

Abstract
Purpose
Age-related macular degeneration (AMD) has a substantial genetic risk component, as evidenced by the risk from common genetic variants uncovered in the first genome-wide association studies. More recently, it has become apparent that rare genetic variants also play an independent role in AMD risk. We sought to determine if rare variants in complement factor H (CFH) played a role in AMD risk.
Methods
We had previously collected DNA from a large population of patients with advanced age-related macular degeneration (A-AMD) and controls for targeted deep sequencing of candidate AMD risk genes. In this analysis, we tested for an increased burden of rare variants in CFH in 1665 cases and 752 controls from this cohort.
Results
We identified 65 missense, nonsense, or splice-site mutations with a minor allele frequency ≤ 1%. Rare variants with minor allele frequency ≤ 1% (odds ratio [OR] = 1.5, P = 4.4 × 10−2), 0.5% (OR = 1.6, P = 2.6 × 10−2), and all singletons (OR = 2.3, P = 3.3 × 10−2) were enriched in A-AMD cases. Moreover, we observed loss-of-function rare variants (nonsense, splice-site, and loss of a conserved cysteine) in 10 cases and serum levels of FH were decreased in all 5 with an available sample (haploinsufficiency). Further, rare variants in the major functional domains of CFH were increased in cases (OR = 3.2; P = 1.4 × 10−3) and the magnitude of the effect correlated with the disruptive nature of the variant, location in an active site, and inversely with minor allele frequency.
Conclusions
In this large A-AMD cohort, rare variants in the CFH gene were enriched and tended to be located in functional sites or led to low serum levels. These data, combined with those indicating a similar, but even more striking, increase in rare variants found in CFI, strongly implicate complement activation in A-AMD etiopathogenesis as CFH and CFI interact to inhibit the alternative pathway.
Keywords: macular degeneration, rare genetic variants, sequencing
Advanced (A-AMD) is a disease with known genetic risk factors. In this paper, we show that rare variants in CFH increase A-AMD risk.
Age-related macular degeneration (AMD) features excessive complement activation, including the release and deposition of proinflammatory fragments in the retina. Genetic evidence also directly implicates the role of complement activation in disease (reviewed in Refs. 1–9). One of the first reproducible genome-wide association study findings was the rs1061170 (Y402H) polymorphism in complement factor H (CFH).10 An intronic single-nucleotide polymorphism (SNP) in CFH, rs1410996, was also independently associated with AMD, with the minor allele conferring a similar risk of disease.11 Common SNPs near the gene for the regulator complement factor I (CFI),12 and in the genes for the complement component 3 (C3)13,14 and the complement protease factor B (CFB)11,15 were also associated with disease. Carrying risk alleles in these genes leads to a lifetime AMD risk of nearly 50%.11,14,16,17
Factor H (FH) is a key inhibitor of the alternative pathway (AP) of complement activation.18,19 It is an abundant (average level, ∼300 μg/mL) serum protein that negatively regulates the C3 convertase of the alternative pathway. To accomplish this task, FH competes with factor B (FB) in binding to C3b and decays both the proconvertase (C3bB) and the active C3 convertase (C3bBb). Further, it serves as a cofactor protein for the serine protease factor I (FI)–mediated cleavage of C3b to hemolytically inactive iC3b. The latter cannot serve as a nidus for formation of the AP's positive feedback/amplification loop, in contrast to C3b.
In the era of next-generation sequencing, AMD has provided one of the first examples of rare variants increasing the risk of a common disease. A rare, penetrant variant in CFH coding for R1210C was associated with AMD with an odds ratio (OR) of approximately 20, representing the strongest risk factor for AMD to date.20 This variant was present in only two individuals (minor allele frequency [MAF] = 0.015%) in the NHLBI 6500 exome sequencing project (6500 ESP). Additionally, a single missense variant in C3, K155Q, was found by three independent groups to be associated with AMD.21–23 Variant K155Q was present at a frequency of 0.4% in European Americans sequenced as part of the 6500 ESP. A diversity of rare and private variants in CFI was also found to be associated with AMD in a large targeted sequencing study.21
Two reports of additional rare CFH variants linked to AMD within families have been published.24,25 In one report using whole-exome sequencing, two different variants segregated with disease in two unrelated families.24 Both variants had similar detrimental effects on function. In the other report, a rare variant in the same domain as the common predisposing variant Y402H was identified in an affected Amish family.25
In this report, our goal was to assess the presence of rare variants in CFH in patients with advanced AMD (A-AMD). The prior observations of a remarkable number of rare variants in CFI in A-AMD,21 the striking association of common variants in CFH with AMD,1–5,10,11 and two reports of rare CFH variants with high penetrance in families24,25 suggested that there should be rare variants with a large effect size in CFH. To this end, we analyzed data from our targeted sequencing study with a focus on the coding region of CFH, especially its functional domains.
Materials And Methods
Samples and Sequencing
We have previously described the sequencing and genotyping of this cohort of 2493 individuals.21 This research was conducted following the tenets of the Declaration of Helsinki. Informed consent was obtained from each subject. Subjects were referred by physicians throughout the US or self-referral and were enrolled in our ongoing AMD study protocols.3,11,12,14,20 Cases had either geographic atrophy (n = 402) or neovascular disease (n = 1282) based on dilated ocular examination and fundus photography. Controls were examined and had no signs of intermediate or advanced AMD in either eye and absence of bilateral early maculopathy. All non-AMD causes of atrophy or neovascular disease were excluded from both groups, including high myopia, ocular histoplasmosis, and angioid streaks. All cases and controls were unrelated. For this study, we retained 2417 individuals for whom genotype information for two common CFH SNPs (Y402H and rs10737680, a proxy for rs1410996) was available (1665 cases and 752 controls, median ages 75.6 ± 7.4 years and 77.2 ± 6.0 years, respectively). We enriched our screening panel for genes involved in the classical, lectin and alternative pathways (including CFH) using targeted genomic capture and sequenced the pooled libraries on a sequencing system (Illumina HiSeq2000; Illumina, Inc., San Diego, CA, USA). We aligned the sequencing data to the hg19 human genome release using the Burrows-Wheeler Aligner v0.59 (https://github.com/lh3/bwa)26 generated sample genotypes using the UnifiedGenotyper of the Genome Analysis Tool Kit v2.18 (https://www.broadinstitute.org/gatk), and then reduced false-positive calls with Variant Quality Score Recalibration (VQSR) tool.27,28 We manually screened the relevant sequencing data for variants that failed VQSR. Review of the sequencing reads in the Integrated Genome Viewer supported the validity of all but two variant calls.29,30 For this study, we reduced the variant call format file to the annotated CFH interval (hg19 chr1:196,621,008-196,716,634), extracted genotypes, and coded them for further analysis.
Statistical Analysis
We identified rare variants by calculating the allele frequency in controls for each variant site and then kept only those variants with a MAF less than a cutoff of either 1% or 0.5%. For each individual, we determined if a rare variant was present and then coded absence or presence of a rare variant as a dominant model using 0/1 indicator variable. The two common variants were coded as an additive model employing a 0/1/2 allele dose variable. We performed logistic regression (Equation 1) predicting disease status as a function of having a rare variant and accounting for the common alleles:
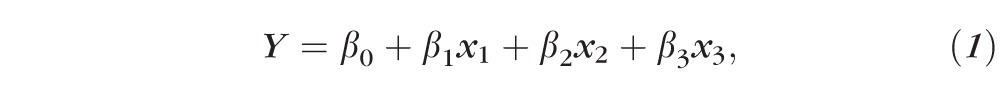 |
where Y is the disease status, x1 is the allele dosage of rs1061170, x2 is the allele dosage of rs10737680, x3 is the indicator variable for the rare variant (assuming a dominant model), β0 is the intercept, β1 is the regression coefficient for rs1061170, β2 is the regression coefficient for rs10737680, and β3 is the regression coefficient for the rare variant. We performed these calculations in a statistical programming language (R, v3.1.1; R Foundation for Statistical Computing, Vienna, Austria) using the logistf package, which performs logistic regression with Firth's correction and estimates the coefficients confidence intervals (CIs) for each predictor. We calculated the significance of the association by the likelihood-ratio test.
We estimated a permutation P value (Ppermute) by randomly reassigning case-control labels for 20,000 iterations, calculating a logistic model each time, and determining how often the estimated risk was greater than or equal to our initial estimate. We calculated the rare variant effects including the four known CFH rare variants (R53C, D90G, P503A, and R1210C; Table 1), as well as removing individuals with the known rare variants in CFH (Supplementary Table S1).20,24,25 We also tested variant significance employing the SNP-set kernel association test (SKAT) with the common variant genotypes as covariates.31
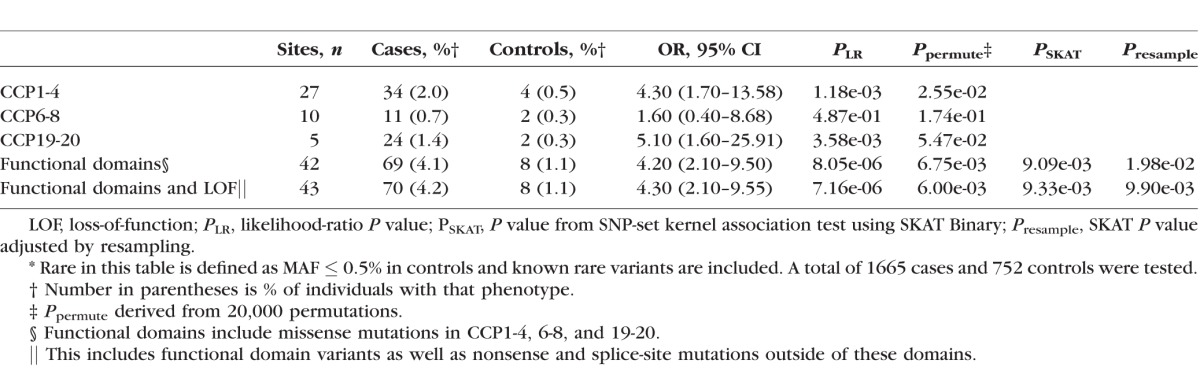
Table 1.
Advanced AMD Cases Are Enriched for Rare* Variants in the Functional Domains of CFH
Quantification of Serum Factor H
Blood samples were collected from five cases. The blood was centrifuged and serum was separated within 30 minutes of collection. The samples were frozen and stored in liquid nitrogen until testing was performed. Antigenic levels of FH and C3 were determined at the National Jewish Center for Immunology and Respiratory Medicine, Diagnostic Immunology and Complement Laboratory.32
Results
Rare CFH Variants Are Associated With A-AMD Risk
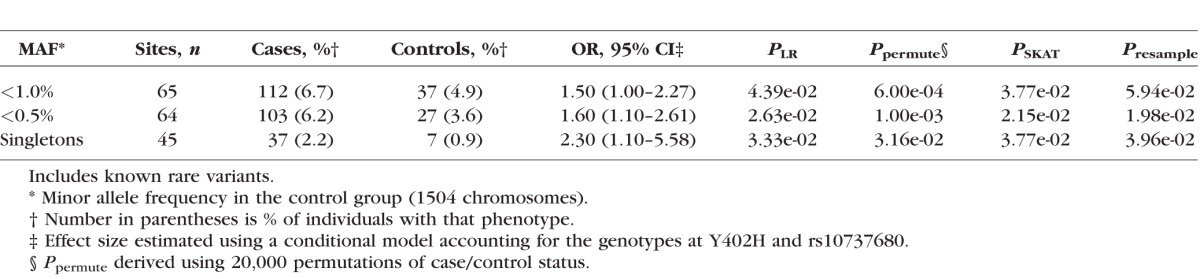
The original study included 2493 individuals.21 We identified 65 rare (control MAF ≤ 1%) missense, nonsense, splice-site, or stop read-through mutations in CFH among cases and controls (Supplementary Table S2). There was an excess of A-AMD cases who carried a variant with a MAF ≤ 1% (OR = 1.5, P = 4.4 × 10−2) compared with controls.
The association between rare variants in CFH with A-AMD became more robust and the risk conferred by these variants increased as the variant frequency decreased (Table 2). For variants with a MAF of 0.5% or less, the association was stronger (OR = 1.6, P = 2.6 × 10−2) than variants with a MAF of ≤1%. Additionally, singleton variants were observed more frequently in these cases (OR = 2.3, P = 3.3 × 10−2). All of these associations with A-AMD were independent of the effect of Y402H and rs10737680, the rs1410996 proxy.
Table 2.
Effect of Rare Variants in CFH on Risk of A-AMD Increases as the Allele Frequency Decreases
More sophisticated alternatives to the burden test that test for variable distribution of rare variants among cases and controls, implemented using SKAT, failed to identify stronger evidence for an association between CFH rare variants and A-AMD. This approach may have been complicated by a similar distribution of neutral variants in cases and controls, and a comparatively low cumulative frequency of rare variants as a class.
Rare Variants in Functional Domains of CFH Are Enriched in A-AMD Cases
The domain of FH that encodes regulatory activity consists of complement control protein (CCP) repeats 1-4 (Supplementary Fig. S1). This region contained a significant excess of rare variants (MAF ≤ 0.5%) in cases (n = 34) compared with controls (n = 4; OR = 4.3, P = 1.2 × 10−3; Table 1), highlighting the critical nature of this particular domain. Cases of A-AMD carried variants that clustered at the interface between CCP repeats 1-4 and C3b, whereas those in controls were consistently found facing away from the C3b interface (Fig. 1). There was a precedent for variants in CCP1 that do not contact the C3b structure to affect decay acceleration (e.g., R53C),24 as FB, the protease that activates the AP, binds the C345C domain of C3b.33,34 Such variants have been observed in atypical hemolytic uremic syndrome (aHUS) cases6–9 and have been shown to be linked to AMD in family studies.24 After removing individuals (n = 4) with one of the two known rare variants in CCP1-4, the enrichment in cases (n = 30) compared with controls (n = 4) was still significant (OR = 4.0, P = 2.6 × 10−3; Supplementary Table S1).
Figure 1.
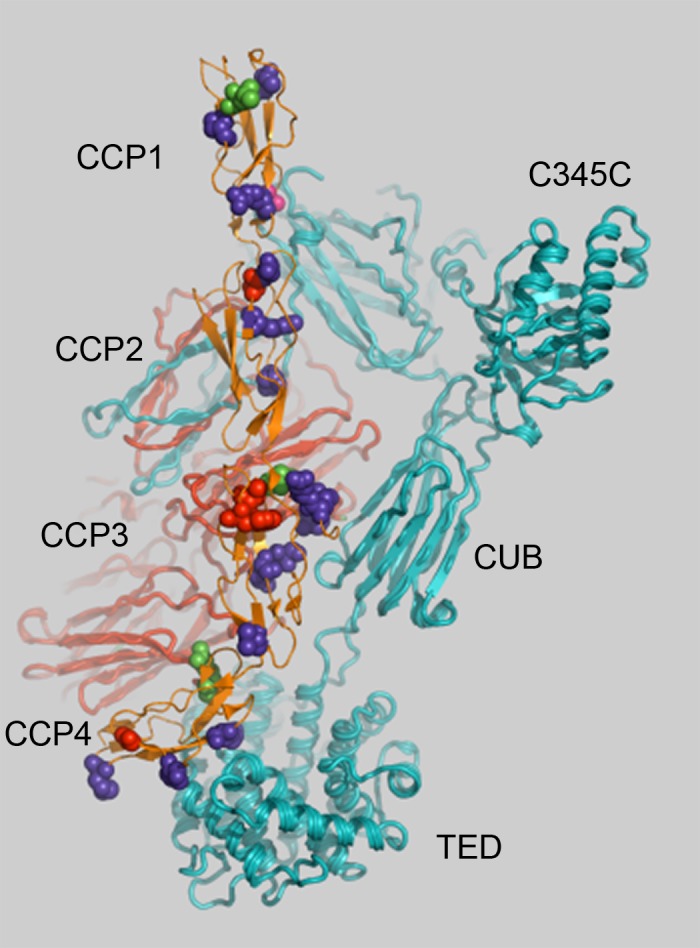
Rare CFH variants within CCP1-4 repeats cluster at the C3b:FH interface.
Variants present only in A-AMD cases are shown in purple or red. Variants that are nonsense or remove a conserved cysteine are shown in red (these are only seen in cases). Variants seen only in controls are shown in green. One variant was observed in both cases and controls (magenta). The variants in repeats CCP3 contact the CUB domain and variants in repeats CCP4 contact the TED of C3b. Complement component 3 α-chain is in teal (foreground) and the β-chain is in red (background). The structure of FH and C3b (PDB ID 2wII) was displayed in Pymol (Delano Scientific). C345C (NTR – Netrin C-terminal domain); CUB, complement C1r/C1s, Uegf, Bmp1; TED, thioester domain.
Cases of A-AMD also have more rare variants in the anionic binding domains located at CCP6-8 and in CCP19-20 than do controls (Table 1; Supplementary Table S1). Complement control protein 7 is the site of the common AMD risk variant Y402H which affects heparin binding.35 Eleven cases carried a rare variant in CCP6-8 compared with two controls, but the difference did not reach significance (OR = 1.6, P = 4.9 × 10−1). One case and no controls had the known risk rare variant, P503A, in CCP6-8.25 Removing that individual did not affect the significance of the test (OR = 1.5, P = 5.7 × 10−1). In control complement protein repeats 19-20, 24 cases carried a variant compared with two controls, corresponding to an OR of 5.1 (P = 3.6 × 10−3). One rare variant in CCP19-20, chr1:196716375 (R1210C), was previously described as a highly penetrant risk variant associated with high risk for A-AMD.20 It was present in 16 cases and one control. The main effect of this variant was large enough that the enrichment in CCP19-20 was not significant after removing individuals with R1210C. Eight cases carried the remaining four rare variants in CCP19-20 compared with one control (OR = 2.0, P = 4.3 × 10−1). The variants found in CCP20 all clustered on the same face as R1210C, known to affect binding to glycosaminoglycans (GAGs; Fig. 2).20,36 There were no variants within the CCP19 or CCP20 domains that directly opposed the C3d binding site in the crystal structure.
Figure 2.
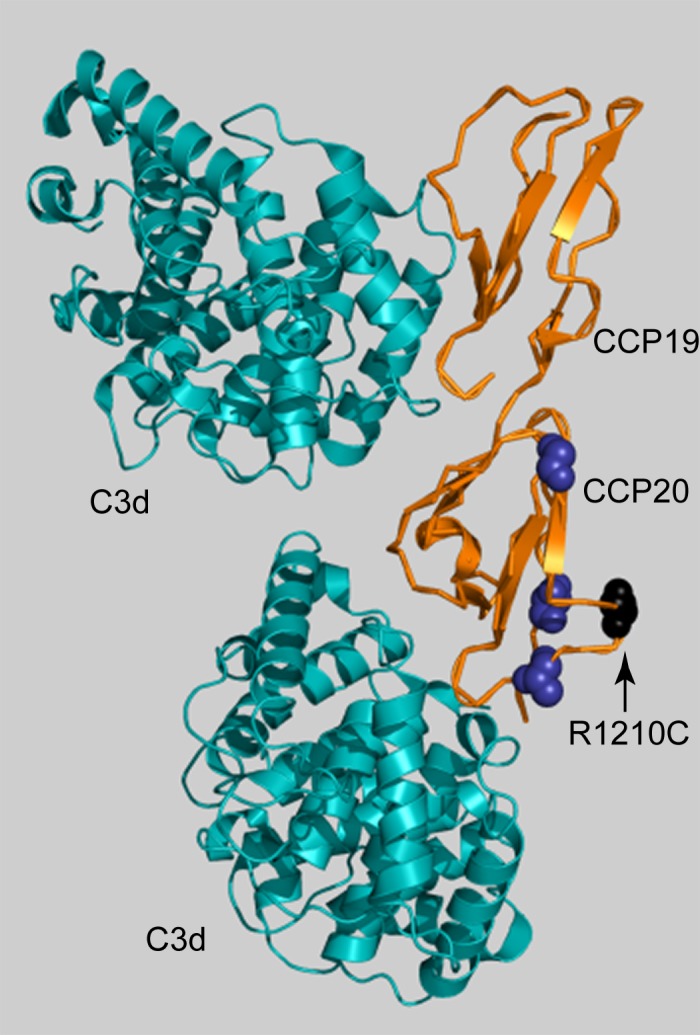
Complement Factor H rare variants in CCP20 cluster near R1210C. Factor H CCP19-20 is in orange; C3d is in teal. Variant R1210C is shown in black (arrow). The crystal structure of CCP19-20 contains two C3d fragments (PDB ID 2XQW) and was displayed in Pymol (Delano Scientific). Mutations seen only in A-AMD cases are shown in dark blue. The CCP19 variant (Q1143E, in one control) is not present in the crystal structure. The rare variants cluster on the same face as R1210C, known to affect heparin/GAG binding.42
We next collapsed the putative function altering and rare variants into a single loss-of-function (LOF) class. We included rare variants (MAF in controls < 0.5%) within the three functional domains as well as those that are predicted to effect the protein (nonsense, loss of a conserved cysteine, splice-site) but outside of a functional domain. This class of LOF, as a group, was associated with an increased risk of A-AMD independent of the common variants (OR = 4.3, P = 7.2 × 10−6, Table 1). We observed an increase in the number of cases that carried a LOF variant compared with controls (4.2% vs. 1.1%). Rare variants in CFH that have been previously reported (R53C,24 D90G,24 P503A,25 and R1210C20) did not account for the enrichment. When individuals with these variants were excluded from the analyses, the LOF variant class remained associated with risk of A-AMD (OR = 3.2, P = 1.4 × 10−3). The common SNPs associated with A-AMD were also significant in this analysis of rare variants and had effect sizes consistent with previously published estimates (Supplementary Table S3).
Cases Exclusively Carry LOF Variants
Factor H consists solely of 20 consecutive CCP domains (“beads on a string”), each having two conserved disulfide bonds (Supplementary Fig. S1). Loss of a conserved cysteine typically leads to a failure of the protein to be secreted because of improper folding. There were 10 cases and 0 controls that carried a variant leading to loss of a conserved cysteine, a nonsense change, or loss of a canonical splice site (Table 3). Each LOF variant was also a singleton in this set of 2417 individuals.
Table 3.
Serum Levels of FH and C3 in Cases Carrying Rare CFH Variants
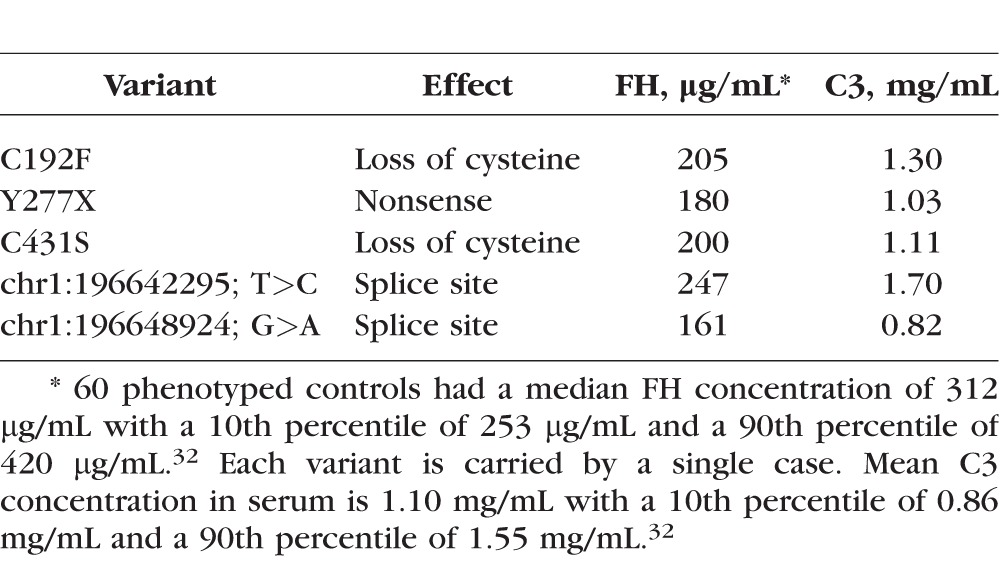
Of these 10 cases, serum samples were available for five individuals (Table 3). All five cases had lower FH serum levels compared with control values (<10th percentile; 10th percentile threshold = 253 μg/mL as determined from FH levels of 60 controls32), indicating haploinsufficiency for FH. Only one individual had a low C3 level and, interestingly, this A-AMD case also had the lowest FH level.
Discussion
In this report, we have demonstrated that a diversity of variants exists in CFH and that the rarest variants were associated with A-AMD cases. We identified an enrichment of rare variants in functional domains and an enrichment of mutations leading to haploinsufficiency. Some of these variants have been reported in aHUS, a disease caused by haploinsufficiency of regulators of the AP or, rarely, by gain-of-function mutations in the components of the AP.6–9 Our findings have confirmed the role of CFH LOF and haploinsufficiency mutations in A-AMD and the overlap with aHUS variants highlights the pleiotropic outcome of AP perturbation. These data also further indicate that rare variant carriers have more severe disease.20,24,37
Genetic risk factors for AMD have spanned the allelic spectrum: associations with risk of AMD have been found for common variants in the CFH,10,11,38–40 C3,13,14 and CFB,11,15 genes, common variants near the CFI12 gene, and for a diversity of rare variants within the CFI gene.21 Additionally, a single rare variant in C3 was shown to be associated with AMD using a variety of study designs21–23 as well as a rare variant in C9.21 Four independent rare variants in CFH have recently been demonstrated to be associated with AMD.20,24,25 Variant R1210C in CCP20 of CFH was the most frequent of the four identified rare variants in the European American population and had a large effect size (OR of approximately 20).20,24 Complement factor H variants R53C and D90G, both in the regulatory domain of CFH, were independently linked to AMD within families.24 In complement control proteins 6-8 (which harbors the common variant Y402H), a rare variant, P503A, was also found to be linked to AMD in an Amish family.25
We observed three of these known rare CFH variants in this sequencing study of advanced A-AMD cases and controls, as well as 61 other variants with a MAF ≤ 0.5%. There was an excess burden, nearly 4-fold, of rare variants in the functional domains of CFH in cases relative to controls. The association between rare variants in CFH and A-AMD was significant when considering the variants with a MAF ≤ 1% in all domains of the protein. However, an important observation was that the effect was stronger with a cutoff of ≤0.5% or by using only singletons. Also, variants in controls were largely in domains of the protein without defined roles in regulatory function or in localizing FH to a site of injury.
The spectrum of allelic frequencies seen in CFH was in contrast to CFI where the enrichment of rare variants was highly significant and the frequency of uncommon variants (MAF 0.5%–2%) much lower.21 The allelic spectrum of CFI is similar to PLD3, a gene recently found to have a burden of rare variants in late onset Alzheimer disease cases. Genes PLD3 and CFI had no variants between 0.5% and 2% MAF in the 6500 ESP dataset.41 Association between a phenotype and rare variants will be more easily identified in genes with less neutral variation (i.e., noise obscuring a signal). In the case of CFH, the most important alterations were best detected by using knowledge of the protein to aggregate variants that affect protein domains of FH known to have functional importance.42
Many of these CFH variants have been reported in aHUS or dense deposit disease (DDD or type II membranoproliferative glomerulonephritis), diseases of the kidney with early age of onset.6–9,42 Additional genetic and environmental factors must exist that affect the risk of developing kidney disease versus retinal disease. Unaffected relatives of aHUS patients carrying the same mutation can have normal renal function or only mild impairment. Penetrance differences in aHUS between carriers of a variant may reflect differing levels of AP activity, susceptibility, and environmental/pathogen exposures.
Taken together, the data indicate that either a rare variant producing a haploinsufficient state or a common variant with a more modest effect will lead to excessive AP activation over a lifetime. Rare variants in CFH were present in 4.2% of cases compared with 1.1% of controls herein, and rare variants in CFI were reported in 7.8% of cases compared with 2.3% in controls.21
In summary, AMD is a disease predominantly mediated by the AP of complement activation. Patients inherit common and rare variants in the AP, particularly its inhibitors, that lead to excessive and undesirable AP activity for a given degree of injury. Herein we have presented data indicating that rare variants act in an autosomal dominant manner. This means that haploinsufficiency of the cofactor protein (FH) or the necessary protease (FI) to inactivate C3b21 is sufficient to allow for excessive inflammation resulting in a chronic degenerative disease of the retina.
Supplementary Material
Acknowledgments
The authors thank the study participants and ophthalmologists who contributed to this study.
Supported in part by NIH Grants R01-EY11309 (JMS), 1R01AR063759-01A1 (SR), 5U01GM092691-04 (SR), 1U01HG007690-01 (SR), F30HL103072 (MT), R01-AI041592 (JPA, ECS), and U54 HL112303 (JPA); the Edward N. & Della L. Thome Memorial Foundation (JPA); the Doris Duke Clinical Scientist Development Award (SR); the Rheumatic Disease Core Center supported by NIH-Arthritis and Musculoskeletal and Skin Diseases P30 AR48335 (JPA, EDOR); the Massachusetts Lions Eye Research Fund, Inc. (JMS); Research to Prevent Blindness Challenge Grant to the New England Eye Center, Department of Ophthalmology, Tufts University School of Medicine (JMS); the American Macular Degeneration Foundation (JMS) and the Macular Degeneration Research Fund of the Ophthalmic Epidemiology and Genetics Service, New England Eye Center, Tufts Medical Center, Tufts University School of Medicine (JMS); and an F30 Ruth L. Kirschstein National Research Service Award (National Heart, Lung and Blood Institute; JMS). The authors alone are responsible for the content and writing of the paper.
Disclosure: M.P. Triebwasser, None; E.D.O. Roberson, None; Y. Yu, None; E.C. Schramm, None; E.K. Wagner, None; S. Raychaudhuri, None; J.M. Seddon, P; J.P. Atkinson, None
References
- 1. Schramm EC,, Clark SJ,, Triebwasser MP,, Raychaudhuri S,, Seddon JM,, Atkinson JP. Genetic variants in the complement system predisposing to age-related macular degeneration: a review. Mol Immunol. 2014. ; 61: 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson DH,, Radeke MJ,, Gallo NB,, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. 2010. ; 29: 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sobrin L,, Seddon JM. Nature and nurture-genes and environment- predict onset and progression of macular degeneration. Prog Retin Eye Res. 2014; 40: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ambati J,, Atkinson JP,, Gelfand BD. Immunology of age-related macular degeneration. Nat Rev Immunol. 2013. ; 13: 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ansari M,, McKeigue PM,, Skerka C,, et al. Genetic influences on plasma CFH and CFHR1 concentrations and their role in susceptibility to age-related macular degeneration. Hum Mol Genet. 2013. ; 22: 4857–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Noris M,, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009. ; 361: 1676–1687. [DOI] [PubMed] [Google Scholar]
- 7. Noris M,, Caprioli J,, Bresin E,, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010; 5: 1844–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rodriguez de Cordoba S,, Hidalgo MS,, Pinto S,, Tortajada A. Genetics of atypical hemolytic uremic syndrome (aHUS). Semin Thromb Hemost. 2014. ; 40: 422–430. [DOI] [PubMed] [Google Scholar]
- 9. Kavanagh D,, Richards A,, Atkinson JP. Complement regulatory genes and hemolytic uremic syndromes. Annu Rev Med. 2008. ; 59: 293–309. [DOI] [PubMed] [Google Scholar]
- 10. Klein RJ,, Zeiss C,, Chew EY,, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005. ; 308: 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maller J,, George S,, Purcell S,, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006. ; 38: 1055–1059. [DOI] [PubMed] [Google Scholar]
- 12. Fagerness JA,, Maller JB,, Neale BM,, Reynolds RC,, Daly MJ,, Seddon JM. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2009. ; 17: 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yates JR,, Sepp T,, Matharu BK,, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007. ; 357: 553–561. [DOI] [PubMed] [Google Scholar]
- 14. Maller JB,, Fagerness JA,, Reynolds RC,, Neale BM,, Daly MJ,, Seddon JM. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. 2007. ; 39: 1200–1201. [DOI] [PubMed] [Google Scholar]
- 15. Gold B,, Merriam JE,, Zernant J,, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006; 38: 458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Seddon JM,, Reynolds R,, Maller J,, Fagerness JA,, Daly MJ,, Rosner B. Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic demographic, and environmental variables. Invest Ophthalmol Vis Sci. 2009. ; 50: 2044–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fritsche LG,, Chen W,, Schu M,, et al. Seven new loci associated with age-related macular degeneration. Nat Genet. 2013; 45: 433–439, 439e1–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lachmann PJ. The amplification loop of the complement pathways. Adv Immunol. 2009. ; 104: 115–149. [DOI] [PubMed] [Google Scholar]
- 19. Bradley DT,, Zipfel PF,, Hughes AE. Complement in age-related macular degeneration: a focus on function. Eye (Lond). 2011. ; 25: 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Raychaudhuri S,, Iartchouk O,, Chin K,, et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet. 2011. ; 43: 1232–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seddon JM,, Yu Y,, Miller EC,, et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet. 2013. ; 45: 1366–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Helgason H,, Sulem P,, Duvvari MR,, et al. A rare nonsynonymous sequence variant in C3 is associated with high risk of age-related macular degeneration. Nat Genet. 2013. ; 45: 1371–1374. [DOI] [PubMed] [Google Scholar]
- 23. Zhan X,, Larson DE,, Wang C,, et al. Identification of a rare coding variant in complement 3 associated with age-related macular degeneration. Nat Genet. 2013. ; 45: 1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu Y,, Triebwasser MP,, Wong EK,, et al. Whole-exome sequencing identifies rare, functional CFH variants in families with macular degeneration. Hum Mol Genet. 2014. ; 23: 5283–5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoffman JD,, CookeBailey JN,, D'Aoust L,, et al. Rare complement factor h variant associated with age-related macular degeneration in the Amish. Invest Ophthalmol Vis Sci. 2014; 55: 4455–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li H,, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009. ; 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McKenna A,, Hanna M,, Banks E,, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010. ; 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DePristo MA,, Banks E,, Poplin R,, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011. ; 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Robinson JT,, Thorvaldsdottir H,, Winckler W,, et al. Integrative genomics viewer. Nat Biotechnol. 2011. ; 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thorvaldsdottir H,, Robinson JT,, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013; 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu MC,, Lee S,, Cai T,, Li Y,, Boehnke M,, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011. ; 89: 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reynolds R,, Hartnett ME,, Atkinson JP,, Giclas PC,, Rosner B,, Seddon JM. Plasma complement components and activation fragments: associations with age-related macular degeneration genotypes and phenotypes. Invest Ophthalmol Vis Sci. 2009. ; 50: 5818–5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pechtl IC,, Kavanagh D,, McIntosh N,, Harris CL,, Barlow PN. Disease-associated N-terminal complement factor H mutations perturb cofactor and decay-accelerating activities. J Biol Chem. 2011. ; 286: 11082–11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Forneris F,, Ricklin D,, Wu J,, et al. Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science. 2010. ; 330: 1816–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Clark SJ,, Ridge LA,, Herbert AP,, et al. Tissue-specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan-binding regions. J Immunol. 2013. ; 190: 2049–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jozsi M,, Heinen S,, Hartmann A,, et al. Factor H and atypical hemolytic uremic syndrome: mutations in the C-terminus cause structural changes and defective recognition functions. J Am Soc Nephrol. 2006. ; 17: 170–177. [DOI] [PubMed] [Google Scholar]
- 37. Duvvari MR,, Saksens NT,, van de Ven JP,, et al. Analysis of rare variants in the CFH gene in patients with the cuticular drusen subtype of age-related macular degeneration. Mol Vis. 2015. ; 21: 285–292. [PMC free article] [PubMed] [Google Scholar]
- 38. Edwards AO,, Ritter R,, III,, Abel KJ,, Manning A,, Panhuysen C,, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005. ; 308: 421–424. [DOI] [PubMed] [Google Scholar]
- 39. Hageman GS,, Anderson DH,, Johnson LV,, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005. ; 102: 7227–7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Haines JL,, Hauser MA,, Schmidt S,, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005. ; 308: 419–421. [DOI] [PubMed] [Google Scholar]
- 41. Cruchaga C,, Karch CM,, Jin SC,, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer's disease. Nature. 2014. ; 505: 550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zipfel PF,, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009. ; 9: 729–740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.