


Abstract
To gain insight into the pathogenesis of adrenocortical carcinoma (ACC) and whether there is progression from normal-to-adenoma-to-carcinoma, we performed genome-wide gene expression, gene methylation, microRNA expression and comparative genomic hybridization (CGH) analysis in human adrenocortical tissue (normal, adrenocortical adenomas and ACC) samples. A pairwise comparison of normal, adrenocortical adenomas and ACC gene expression profiles with more than four-fold expression differences and an adjusted P-value < 0.05 revealed no major differences in normal versus adrenocortical adenoma whereas there are 808 and 1085, respectively, dysregulated genes between ACC versus adrenocortical adenoma and ACC versus normal. The majority of the dysregulated genes in ACC were downregulated. By integrating the CGH, gene methylation and expression profiles of potential miRNAs with the gene expression of dysregulated genes, we found that there are higher alterations in ACC versus normal compared to ACC versus adrenocortical adenoma. Importantly, we identified several novel molecular pathways that are associated with dysregulated genes and further experimentally validated that oncostatin m signaling induces caspase 3 dependent apoptosis and suppresses cell proliferation. Finally, we propose that there is higher number of genomic changes from normal-to-adenoma-to-carcinoma and identified oncostatin m signaling as a plausible druggable pathway for therapeutics.
INTRODUCTION
Adrenocortical carcinoma (ACC) is a rare endocrine malignancy with an annual incidence of ∼0.7–2 cases per million and a 5-year survival rate lower than 35% in patients with locally advanced and metastatic disease (1–3). Currently the adrenolytic mitotane and/or systemic chemotherapy with etoposide, doxorubicin and cisplatin for locally advanced and metastatic ACC result in very limited therapeutic benefit (4,5). For this reason, elucidation of important molecular events that are involved in ACC initiation and progression could help in identifying druggable targets for therapy. Several genome-wide gene expression profiling studies of ACC have been reported and are useful for the molecular classification of ACC into three groups (IGF, TP53 and WNT/β catenin), which correlate with patient survival (6–10). In addition, alterations in gene copy number, DNA methylation and microRNA (miRNA) expression have been observed in ACC (11–16). Several genes with altered DNA methylation status and a CpG hypermethylated phenotype in ACC have been reported (13,14). MicroRNA studies in ACC have identified consistently dysregulated miRNAs, such as miR-483–3p and miR-210, and an association with disease aggressiveness and poor prognosis (17,18). Germline mutations in TP53 occur in pediatric and adult patients with ACC, in addition to somatic TP53 inactivating mutations (19–23). Molecular pathways such as IGF signaling have been found to be dysregulated in ACC (16,24). Alterations in the WNT signaling pathway are frequently found in ACC, with abnormal accumulation and somatic activating mutations in the CTNNB1 gene (15,25). A recent study proposed novel candidate driver genes, such as ZNRF3, DAXX, TERT and MED12, using integrated genomic analysis with SNP array analysis and whole-exome sequencing (15). While the studies outlined above have offered important insight into the molecular basis of ACC, we still have very little knowledge about druggable targets/pathways that are dysregulated in ACC.
Although some investigators have suggested that there could be a multistep progression in ACC, from normal to adenoma to carcinoma, a genome-wide analysis of these tissues has not been performed to characterize the molecular changes. This is especially important, given that combined genetic alterations in IGF and WNT signaling in mouse models lead to tumorigenesis but are mutually exclusive in human ACC, thus mouse models do not completely recapitulate a multistep process in ACC occurrence (16).
Three major mechanisms of gene expression regulation in cancer are copy number changes, differential microRNA expression and gene CpG methylation status. Alterations in any of these mechanisms can lead to dysregulated gene expression and pathways that lead to cancer initiation and progression. While the primary genetic events, such as inactivating mutations in TP53 and increased signaling in the IGF and WNT pathways, have been characterized in ACC and an integrated genome atlas of ACC has been developed, a comprehensive and integrated analysis of the modes of gene expression regulation of dysregulated genes in normal, adrenocortical adenoma and ACC has not been performed. Such an investigation can shed light on the hypothesis that ACC is a multistep cancer and that gene expression changes are cumulative and can be driven by copy number changes, differential miRNA expression and gene CpG methylation, alone or in combination. In this study, we performed an integrated analysis of differentially expressed genes in normal, adrenocortical adenoma and ACC tissue with miRNA and methylation profiling and comparative genomic hybridization (CGH) in a reference and validation set of 124 human adrenocortical samples.
MATERIALS AND METHODS
Tumor samples
Human adrenocortical tissue samples were collected according to an institutional review board–approved clinical protocol after written informed consent was obtained (NCT01005654 and NCT01348698). We included 20 human ACCs, 75 benign tumor samples and 21 normal adrenal cortex samples in two independent cohorts in this study. All diagnoses were confirmed by an endocrine pathologist. ACC was diagnosed based on the Weiss criteria and tumor samples were confirmed to contain ≥80% tumor cells/nuclei. Tumor samples were obtained at surgical resection while normal adrenal glands were collected at the time of nephrectomy for organ donation and immediately snap frozen and stored at −80°C.
Genome-wide gene expression analysis
Frozen tissues were serially sectioned, and total RNA was extracted using TRIzol reagent (Invitrogen) and purified using an RNeasy Mini Kit (Qiagen). One microgram of total RNA was used for amplification and labeling with the MessageAmp aRNA kit (Ambion, Inc.). Fragmented and labeled RNA (12 μg) was hybridized to a gene chip (Affymetrix Human Genome U133 plus 2.0 GeneChip) for 16 h at 45°C. The gene chip arrays were stained and washed (Affymetrix Fluidics Station 400) according to the manufacturer's protocol. The probe intensities were measured using an argon laser confocal scanner (GeneArray scanner; Hewlitt-Packard). The array signal intensities were further analyzed by the Agilent Genespring GX software (version 12.1; Agilent Technologies). Significant differentially expressed genes in each group of comparisons were identified using an adjusted Student's t-test (P < 0.05) corrected for multiple comparisons with the false discovery rate (FDR). The results were used to run principal component analysis to reveal the clusters across all the samples. The gene list was further narrowed by applying the fold-change filtering criteria. Genes that exhibited at least a fourfold difference were selected for further downstream analysis.
DNA methylation profiling
Serial sections were made from the same tissues and used for DNA methylation profiling as explained in our earlier studies (26). After data acquisition and normalization, the M-values were used for the statistical testing (ANOVA); a Benjamini–Hochberg-adjusted P-value < 0.05 for multiple comparisons and an absolute β-value difference greater than 0.2 were applied for all pairwise comparisons.
Comparative genomic hybridization array analysis
For the SNP-CGH array experiment, Illumina HumanCytoSNP-12 v2.1 BeadChip was used. Data generated were then analyzed by using the ASCAT method described by Van Loo et al. (27). We manually scanned for the copy number status of differentially expressed genes using Nexus software and considered only the genes that underwent copy number aberrations in at least 50% of the patients. For correlation analysis, we used the log2-centered ratio of copy number changes in each pair of comparisons.
miRNA expression array analysis
From the above-mentioned cohort, we selected 26 benign adrenocortical tumors, 10 ACCs and a pool of 21 normal adrenal cortices, subjected them to miRNA expression array using a Exiqon miRCURY LNA miRNA array (v. 11.0) kit and performed the array as described earlier (18). After normalization, the summarized log2 ratios were used in moderated t-statistics and P-value calculation using the limma package in R/Bioconductor, with adjustment for FDR using the Benjamini–Hochberg method. To increase the confidence level of prediction algorithms, we manually subjected candidate genes to GOmir (http://bioacademy.gr/bioinformatics/projects/GOmir/) and considered only the miRNAs that were predicted by more than five databases. The expression status of such miRNAs was examined in three pairs of comparisons.
Molecular pathway analysis
Functional pathway analysis was performed using Ingenuity Pathway Analysis (IPA). For all differentially expressed genes, GO terms derived from the ‘Biological Process’ category were obtained using IPA, and these terms were organized manually into functional categories. For each category, the corresponding genes were uploaded to the STRING database to get insight into the molecular network (28).
Cell culture and reagents
The NCI-H295R adrenocortical cell line (ATCC) were grown and maintained in Dulbecco's modified Eagle's medium media supplemented with 1% insulin transferrin selenium (BD Biosciences) and 2.5% Nu-Serum I (BD Biosciences) in a standard humidified incubator at 37 °C in a 5% CO2 atmosphere. The BD140A ACC cell line was kindly provided by Drs Kimberly Bussey and Michael Demeure (TGen, Pheonix, AZ, USA), and it was cultured in RPMI media supplemented with 10% FBS (Invitrogen), 1% penicillin-streptomycin and 1% L-glutamate.
Caspase-Glo® 3/7 apoptosis assay
To investigate the antiproliferative effect of oncostatin M, caspase 3/7 activity was measured as an early indicator of apoptosis, using a Caspase-Glo® 3/7 apoptosis assay kit (Promega). 2 × 104 cells were seeded on 96-well plates and incubated for 18 h at 37°C. The cells were then serum starved for 18–24 h and then stimulated with different concentrations of oncostatin M for 24 h at 37°C. The caspase assay was then performed according to the manufacturer's instructions.
Western blot analysis
Total protein lysates were subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes and immunostained with the following antibodies overnight at 4°C: anti-p-Stat1 (1:1000, Cell Signaling Technology); anti-Stat1 (1:1000, Cell Signaling Technology), anti-Caspase 3 (1:1000, Cell Signaling Technology); anti-PARP (1:5000, Santa Cruz Biotechnology). Anti-human β-actin (1:5000, Santa Cruz Biotechnology) was used as a loading control. Membranes were incubated with appropriate secondary horseradish peroxidase-conjugated IgG (anti-rabbit 1:3000, Cell Signaling Technology, or anti-mouse 1:10,000, Santa Cruz Biotechnology) and proteins were detected using enhanced chemiluminescence (ECL, Pierce Biotechnology).
Statistical analysis
Most of the statistical analysis was performed using Partek Genomic Suite software unless otherwise specifically mentioned. A paired t-test was used to compare the significance between each comparison. A Pearson correlation coefficient was used to assess the relationship between gene expression and methylation/copy number/miRNA profile on representative genes.
RESULTS
Genome-wide expression profiling of normal adrenal cortex, adrenocortical adenoma and adrenocortical carcinoma (ACC) tumors shows cumulative differences in ACC
Genome-wide gene expression studies were conducted in two independent cohorts to identify differentially expressed genes. The initial reference cohort comprised 2 normal adrenal cortex samples, 75 benign adrenal tumor samples and 10 ACC tumor samples. The genome-wide gene expression analysis was further validated in a cohort that included 17 normal adrenal cortex samples, 10 benign adrenal tumor samples and 10 ACC tumors samples. We performed three pairwise comparisons: adrenocortical adenoma versus normal adrenal cortex; ACC versus adrenocortical adenoma; and ACC versus normal adrenal cortex. We selected genes that were significantly differentially expressed (adjusted P < 0.05) and that demonstrated a more-than-four-fold expression difference in each comparison (Figure 1A). There were 23 dysregulated genes between normal adrenal cortex and adrenocortical adenomas samples. In contrast, there were 808 dysregulated genes between ACC and adrenocortical adenoma samples, and 1085 dysregulated genes between ACC and normal adrenal cortex (Figure 1A). There was overlap in about 36% (683/1893) of the differentially expressed genes (Figure 1A). The majority of dysregulated genes were downregulated (Supplementary Figure S1B and C). Next, we analyzed genome-wide differential methylation in each pairwise comparison and observed very few differences between normal adrenal cortex and adrenocortical adenomas (Figure 1B). In contrast, there were 156 differentially methylated genes in ACC versus adrenocortical adenoma. A similar analysis to examine the copy number alterations showed that 5276 chromosomal loci had significant loss/gain in adrenocortical adenomas, as compared with normal adrenal cortex, whereas 35 747 chromosomal loci had copy number loss/gain in ACC, as compared with adrenocortical adenomas. Interestingly, there was an overlap of 2439 regions in these two comparisons. Our pairwise genome-wide comparisons between normal, adrenocortical adenoma and ACC suggest that ACC is associated with cumulative methylation changes and copy number alterations.
Figure 1.
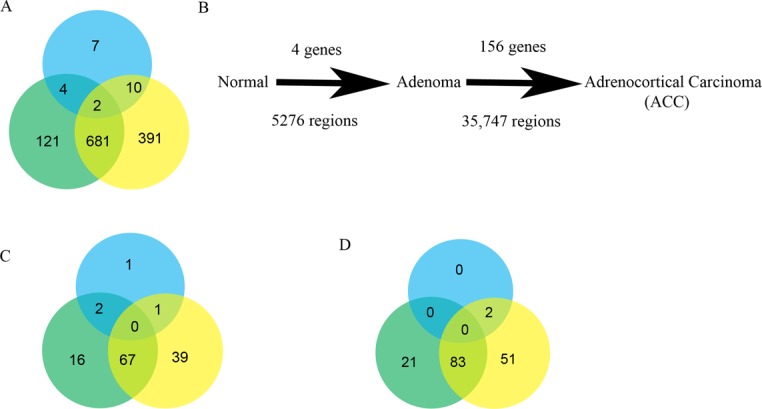
Genome-wide gene expression and regulation in adrenocortical tissue samples. (A) Venn diagram of total number of differentially expressed genes with an adjusted P-value < 0.05 and a four-fold difference in the pairwise comparison of adrenocortical adenoma versus normal adrenal cortex (blue), ACC versus adrenocortical adenoma (green) and ACC versus normal adrenal cortex (yellow). (B) Schematic representation of the pairwise comparison, from normal adrenal cortex to adrenocortical adenoma to ACC, displaying significantly altered global changes in methylation (top) and copy number aberrations (bottom). Venn diagrams represent the total number of dysregulated genes that are involved in cell proliferation (C) and cell invasion/migration (D) in each comparison.
To gain a better insight into the molecular functions of dysregulated genes in ACC, we constructed a functional network for key cancer associated processes, such as cellular proliferation (Supplementary Figure S1C) and cell migration and invasion (Supplementary Figure S1D), in each comparison of the dysregulated genes. Interestingly, not many genes were involved in these functional processes in the adrenocortical adenoma versus normal adrenal cortex comparison, as compared with ACC versus adrenocortical adenoma and ACC versus normal adrenal cortex comparisons (Figure 1C and D). Fifty-three percent (67/125) and 52.8% (83/157) genes are found in common for cell proliferation and cell migration/invasion between ACC versus adrenocortical adenoma and ACC versus normal adrenal cortex, respectively (Figure 1C and D).
To determine whether the functional status of the tumors could impact the gene expression profiles, we subclassified our cohort depending upon the functional status of the tumors and did a pairwise comparison of ACC-adenoma, ACC-Conn's adenoma and ACC-Cushing's adenoma. On the Principal Component Analysis plot, there were separate normal and ACC clusters with clustering of non-functioning, Conn's and Cushing's adenomas together (Supplementary Figure S1F). Approximately 50% of the dysregulated genes were found to be common between the three groups with few genes that were unique in each group suggesting that the functional status of the tumors could be important but plays a minor role in the context of finding significantly dysregulated genes/pathways in ACC (Supplementary Figure S1G).
Epigenetic dysregulation of genes by CpG methylation in ACC
A comprehensive analysis of the methylation status of multiple CpG islands within the genomic loci of the differentially expressed genes was performed. There was significant inverse association between the gene expression status and methylation levels of 52 and 92 genes, respectively, in the ACC versus adrenocortical adenomas and ACC versus normal adrenal cortex comparisons (Supplementary Figure S2A and B). Methylated CpG sites were distributed throughout the gene loci, including upstream of transcription start site (TSS), 5′UTR, body and exonic regions (Figure 2A and B). In addition, the gene methylation differences were higher in the normal adrenal cortex versus ACC comparison and the adrenocortical adenoma versus ACC comparison (Figure 2A and B), suggesting cumulative epigenetic alterations in ACC. For example, cytochrome P450 1B1 (CYP1B1), a key enzyme in steroid metabolism, was hypermethylated and downregulated in ACC gene expression in the initial (r = −0.52, Figure 2C) and validation (r = −0.70, Supplementary Figure S2C) cohorts. To better understand whether CYP1B1 was epigenetically regulated in ACC, we treated the ACC cell line, H295R, with decitabine (a global methylation inhibitor) and analyzed the expression of CYP1B1 (Figure 2D). The gene expression of CYP1B1 was dramatically increased upon decitabine treatment, as compared with the untreated control. This finding confirms that the aberrant DNA methylation observed in this analysis likely has a functional impact on gene expression status in ∼6.4% (52/808 genes) and 8.4% (92/1085 genes), respectively, of the dysregulated genes in the ACC versus adrenocortical adenoma and the ACC versus normal adrenal cortex comparisons (Table 1).
Figure 2.
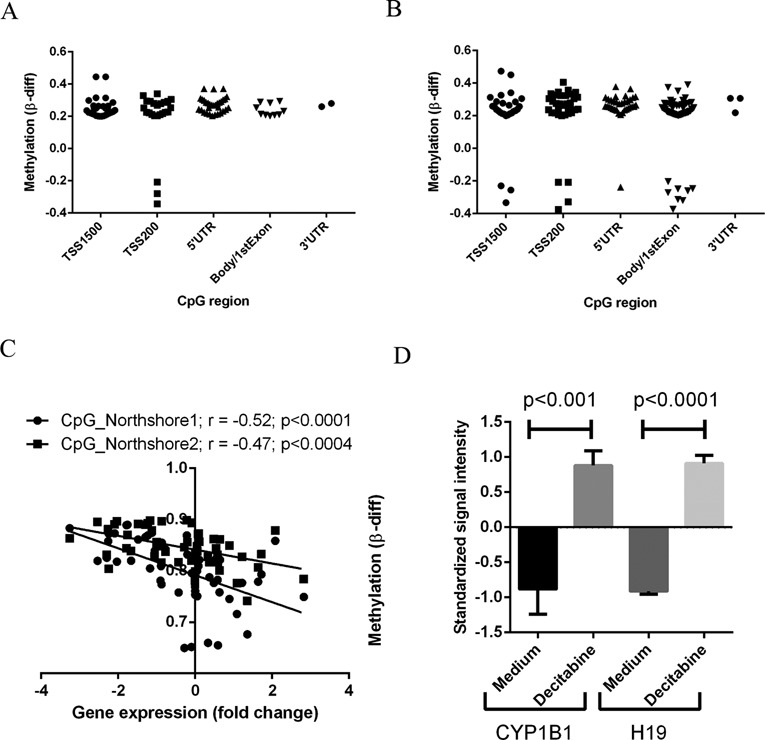
Gene-specific differential CpG methylation in ACC. Scatter plots showing the distribution of CpG sites within the dysregulated genes that are differentially methylated in comparisons of ACC versus adrenocortical adenoma (A) and ACC versus normal adrenal cortex (B) samples. (C) Correlation analysis showing methylation (β-value differences between ACC and adrenocortical adenoma) at two different CpG islands and gene expression (fold change) in CYP1B1. (D) CYP1B1 mRNA expression in ACC cells (H295R) treated with and without decitabine (10 μM) for 24 h. H19 is used as positive control as its expression is known to be regulated by CpG methylation status and we saw an increase with decitabine treatment. Y axis shows the standardized signal intensity from Affymetrix Human Genome U133 plus 2.0 GeneChip of the H295R cell line with and without decitabine treatment.
Table 1. Different modes of gene regulation in ACC (%).
| Adenoma versus normal | ACC versus adenoma | ACC versus normal | |
|---|---|---|---|
| Methylation | 0 | 6.4 | 8.4 |
| miRNA | 0 | 14.7 | 16.6 |
| Copy number | 0 | 5.9 | 9.3 |
The percentage of each mode of gene regulation mechanism in each pairwise comparison was tabulated.
Dysregulated genes and DNA copy number analysis in ACC
We next performed an analysis of genome-wide CGH array data in our cohort to assess the impact of copy number aberrations, focusing on the differentially expressed genes in ACC. Variations in gene copy number were more frequent in the ACC versus normal adrenal cortex, as compared with the ACC versus adrenocortical adenoma cases and were directly associated with the gene expression level differences (Figure 3A and B). Some of the top differentially expressed genes that had chromosomal aberrations and an association with gene expression were in several loci, including 2q22, 6p12, 8q11, 11q12–13, 12q13, 17p11 and 20q11 (Table 2). For instance, CDKN1C, which is a cyclin-dependent kinase 1 inhibitor, had frequent copy number loss and downregulation in ACC in both the reference (r = 0.47, Figure 3C) and validation (r = 0.68, Supplementary Figure S3A) cohorts. Similarly, CCAAT/enhancer binding protein δ (C/EBPδ), a tumor suppressor gene that was identified in lymphoid leukemia was also downregulated in ACC and had copy number loss (Figure 3D and Supplementary Figure S3B). Several other regions that encompass tumor suppressor genes, such as CD81, TSSC4, TSSC6 and PHLDA2, were found to have concurrent loss with CDKN1C suggesting it is an important regulator (Supplementary Figure S3C). Possible gene expression regulation through copy number alterations accounted for 5.9% (48/808 genes) and 9.3% (101/1085), respectively, of dysregulated genes in the ACC versus adrenocortical adenoma and ACC versus normal adrenal cortex comparisons (Table 1).
Figure 3.
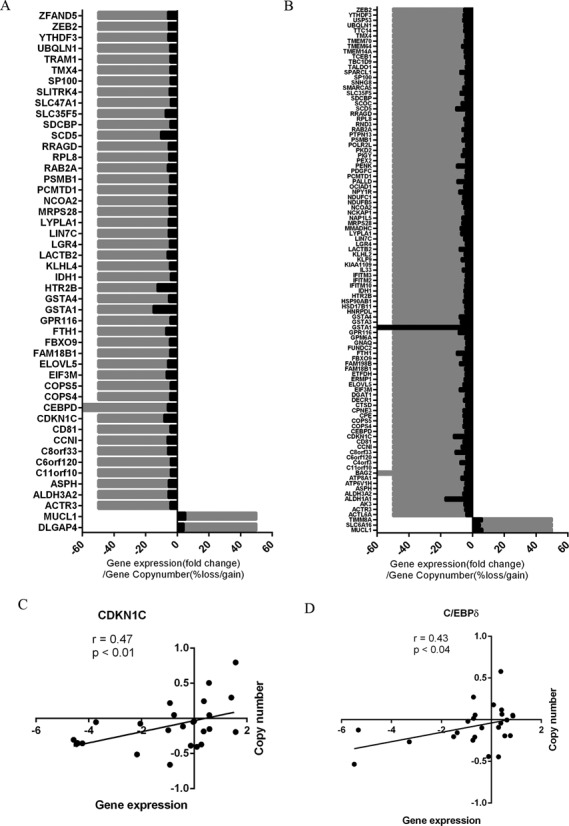
Copy number alterations associated with gene expression differences in ACC. Association of gene expression (black) and overall frequency of copy number alterations (gray) observed in ACC versus adrenocortical adenoma (A) and ACC versus normal adrenal cortex (B) samples. Correlation scatter plots of gene expression (fold change) and copy number (log2-centered ratio) of individual samples for cyclin-dependent kinase inhibitor, CDKN1C (C) and CCAAT/enhancer binding protein δ, C/EBPδ (D).
Table 2. Top listed genes that had copy number variation in ACC.
| Gene | Gene symbol | Fold change | Cytogenic location | Copy number alteration | % of frequency |
|---|---|---|---|---|---|
| ACC tumor versus benign tumor | |||||
| Mucin-Like 1 | MUCL1 | 5.1313105 | 12q13.2 | Gain | 50 |
| Discs, large (Drosophila) homolog-associated protein 4 | DLGAP4 | 4.066083 | 20q11.23 | Gain | 50 |
| ACC tumor versus normal | |||||
| Mucin-Like 1 | MUCL1 | 6.0820823 | 12q13.2 | Gain | 50 |
| Translocase of inner mitochondrial membrane 8 homolog A | TIMM8A | 6.052325 | xq22.1 | Gain | 50 |
| ACC tumor versus benign tumor | |||||
| dystonin | DST | −4.543177 | 6p12.1 | Loss | −62.5 |
| G protein-coupled receptor 116 | GPR116 | −4.2464156 | 6p12.3 | Loss | −62.5 |
| Ferritin, heavy polypeptide 1 | FTH1 | −6.976058 | 11q12.3 | Loss | −50 |
| Eukaryotic translation initiation factor 3, subunit M | EIF3M | −6.6611166 | 11p13 | Loss | −50 |
| Aldehyde dehydrogenase 3 family, member A2 | ALDH3A2 | −5.379404 | 17p11.2 | Loss | −50 |
| Solute carrier family 47 (multidrug and toxin extrusion), member 1 | SLC47A1 | −4.1039605 | 17p11.2 | Loss | −50 |
| ACC tumor versus normal | |||||
| dystonin | DST | −5.070555 | 6p12.1 | Loss | −62.5 |
| CCAAT/enhancer binding protein (C/EBP), delta | CEBPD | −6.8873086 | 8q11.21 | Loss | −50 |
| G protein-coupled receptor 116 | GPR116 | −6.809791 | 6p12.3 | Loss | −50 |
| Aldehyde dehydrogenase 3 family, member A2 | ALDH3A2 | −6.7790155 | 17p11.2 | Loss | −50 |
| Zinc finger E-Box binding homeobox 2 | ZEB2 | −6.7785573 | 2q22.3 | Loss | −50 |
| RAB2A, member RAS oncogene family | RAB2A | −6.750401 | 8q12.1 | Loss | −50 |
The cytogenic location, fold change of gene expression with the percentage of copy number alteration for the top listed genes in each comparison was tabulated.
Dysregulated genes and miRNA differential expression in ACC
To identify the differential expressed miRNAs that could potentially target the dysregulated genes in ACC, we integrated miRNA expression profiling with the gene expression in the same cohort. As with the gene methylation and copy number analyses, a reverse screening strategy was applied for identifying the putative miRNAs by restricting our analysis to differentially expressed genes in ACC. Therefore, we subjected all the dysregulated genes to multiple prediction algorithms independently to identify the miRNAs that are predicted to bind the 3′UTR region of the target gene and examined the expression status of each of those candidate miRNAs in our pairwise comparison. We observed that, similar to the methylation and copy number analysis, more genes were predicted to be targeted by differentially expressed miRNAs in ACC versus normal adrenal cortex, as compared with ACC versus adrenocortical adenomas, with an inverse association with their target gene expression (Figure 4A and B). In particular, miR-9, miR-25, miR-124, miR-183, miR-185 and miR-206 were overexpressed in ACC and were associated with downregulated gene expression of at least 10 candidate genes individually (Figure 4). For example, miR-30 family members were downregulated and predicted to regulate the expression of FAM133A, a gene yet to be functionally characterized. Interestingly, we noticed that several miRNAs were predicted to regulate multiple dysregulated genes and multiple miRNAs were predicted to target the same gene, suggesting redundant gene regulation (Figure 4). Integration of gene expression data of two representative genes, EGR1 and RhoB, with their target miRNA expression, miR-124 and miR-183, respectively, showed a significant negative Pearson correlation coefficient (Supplementary Figure S4A and B). This analysis indicates that gene regulation by dysregulated miRNAs in ACC accounts for up to 14.7% (119/808 genes) and 16.68% (181/1085 genes), respectively, of dysregulated genes in ACC versus adrenocortical adenoma and in ACC versus normal adrenal cortex. This analysis also supports that cumulative genomic events are associated with ACC.
Figure 4.
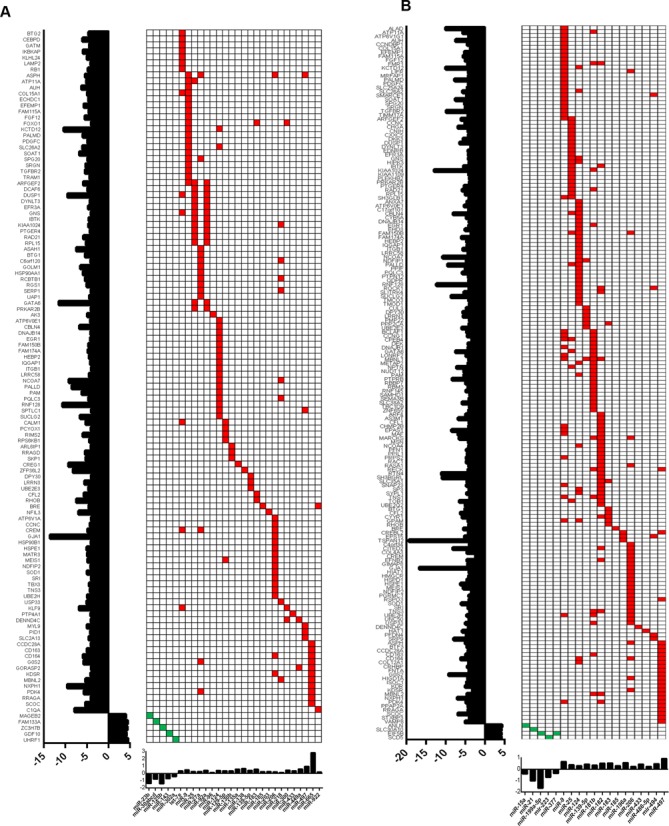
MiRNA expression profiles and gene targets in ACC. Heatmap of the miRNA expression profiles (red, upregulated miRNAs; green, downregulated miRNAs) and the predicted target genes, which are dysregulated in ACC versus adrenocortical adenoma (A) and ACC versus normal adrenal cortex (B). The expression of each target gene is represented in fold change and shown on the left side (y-axis) of the heatmap, while the fold change of each candidate miRNA expression is shown on the x-axis at the bottom of the heatmap.
Functional network analysis based on gene regulation mechanisms
In order to identify the proportion of genes that were regulated by multiple mechanisms, we performed a cumulative comparison among gene methylation, copy number and miRNA profiling. We found that many dysregulated genes in ACC were regulated by more than one gene regulation mechanism, suggesting that they are tightly regulated (Figure 5A and B). Several genes including GATA binding protein 6 (GATA6), G0/G1 switch 2 (G0S2), Meis homeobox 1 (MEIS1), Nuclear receptor coactivator 7 (NCOA7), Potassium channel tetramerization domain containing 12 (KCTD12) and Family with sequence similarity 115 member A (FAM115A) were found to be hypermethylated as well as targeted by miRNAs. Genes such as aspartate β-carboxylase (ASPH) and C/EBPδ were found to have both gene copy number changes and differential expression in the target miRNAs. Collectively, these three modes of gene regulatory mechanisms accounted for about 30% of the differentially expressed genes in ACC. To identify the molecular pathways that are significantly altered by these three modes of gene regulation, we performed IPA on the genes that were potentially regulated by these mechanisms independently and found that oncostatin M signaling, retinoic acid receptor activation (RXR) and PI3K/AKT and CDC42 signaling pathways were among the top pathways altered in ACC (Table 3). We observed that multiple genes were associated within these pathways and were regulated by more than one mechanism (Figure 5C). In particular, we found that genes that are involved in oncostatin M signaling, such as IL6ST (GP130), JAK1 and EPAS1, were downregulated and hypermethylation and/or targeted at the 3′UTR region by specifically upregulated miRNAs (Figure 5C). Further examination of the gene expression levels of the molecules that are involved in this pathway revealed that TIMP-3, MT2A and ELK-1, which are downstream of oncostatin M, were also downregulated but showed less than four-fold difference in each comparison therefore these genes were filtered out in our selection criteria (Figure 5A).
Figure 5.
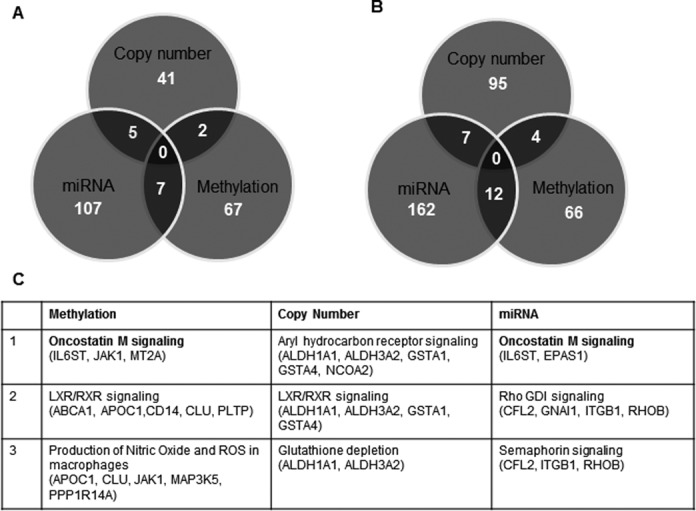
Integrated analysis of gene regulation and gene-specific CpG methylation, copy number alterations and differentially expressed miRNAs in ACC. Venn diagrams show the total number of genes that are regulated by each mechanism: gene-specific CpG methylation, copy number alterations and differentially expressed miRNAs in ACC versus adrenocortical adenoma (A) and ACC versus normal adrenal cortex (B). (C) The top molecular pathways that are altered by the three modes of gene expression regulation mechanisms.
Table 3. List of canonical pathways that are altered in ACC.
| Ingenuity Canonical Pathways | -log(P-value) | Ratio | Molecules |
|---|---|---|---|
| Methylation | |||
| LXR/RXR activation | 3.94E + 00 | 3.91E − 02 | APOC1, CD14, PLTP, CLU, ABCA1 |
| 3-phosphoinositide synthesis and degradation | 2.63E + 00 | 2.67E − 02 | MTMR6, PTPN13, PPTC7, PPP1R14A |
| Oncostatin M signaling | 2.15E + 00 | 5.88E − 02 | IL6ST, JAK1 |
| PI3K/AKT signaling | 1.92E + 00 | 2.34E − 02 | JAK1, ITGA2, MAP3K5 |
| Cdc42 signaling | 1.60E + 00 | 1.78E − 02 | ITGA2, HLA-DPB1, CLIP1 |
| ERK/MAPK signaling | 1.47E + 00 | 1.57E − 02 | ITGA2, PPP1R14A, RAPGEF4 |
| Copy number | |||
| Aryl hydrocarbon receptor signaling | 4.55E + 00 | 3.57E − 02 | NCOA2, ALDH3A2, GSTA4, RARA, GSTA1 |
| LPS/IL-1 mediated inhibition of RXR function | 2.63E + 00 | 1.83E − 02 | ALDH3A2, GSTA4, RARA, GSTA1 |
| EIF2 signaling | 1.09E + 00 | 1.08E − 02 | RPL8, EIF3M |
| Axonal guidance signaling | 1.01E + 00 | 6.94E − 03 | ACTR3, SDCBP, COPS5 |
| Glucocorticoid receptor signaling | 8.43E − 01 | 7.66E − 03 | CDKN1C, NCOA2 |
| miRNA | |||
| Protein ubiquitination pathway | 3.90E + 00 | 3.14E − 02 | HSP90B1, UBE2H, HSPE1, HSP90AA1, UBE2E3, SKP1, DNAJB14, USP33 |
| Nitric oxide signaling | 3.54E + 00 | 5.05E − 02 | CALM1 (includes others), HSP90B1, PRKAR2B, HSP90AA1, PDGFC |
| PI3K/AKT signaling | 3.11E + 00 | 4.07E − 02 | ITGB1, RPS6KB1, HSP90B1, FOXO1, HSP90AA1 |
| IL-8 signaling | 2.35E + 00 | 2.73E − 02 | MYL9, RPS6KB1, RHOB, IQGAP1, PDGFC |
| Aryl hydrocarbon receptor signaling | 2.04E + 00 | 2.86E − 02 | RB1, HSP90B1, NCOA7, HSP90AA1 |
| Cdc42 signaling | 1.78E + 00 | 2.40E − 02 | ITGB1, MYL9, CFL2, IQGAP1 |
| mTOR signaling | 1.01E + 00 | 1.60E − 02 | RPS6KB1, RHOB, PDGFC |
| Oncostatin M signaling | 1.17E + 00 | 5.70E − 02 | IL6ST, EPAS1 |
The lists of genes that are potentially regulated by methylation, copy number and miRNA and are involved in each molecular pathway are tabulated with its significance as obtained from IPA network analysis.
Oncostatin M signaling inhibits cellular proliferation and induces apoptosis in ACC cells
Since our genome-wide integrated analysis suggested that multiple molecules within the oncostatin M signaling pathway are downregulated and are associated with epigenetic and miRNA-based regulation in ACC (Figure 6A), we examined the effect of oncostatin M in two adrenocortical cell lines, NCI-H295R and BD140A. We found that oncostatin M inhibits ACC cell proliferation (Figure 6B and C) and stimulates the activity of the caspase 3/7 enzymes in a dose-dependent manner in both cell lines, suggesting it induces caspase 3 dependent apoptosis (Figure 6D and E). We then examined the status of JAK-STAT signaling, as it is the canonical pathway involved in oncostatin M mediated signaling in immune cells and observed a dose-dependent increase in phosphoStat1 levels in NCI H295R cells (Figure 6F). Also, oncostatin M promoted the cleavage of both full-length PARP and caspase 3 at higher doses, suggesting that it induces apoptosis in these cells (Figure 6F). Taken together, we were able to show that the oncostatin M pathway identified through our genome-wide integrated analysis could inhibit ACC cell growth.
Figure 6.
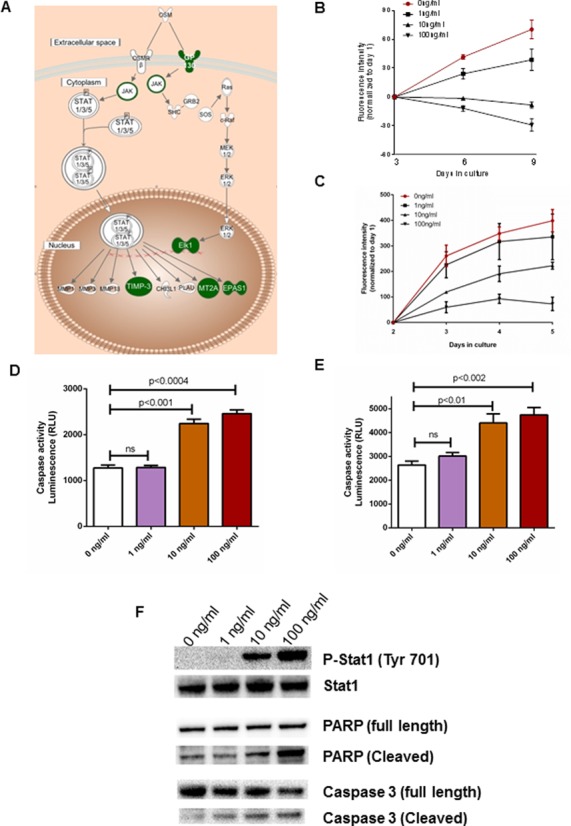
Oncostatin M induces apoptosis in ACC cells. (A) Schematic representation of a canonical oncostatin M signaling pathway. Downregulated genes from the oncostatin M signaling pathway identified by the genome-wide integrated analysis are highlighted in green. Oncostatin M inhibits cell proliferation (B and C) and apoptosis (D and E) by increased caspase 3/7 activity in a dose-dependent manner in NCI-H295R (B and D) and BD140A cells (C and E). It induces apoptosis by stimulating the JAK-STAT signaling cascade and cleavage of PARP and caspase 3 in ACC NCI-H295R cells (F).
DISCUSSION
In this study we show that an increasing number of differentially expressed genes, differentially methylated genes, copy number variations and differentially expressed miRNAs are found in normal adrenal cortex versus adrenocortical adenoma versus ACC. The underlying candidate mechanisms of gene expression regulation were analyzed by assessing gene-specific CpG methylation changes, copy number alterations and microRNA expression in two independent cohorts. Importantly, our integrated analysis was driven by gene expression differences, which excluded changes that were not expected to impact gene expression. Our genome-wide integrated analysis data show that ACC tumors most likely result from pangenomic cumulative changes, if a stepwise progression from normal adrenal cortex to adrenocortical adenoma to ACC truly occurs and from novel pathways that are involved in ACC pathobiology.
A recent study using a four-stage cell model of isogenically matched normal, immortalized, transformed and metastatic human cells proposed that, in a sequential multistep process of cancer initiation, downregulation of many genes is a common event (29). Our genome-wide pairwise analysis of ACC, adrenocortical adenoma and normal adrenal cortex showed that the majority of the differentially expressed genes are downregulated. These data, therefore, suggest that ACC may develop in a multistep process. This hypothesis is supported by a case study in which an adrenocortical tumor was found to consist of two parts, with a central malignant and a distinct peripheral benign component, suggesting a multistep process (30).
There is growing evidence that hypermethylation of CpG islands located in tumor suppressor genes is one of the most common mechanisms for gene silencing in cancer initiation and progression (31,32). Several studies have shown that the downregulation of cytochrome P450 members is associated with the risk of cancers such as colorectal, breast and renal cancer. One study, in a Cyp1b1-deficient mouse model, showed that CYP1B1, a member of the cytochrome P450 family, exerts an opposing effect on intestinal tumorigenesis (33). However, the functional characterization of CYP1B1 in ACC is unclear, other than its role in steroidogenesis.
As with colorectal cancer, which is thought to follow a multistep model of cancer initiation and is known to be associated with aneuploidy and extensive genomic aberrations (34), progression to ACC seems to undergo a vast amount of allelic imbalance associated with altered gene expression. A well-known cell cycle inhibitory regulator, CDKN1C and tumor suppressor transcription factor such as C/EBPδ that play critical role in several other cancers were found to be dysregulated in ACC (35–38). Our integrated analysis sheds light on novel candidate tumor suppressor genes that are implicated in other cancer, such as CD81, TSSC4, TSSC6 and PHLDA2 which are lost in ACC due to copy number variations (39,40). Since the tumor suppressor gene TP53 has been shown to be a candidate driver gene for malignancy, we performed target DNA sequencing for damaging mutations within the open reading frame of TP53 in adrenocortical adenoma and ACC tumors. Consistent with earlier studies, we found loss-of-function mutations in exon 5 (G12219A) and exon 6 (A12749G) in 9 out of 10 tested ACC tumors (15). Since we observed a significant number of copy number aberrations between normal adrenal cortex and benign adrenocortical tumor samples, we believe that these changes with further accumulation of events may lead to malignancy.
Several studies have suggested that miRNAs play a key role in the development of cancer (41,42). Our analysis provides an unbiased approach for identifying critical miRNAs in ACC on a genome-wide scale, as we focused primarily on the miRNAs that are inversely associated with differentially expressed genes. Several miRNAs, such as miR-183, miR-9 and miR-25, which are known to promote cell proliferation and invasion in other cancers, were found to be upregulated in ACC (15,43,44). In addition, we identified miR-665, a novel miRNA that has not yet been fully characterized and may play a role in the development of ACC. Given the pleiotropic nature of miRNAs, it was not surprising to find that oncogenic miRNAs, such as miR-138, and tumor suppressor miRNAs, such as miR-126 and miR-497, were downregulated and upregulated, respectively, in ACC and targeted many differentially expressed genes.
In the current study, we identified new signaling pathways, such as oncostatin M, CDC42 and EIF signaling, which can play critical roles in ACC. Moreover, enzymes that are involved in ubiquitination were dysregulated and targeted by differentially expressed miRNAs in ACC. This is an interesting finding that may have therapeutic relevance, as targeting protein ubiquitination is an emerging therapeutic strategy for cancer therapy (45). Oncostatin M signaling has been shown to play a dual role in cancer progression. For instance, it can stimulate an antiproliferative effect in breast cancer cells and is also known to induce differentiation of breast cancer cells (46,47). In contrast, it could promote cell proliferation in ovarian, prostate and liver cancers (48,49). However, in the context of ACC, it appears to inhibit cell proliferation and induce apoptosis in a caspase-dependent manner, suggesting that this pathway could be a potential therapeutic target for ACC.
Despite several novel findings, our study has some limitations. Firstly, our integrated analysis is primarily driven by gene expression difference, as only the significantly differentially expressed genes were considered for examining the three modes of gene expression regulation. This approach was used, however, because we were interested in identifying primary genetic/genomic changes that may account for the differentially expressed genes, with a focus on the three modes of gene regulation that have been shown to be involved in carcinogenesis. Secondly, the overall percentages for each mode of gene expression regulation reported here apply to only genes markedly dysregulated, exhibiting at least four-fold expression differences. While this study was done to focus on highly relevant candidate genes, it may have excluded relevant genes in ACC. In addition, if there are any driver mutations present in small subset of tumors that may have unique gene expression signature, our stringent criteria may have excluded them. It is likely that the functioning tumor may have different pathogenesis compared to non-functioning tumors. However, the PCA plot based on the functional status of adenomas separated normal and ACC with clustering of both functioning and non-functioning adenomas together (Supplementary Figure S1F). Although we observed a higher number of genomic changes in the paired comparison of normal, adenoma and ACC, it only suggests that ACC may be a stepwise process. We cannot exclude the possibility that the pathogenesis of adenomas is different from ACC and therefore does not arise from an adenoma. Recent studies by Assie et al. determined that ZNRF3, CTNNB1, CDKN2A and RB1 were frequently altered with either damaging mutations or homozygous deletions (15). Corroborative to these studies, we observed copy number loss of these genes; however, these genes had a less than four-fold expression difference and therefore were not included in our analysis, as we did not perform whole-exome sequencing of the tissue samples to study mutations in candidate genes other than TP53.
In conclusion, our integrated analysis of gene expression and regulation suggests that methylation, miRNAs and copy number aberrations are increased in our pairwise comparison of normal adrenal cortex, adrenocortical adenoma and ACC. This study provides new insights into dysregulated pathways that may lead to ACC initiation and or progression.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural research program of the Center for Cancer Research; National Cancer Institute; National Institutes of Health [1 ZIA BC011275 05]. Funding for open access charge: Intramural research program of the Center for Cancer Research; National Cancer Institute; National Institutes of Health [1 ZIA BC011275 05].
Conflict of interest statement. None declared.
REFERENCES
- 1.Kebebew E., Reiff E., Duh Q.Y., Clark O.H., McMillan A. Extent of disease at presentation and outcome for adrenocortical carcinoma: Have we made progress. World J. Surg. 2006;30:872–878. doi: 10.1007/s00268-005-0329-x. [DOI] [PubMed] [Google Scholar]
- 2.Luton J.P., Cerdas S., Billaud L., Thomas G., Guilhaume B., Bertagna X., Laudat M.H., Louvel A., Chapuis Y., Blondeau P., et al. Clinical-features of adrenocortical carcinoma, prognostic factors, and the effect of mitotane therapy. New Engl. J. Med. 1990;322:1195–1201. doi: 10.1056/NEJM199004263221705. [DOI] [PubMed] [Google Scholar]
- 3.Allolio B., Fassnacht M. Clinical review: Adrenocortical carcinoma: Clinical update. J. Clin. Endocr. Metab. 2006;91:2027–2037. doi: 10.1210/jc.2005-2639. [DOI] [PubMed] [Google Scholar]
- 4.Schteingart D.E., Doherty G.M., Gauger P.G., Giordano T.J., Hammer G.D., Korobkin M., Worden F.P. Management of patients with adrenal cancer: recommendations of an international consensus conference. Endocr. Relat. Cancer. 2005;12:667–680. doi: 10.1677/erc.1.01029. [DOI] [PubMed] [Google Scholar]
- 5.Fassnacht M., Johanssen S., Quinkler M., Bucsky P., Willenberg H.S., Beuschlein F., Terzolo M., Mueller H.H., Hahner S., Allolio B., et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a Revised TNM Classification. Cancer. 2009;115:243–250. doi: 10.1002/cncr.24030. [DOI] [PubMed] [Google Scholar]
- 6.Suh I., Guerrero M.A., Kebebew E. Gene-expression profiling of adrenocortical carcinoma. Expert Rev. Mol. Diagn. 2009;9:343–351. doi: 10.1586/erm.09.13. [DOI] [PubMed] [Google Scholar]
- 7.de Reynies A., Assie G., Rickman D.S., Tissier F., Groussin L., Rene-Corail F., Dousset B., Bertagna X., Clauser E., Bertherat J. Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J. Clin. Oncol. 2009;27:1108–1115. doi: 10.1200/JCO.2008.18.5678. [DOI] [PubMed] [Google Scholar]
- 8.Giordano T.J., Kuick R., Else T., Gauger P.G., Vinco M., Bauersfeld J., Sanders D., Thomas D.G., Doherty G., Hammer G. Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin. Cancer Res. 2009;15:668–676. doi: 10.1158/1078-0432.CCR-08-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laurell C., Velazquez-Fernandez D., Lindsten K., Juhlin C., Enberg U., Geli J., Hoog A., Kjellman M., Lundeberg J., Hamberger B., et al. Transcriptional profiling enables molecular classification of adrenocortical tumours. Eur. J. Endocrinol. 2009;161:141–152. doi: 10.1530/EJE-09-0068. [DOI] [PubMed] [Google Scholar]
- 10.Lombardi C.P., Raffaelli M., Pani G., Maffione A., Princi P., Traini E., Galeotti T., Rossi E.D., Fadda G., Bellantone R. Gene expression profiling of adrenal cortical tumors by cDNA macroarray analysis. Results of a preliminary study. Biomed. Pharmacother. 2006;60:186–190. doi: 10.1016/j.biopha.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 11.Barreau O., de Reynies A., Wilmot-Roussel H., Guillaud-Bataille M., Auzan C., Rene-Corail F., Tissier F., Dousset B., Bertagna X., Bertherat J., et al. Clinical and pathophysiological implications of chromosomal alterations in adrenocortical tumors: an integrated genomic approach. J. Clin. Endocrinol. Metab. 2012;97:E301–E311. doi: 10.1210/jc.2011-1588. [DOI] [PubMed] [Google Scholar]
- 12.Stephan E.A., Chung T.H., Grant C.S., Kim S., Von Hoff D.D., Trent J.M., Demeure M.J. Adrenocortical carcinoma survival rates correlated to genomic copy number variants. Mol. Cancer Ther. 2008;7:425–431. doi: 10.1158/1535-7163.MCT-07-0267. [DOI] [PubMed] [Google Scholar]
- 13.Rechache N.S., Wang Y., Stevenson H.S., Killian J.K., Edelman D.C., Merino M., Zhang L., Nilubol N., Stratakis C.A., Meltzer P.S., et al. DNA methylation profiling identifies global methylation differences and markers of adrenocortical tumors. J. Clin. Endocrinol. Metab. 2012;97:E1004–E1013. doi: 10.1210/jc.2011-3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fonseca A.L., Kugelberg J., Starker L.F., Scholl U., Choi M., Hellman P., Akerstrom G., Westin G., Lifton R.P., Bjorklund P., et al. Comprehensive DNA methylation analysis of benign and malignant adrenocortical tumors. Genes Chromosomes Cancer. 2012;51:949–960. doi: 10.1002/gcc.21978. [DOI] [PubMed] [Google Scholar]
- 15.Assie G., Letouze E., Fassnacht M., Jouinot A., Luscap W., Barreau O., Omeiri H., Rodriguez S., Perlemoine K., Rene-Corail F., et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014;46:607–612. doi: 10.1038/ng.2953. [DOI] [PubMed] [Google Scholar]
- 16.Heaton J.H., Wood M.A., Kim A.C., Lima L.O., Barlaskar F.M., Almeida M.Q., Fragoso M.C., Kuick R., Lerario A.M., Simon D.P., et al. Progression to adrenocortical tumorigenesis in mice and humans through insulin-like growth factor 2 and beta-catenin. Am. J Pathol. 2012;181:1017–1033. doi: 10.1016/j.ajpath.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duregon E., Rapa I., Votta A., Giorcelli J., Daffara F., Terzolo M., Scagliotti G.V., Volante M., Papotti M. MicroRNA expression patterns in adrenocortical carcinoma variants and clinical pathologic correlations. Hum. Pathol. 2014;45:1555–1562. doi: 10.1016/j.humpath.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 18.Patterson E.E., Holloway A.K., Weng J., Fojo T., Kebebew E. MicroRNA profiling of adrenocortical tumors reveals miR-483 as a marker of malignancy. Cancer. 2011;117:1630–1639. doi: 10.1002/cncr.25724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohgaki H., Kleihues P., Heitz P.U. p53 mutations in sporadic adrenocortical tumors. Int. J. Cancer. 1993;54:408–410. doi: 10.1002/ijc.2910540310. [DOI] [PubMed] [Google Scholar]
- 20.Reincke M., Karl M., Travis W.H., Mastorakos G., Allolio B., Linehan H.M., Chrousos G.P. p53 mutations in human adrenocortical neoplasms: immunohistochemical and molecular studies. J. Clin. Endocrinol. Metab. 1994;78:790–794. doi: 10.1210/jcem.78.3.8126158. [DOI] [PubMed] [Google Scholar]
- 21.Wagner J., Portwine C., Rabin K., Leclerc J.M., Narod S.A., Malkin D. High frequency of germline p53 mutations in childhood adrenocortical cancer. J. Natl. Cancer Inst. 1994;86:1707–1710. doi: 10.1093/jnci/86.22.1707. [DOI] [PubMed] [Google Scholar]
- 22.Raymond V.M., Else T., Everett J.N., Long J.M., Gruber S.B., Hammer G.D. Prevalence of germline TP53 mutations in a prospective series of unselected patients with adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2013;98:E119–E125. doi: 10.1210/jc.2012-2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herrmann L.J., Heinze B., Fassnacht M., Willenberg H.S., Quinkler M., Reisch N., Zink M., Allolio B., Hahner S. TP53 germline mutations in adult patients with adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2012;97:E476–E485. doi: 10.1210/jc.2011-1982. [DOI] [PubMed] [Google Scholar]
- 24.Libe R., Fratticci A., Bertherat J. Adrenocortical cancer: pathophysiology and clinical management. Endocr. Relat. Cancer. 2007;14:13–28. doi: 10.1677/erc.1.01130. [DOI] [PubMed] [Google Scholar]
- 25.Tissier F., Cavard C., Groussin L., Perlemoine K., Fumey G., Hagnere A.M., Rene-Corail F., Jullian E., Gicquel C., Bertagna X., et al. Mutations of beta-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005;65:7622–7627. doi: 10.1158/0008-5472.CAN-05-0593. [DOI] [PubMed] [Google Scholar]
- 26.Howard B., Wang Y., Xekouki P., Faucz F.R., Jain M., Zhang L., Meltzer P.G., Stratakis C.A., Kebebew E. Integrated analysis of genome-wide methylation and gene expression shows epigenetic regulation of CYP11B2 in aldosteronomas. J. Clin. Endocrinol. Metab. 2014;99:E536–E543. doi: 10.1210/jc.2013-3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Loo P., Nordgard S.H., Lingjaerde O.C., Russnes H.G., Rye I.H., Sun W., Weigman V.J., Marynen P., Zetterberg A., Naume B., et al. Allele-specific copy number analysis of tumors. Proc. Natl. Acad. Sci. U.S.A. 2010;107:16910–16915. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C., et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Danielsson F., Skogs M., Huss M., Rexhepaj E., O'Hurley G., Klevebring D., Ponten F., Gad A.K., Uhlen M., Lundberg E. Majority of differentially expressed genes are down-regulated during malignant transformation in a four-stage model. Proc. Natl. Acad. Sci. U.S.A. 2013;110:6853–6858. doi: 10.1073/pnas.1216436110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernard M.H., Sidhu S., Berger N., Peix J.L., Marsh D.J., Robinson B.G., Gaston V., Le Bouc Y., Gicquel C. A case report in favor of a multistep adrenocortical tumorigenesis. J. Clin. Endocrinol. Metabo. 2003;88:998–1001. doi: 10.1210/jc.2002-021117. [DOI] [PubMed] [Google Scholar]
- 31.Sato N., Parker A.R., Fukushima N., Miyagi Y., Iacobuzio-Donahue C.A., Eshleman J.R., Goggins M. Epigenetic inactivation of TFPI-2 as a common mechanism associated with growth and invasion of pancreatic ductal adenocarcinoma. Oncogene. 2005;24:850–858. doi: 10.1038/sj.onc.1208050. [DOI] [PubMed] [Google Scholar]
- 32.Esteller M. Epigenetics in cancer. New Engl. J. Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 33.Halberg R.B., Larsen M.C., Elmergreen T.L., Ko A.Y., Irving A.A., Clipson L., Jefcoate C.R. Cyp1b1 exerts opposing effects on intestinal tumorigenesis via exogenous and endogenous substrates. Cancer Res. 2008;68:7394–7402. doi: 10.1158/0008-5472.CAN-07-6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yim K.L. Microsatellite instability in metastatic colorectal cancer: a review of pathology, response to chemotherapy and clinical outcome. Med. Oncol. 2012;29:1796–1801. doi: 10.1007/s12032-011-0050-6. [DOI] [PubMed] [Google Scholar]
- 35.Larson P.S., Schlechter B.L., King C.L., Yang Q., Glass C.N., Mack C., Pistey R., de Las Morenas A., Rosenberg C.L. CDKN1C/p57kip2 is a candidate tumor suppressor gene in human breast cancer. BMC Cancer. 2008;8:68. doi: 10.1186/1471-2407-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Algar E.M., Muscat A., Dagar V., Rickert C., Chow C.W., Biegel J.A., Ekert P.G., Saffery R., Craig J., Johnstone R.W., et al. Imprinted CDKN1C is a tumor suppressor in rhabdoid tumor and activated by restoration of SMARCB1 and histone deacetylase inhibitors. PLoS One. 2009;4:e4482. doi: 10.1371/journal.pone.0004482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agrawal S., Hofmann W.K., Tidow N., Ehrich M., van den Boom D., Koschmieder S., Berdel W.E., Serve H., Muller-Tidow C. The C/EBPdelta tumor suppressor is silenced by hypermethylation in acute myeloid leukemia. Blood. 2007;109:3895–3905. doi: 10.1182/blood-2006-08-040147. [DOI] [PubMed] [Google Scholar]
- 38.Balamurugan K., Wang J.M., Tsai H.H., Sharan S., Anver M., Leighty R., Sterneck E. The tumour suppressor C/EBPdelta inhibits FBXW7 expression and promotes mammary tumour metastasis. EMBO J. 2010;29:4106–4117. doi: 10.1038/emboj.2010.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoo T.H., Ryu B.K., Lee M.G., Chi S.G. CD81 is a candidate tumor suppressor gene in human gastric cancer. Cell Oncol (Dordr) 2013;36:141–153. doi: 10.1007/s13402-012-0119-z. [DOI] [PubMed] [Google Scholar]
- 40.Lee M.P., Brandenburg S., Landes G.M., Adams M., Miller G., Feinberg A.P. Two novel genes in the center of the 11p15 imprinted domain escape genomic imprinting. Hum. Mol. Genet. 1999;8:683–690. doi: 10.1093/hmg/8.4.683. [DOI] [PubMed] [Google Scholar]
- 41.Jasinski-Bergner S., Mandelboim O., Seliger B. The role of microRNAs in the control of innate immune response in cancer. J. Natl. Cancer Inst. 2014;106 doi: 10.1093/jnci/dju257. doi:10.1093/jnci/dju257. [DOI] [PubMed] [Google Scholar]
- 42.Adams B.D., Kasinski A.L., Slack F.J. Aberrant regulation and function of microRNAs in cancer. Current Biol. 2014;24:R762–R776. doi: 10.1016/j.cub.2014.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou L., Zhang W.G., Wang D.S., Tao K.S., Song W.J., Dou K.F. MicroRNA-183 is involved in cell proliferation, survival and poor prognosis in pancreatic ductal adenocarcinoma by regulating Bmi-1. Oncol. Rep. 2014;32:1734–1740. doi: 10.3892/or.2014.3374. [DOI] [PubMed] [Google Scholar]
- 44.Li B.S., Zuo Q.F., Zhao Y.L., Xiao B., Zhuang Y., Mao X.H., Wu C., Yang S.M., Zeng H., Zou Q.M., et al. MicroRNA-25 promotes gastric cancer migration, invasion and proliferation by directly targeting transducer of ERBB2, 1 and correlates with poor survival. Oncogene. 2014:2556–2565. doi: 10.1038/onc.2014.214. [DOI] [PubMed] [Google Scholar]
- 45.Pal A., Young M.A., Donato N.J. Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer Res. 2014;74:4955–4966. doi: 10.1158/0008-5472.CAN-14-1211. [DOI] [PubMed] [Google Scholar]
- 46.Douglas A.M., Goss G.A., Sutherland R.L., Hilton D.J., Berndt M.C., Nicola N.A., Begley C.G. Expression and function of members of the cytokine receptor superfamily on breast cancer cells. Oncogene. 1997;14:661–669. doi: 10.1038/sj.onc.1200882. [DOI] [PubMed] [Google Scholar]
- 47.Douglas A.M., Grant S.L., Goss G.A., Clouston D.R., Sutherland R.L., Begley C.G. Oncostatin M induces the differentiation of breast cancer cells. Int. J. Cancer. 1998;75:64–73. doi: 10.1002/(sici)1097-0215(19980105)75:1<64::aid-ijc11>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 48.Taga T., Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 1997;15:797–819. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- 49.Hoffman R.C., Moy F.J., Price V., Richardson J., Kaubisch D., Frieden E.A., Krakover J.D., Castner B.J., King J., March C.J., et al. Resonance assignments for Oncostatin M, a 24-kDa alpha-helical protein. J. Biomol. NMR. 1996;7:273–282. doi: 10.1007/BF00200429. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.