 6E of the companion paper) and a fully bound state with the arm swung up (Figure 6F).
Since a gp32 monomer bound to a ssDNA lattice that is essentially one monomer site size in length is unable to shuffle along the lattice, each DNA probe position must have a one-to-one correspondence to a position on the binding surface of the protein. However, this is not true for proteins bound to longer lattices that can bind several contiguous gp32 molecules, because an isolated gp32 monomer does not maintain a fixed position on the lattice as the input gp32 protein concentration is increased. Thus initially, at very low binding density, positional entropy prevails and the protein binds essentially at random, assuming that the value of the monomer binding constant (K) is not base sequence dependent. (This is generally the case for ssDNA of heterogeneous sequence.) Then, as the input protein binding density increases, cooperativity comes into play and the proteins rearrange on the lattice.
These rearrangements can be modeled on a conditional probability basis using finite (or infinite) lattice binding theory (3,18,22), with the most probable arrangements at various concentrations being defined by the values of n, K and ω used. Eventually, for values of ω large relative to n, binding cooperativity overcomes the ‘negativity cooperativity’ introduced by overlap binding and the lattice is saturated. (For finite lattice calculations relevant to the 25-mer lattices considered in Figures 1–4 of this paper, see Supplementary Figure S1 and related discussion in the Supplementary Data submitted with this paper.) Then, for lattices that contain an integral number of protein monomer binding sites, each protein is again fixed in position with a one-to-one correspondence between probe position on the DNA and a discrete complementary position on the binding surface of the protein. To map this situation completely requires that data be taken on constructs with probes located at each lattice position under a protein monomer that is fixed in position, ideally bound at a central position on a saturated lattice (we are currently completing such a ‘saturation mapping’ study, using both ssDNA lattices of heterogeneous base sequence and lattices containing sequences where binding is ‘preferred’ [primarily oligo(dT) sequences], D. Jose, K. Meze and B. Camel, unpublished data).In the studies reported here we have not performed such ‘saturation mapping’ for a ssDNA lattice completely covered with contiguously bound gp32 monomers, but important structural aspects of the binding interaction that go beyond the thermodynamic values of cooperative binding parameters and that significantly modulate binding can already be inferred from the data we have obtained. These insights and conjectures are summarized in the schematic model for cooperative gp32 binding to a ssDNA lattice depicted in Figure 7, which again draws on, and is consistent with, earlier models put forward by our laboratory and others (10,24).
In Figure 7A we show a schematic of a gp32 monomer free in solution, together with a 25-mer ssDNA lattice to which three such monomers can bind in the cooperative binding mode. The three domains of the free and intact gp32 monomer are indicated: (i) the core nucleic acid binding domain, including the tight di-nucleotide binding site located near the N-terminus of the protein, and the positively charged binding cleft located near the C-terminus of the protein, which is shielded from interaction with the nucleic acid in this conformation of the gp32 monomer by the negatively charged C-terminal ‘arm’; (ii) the N-terminal domain that interacts with a binding site on the core domain of a contiguously bound gp32 monomer to achieve cooperative binding; and (iii) the C-terminal domain of the protein that shields the binding cleft in the isolated binding mode and can also fluctuate into a binding position on the ‘top’ of the core binding domain (and presumably at the anion binding site) to permit nucleic acid access to the binding cleft.
On the right in Figure 7B–D we show three possible binding modes for a cluster of three gp32 molecules bound contiguously and cooperatively to the ssDNA lattice. Note that the ‘interior’ interaction interfaces of the subunits are fully cooperatively bound and stabilized by the ‘flipped-up’ C-terminal arms and by the interaction of the N-terminal domains with their complementary binding sites on the adjacent gp32 monomer. In Figure 7B we show the bound block of three gp32 monomers in an ‘unstable’ configuration, with the N-terminus of the right-hand gp32 monomer fluctuated into a non-binding position and the binding of the C-terminal side of the left-hand gp32 monomer destabilized by the C-terminal arm in the ‘down’ position, in which it blocks the DNA binding cleft of the core domain. Comparison with Figure 7C shows that both of the ‘outside’ interaction surfaces of this cooperatively bound gp32 cluster can fluctuate into more stable binding positions that permit interactions with another incoming gp32 monomer on the 3′-side of the lattice, and may prevent interactions with other proteins (such as the gp59 helicase loading protein and the gp61 primase protein of the primosome helicase) of the replication complex that we propose will attack and destabilize the gp32 cluster from the 3′-end of the lattice in the course of lagging strand synthesis by the T4 DNA replication complex (D. Jose, S. Weitzel and W. Lee, unpublished results).
This model suggests that additional gp32 monomers can only be added to the cooperatively bound gp32 cluster from the right-hand (3′) side of the ssDNA lattice, with such addition serving to drive the freely sliding cluster to the left and eventually into a fully stabilized position at the left-hand (5′) end of the ssDNA lattice, thus providing the bias that results in the preferential coverage of our base analogue probes at the 5′-end of our ssDNA constructs, as shown in this paper (see Figure 5). Of course, in addition the gp32 monomer located at the 5′-end of the ssDNA lattice as a part of a cooperatively bound gp32 cluster may be further stabilized in this position by additional binding interactions with the end of the lattice, thus further accounting for the preference for 5′-end binding for contiguously-bound clusters of gp32 molecules. These binding subtleties likely all play a role in controlling the equilibria and dynamics of the clusters of gp32 protein that bind to, and dissociate from, the transient ssDNA sequences that are formed by the primosome helicase and exploited by the replication polymerase—aided and abetted by the other regulatory cofactor proteins of the T4 DNA replication system.
The central protein sub-assemblies used by bacteriophage T4 to discharge its core DNA replication functions resemble closely those used by most of the higher organisms whose replication systems that have been studied to date. Therefore this complex serves as an excellent model system to study the ‘core’ processes of DNA replication. However, the molecular mechanisms of many aspects of the movement of the ‘trombone’ (25) version of the replication complex through the genome in the course of DNA replication are still not understood, and some of these aspects may be further clarified by using the base analog technique we have employed here to map the interactions of gp32 proteins in various binding modes with the underlying DNA at single nucleotide resolution. Thus the finding that clusters of cooperatively bound gp32 proteins bind preferentially at the 5′-end of ssDNA lattices, even though initial isolated binding is random, may have significant biological implications. Since it is known that the helicase-primase complex (primosome complex) moves in the 5′→3′ direction and the polymerase moves in the 3′→5′ direction on the lagging strand, it may be that the weaker binding of gp32 molecule at the 3′-end of gp32 clusters could provide a point of attack to facilitate the continuous removal of gp32 clusters by the advancing DNA replication polymerase in lagging strand synthesis. Our present studies also suggest (consistent with earlier studies (17)) that, at least, small clusters of cooperatively bound gp32 molecules can freely move across ssDNA lattices, likely by a ‘block’ sliding mechanism, to reach preferred lattice binding positions, such as at the 5′-end of the ssDNA lattice or other important binding sites. Additional equilibrium and dynamic studies using these mapping approaches should further illuminate these molecular mechanisms and their roles in the regulation and integration of the processes of genome function and expression.
Supplementary Material
Acknowledgments
We are grateful to our colleagues in both the von Hippel and the Marcus laboratories for many useful discussions of this work.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [GM-15792 to P.H.v.H, in part]. Funding for open access charge: NIH [GM-15792].
Conflict of interest statement. None declared.
REFERENCES
- 1.Jose D., Weitzel S.E., Baase W.A., von Hippel P.H. Mapping the interactions of the single-stranded DNA binding protein of bacteriophage T4 (gp32) with DNA lattices at single nucleotide resolution: gp32 monomer binding. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv817. doi:10.1093/nar/gkv817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shamoo Y., Friedman A.M., Parsons M.R., Konigsberg W.H., Steitz T.A. Crystal structure of a replication fork single-stranded DNA binding protein (T4 gp32) complexed to DNA. Nature. 1995;376:362–366. doi: 10.1038/376362a0. [DOI] [PubMed] [Google Scholar]
- 3.von Hippel P.H., Kowalczykowski S.C., Lonberg N., Newport J.W., Paul L.S., Stormo G.D., Gold L. Autoregulation of gene expression. Quantitative evaluation of the expression and function of the bacteriophage T4 gene 32 (single-stranded DNA binding) protein system. J. Mol. Biol. 1982;162:795–818. doi: 10.1016/0022-2836(82)90548-4. [DOI] [PubMed] [Google Scholar]
- 4.Russel M., Gold L., Morrissett H., O'Farrell P.Z. Translational, autogenous regulation of gene 32 expression during bacteriophage T4 infection. J. Biol. Chem. 1976;251:7263–7270. [PubMed] [Google Scholar]
- 5.Johnson N.P., Baase W.A., von Hippel P.H. Low-energy circular dichroism of 2-aminopurine dinucleotide as a probe of local conformation of DNA and RNA. Proc. Natl. Acad. Sci. U.S.A. 2004;101:3426–3431. doi: 10.1073/pnas.0400591101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jose D., Datta K., Johnson N.P., von Hippel P.H. Spectroscopic studies of position-specific DNA ‘breathing’ fluctuations at replication forks and primer-template junctions. Proc. Natl. Acad. Sci. U.S.A. 2009;106:4231–4236. doi: 10.1073/pnas.0900803106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Datta K., Johnson N.P., Villani G., Marcus A.H., von Hippel P.H. Characterization of the 6-methyl isoxanthopterin (6-MI) base analog dimer, a spectroscopic probe for monitoring guanine base conformations at specific sites in nucleic acids. Nucleic Acids Res. 2012;40:1191–1202. doi: 10.1093/nar/gkr858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jensen D.E., Kelly R.C., von Hippel P.H. DNA ‘melting’ proteins. II. Effects of bacteriophage T4 gene 32-protein binding on the conformation and stability of nucleic acid structures. J. Biol. Chem. 1976;251:7215–7228. [PubMed] [Google Scholar]
- 9.Kelly R.C., Jensen D.E., von Hippel P.H. DNA ‘melting’ proteins. IV. Fluorescence measurements of binding parameters for bacteriophage T4 gene 32-protein to mono-, oligo-, and polynucleotides. J. Biol. Chem. 1976;251:7240–7250. [PubMed] [Google Scholar]
- 10.Kowalczykowski S.C., Lonberg N., Newport J.W., von Hippel P.H. Interactions of bacteriophage T4-coded gene 32 protein with nucleic acids. I. Characterization of the binding interactions. J. Mol. Biol. 1981;145:75–104. doi: 10.1016/0022-2836(81)90335-1. [DOI] [PubMed] [Google Scholar]
- 11.Jose D., Weitzel S.E., von Hippel P.H. Breathing fluctuations in position-specific DNA base pairs are involved in regulating helicase movement into the replication fork. Proc. Natl. Acad. Sci. U.S.A. 2012;109:14428–14433. doi: 10.1073/pnas.1212929109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Datta K., Johnson N.P., LiCata V.J., von Hippel P.H. Local conformations and competitive binding affinities of single- and double-stranded primer-template DNA at the polymerization and editing active sites of DNA polymerases. J. Biol. Chem. 2009;284:17180–17193. doi: 10.1074/jbc.M109.007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Datta K., Johnson N.P., von Hippel P.H. DNA conformational changes at the primer-template junction regulate the fidelity of replication by DNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 2010;107:17980–17985. doi: 10.1073/pnas.1012277107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Datta K., von Hippel P.H. Direct spectroscopic study of reconstituted transcription complexes reveals that intrinsic termination is driven primarily by thermodynamic destabilization of the nucleic acid framework. J. Biol. Chem. 2008;283:3537–3549. doi: 10.1074/jbc.M707998200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newport J.W., Lonberg N., Kowalczykowski S.C., von Hippel P.H. Interactions of bacteriophage T4-coded gene 32 protein with nucleic acids. II. Specificity of binding to DNA and RNA. J. Mol. Biol. 1981;145:105–121. doi: 10.1016/0022-2836(81)90336-3. [DOI] [PubMed] [Google Scholar]
- 16.Lonberg N., Kowalczykowski S.C., Paul L.S., von Hippel P.H. Interactions of bacteriophage T4-coded gene 32 protein with nucleic acids. III. Binding properties of two specific proteolytic digestion products of the protein (G32P*I and G32P*III) J. Mol. Biol. 1981;145:123–138. doi: 10.1016/0022-2836(81)90337-5. [DOI] [PubMed] [Google Scholar]
- 17.Lohman T.M., Kowalczykowski S.C. Kinetics and mechanism of the association of the bacteriophage T4 gene 32 (helix destabilizing) protein with single-stranded nucleic acids. Evidence for protein translocation. J. Mol. Biol. 1981;152:67–109. doi: 10.1016/0022-2836(81)90096-6. [DOI] [PubMed] [Google Scholar]
- 18.Epstein I.R. Cooperative and non-cooperative binding of large ligands to a finite one-dimensional lattice. A model for ligand-oligonucleotide interactions. Biophys. Chem. 1978;8:327–339. doi: 10.1016/0301-4622(78)80015-5. [DOI] [PubMed] [Google Scholar]
- 19.Hawkins M.E., Pfleiderer W., Balis F.M., Porter D., Knutson J.R. Fluorescence properties of pteridine nucleoside analogs as monomers and incorporated into oligonucleotides. Anal. Biochem. 1997;244:86–95. doi: 10.1006/abio.1996.9879. [DOI] [PubMed] [Google Scholar]
- 20.Anderson R.A., Coleman J.E. Physiochemical properties of DNA binding proteins: gene 32 protein of T4 and Escherichia coli unwinding protein. Biochemistry. 1975;14:5485–5491. doi: 10.1021/bi00696a017. [DOI] [PubMed] [Google Scholar]
- 21.McGhee J.D. Theoretical calculations of the helix-coil transition of DNA in the presence of large, cooperatively binding ligands. Biopolymers. 1976;15:1345–1375. doi: 10.1002/bip.1976.360150710. [DOI] [PubMed] [Google Scholar]
- 22.McGhee J.D., von Hippel P.H. Theoretical aspects of DNA-protein interactions: co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974;86:469–489. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- 23.Gold L., O'Farrell P.Z., Russel M. Regulation of gene 32 expression during bacteriophage T4 infection of Escherichia coli. J. Biol. Chem. 1976;251:7251–7262. [PubMed] [Google Scholar]
- 24.Rouzina I., Pant K., Karpel R.L., Williams M.C. Theory of electrostatically regulated binding of T4 gene 32 protein to single- and double-stranded DNA. Biophys. J. 2005;89:1941–1956. doi: 10.1529/biophysj.105.063776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alberts B.M. Prokaryotic DNA replication mechanisms. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1987;317:395–420. doi: 10.1098/rstb.1987.0068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.