

Abstract
Purpose
Mutations in the gene encoding collagen type IV alpha 1 (COL4A1) cause multisystem disorders including anterior segment dysgenesis (ASD) and optic nerve hypoplasia. The penetrance and severity of individual phenotypes depends on genetic context. Here, we tested the effects of a Col4a1 mutation in two different genetic backgrounds to compare how genetic context influences ocular dysgenesis, IOP, and progression to glaucoma.
Methods
Col4a1 mutant mice maintained on a C57BL/6J background were crossed to either 129S6/SvEvTac or CAST/EiJ and the F1 progeny were analyzed by slit-lamp biomicroscopy and optical coherence tomography. We also measured IOPs and compared tissue sections of eyes and optic nerves.
Results.
We found that the CAST/EiJ inbred strain has a relatively uniform and profound suppression on the effects of Col4a1 mutation and that mutant CASTB6F1 mice were generally only very mildly affected. In contrast, mutant 129B6F1 mice had more variable and severe ASD and IOP dysregulation that were associated with glaucomatous signs including lost or damaged retinal ganglion cell axons and excavation of the optic nerve head.
Conclusions.
Ocular defects in Col4a1 mutant mice model ASD and glaucoma that are observed in a subset of patients with COL4A1 mutations. We demonstrate that different inbred strains of mice give graded severities of ASD and we detected elevated IOP and glaucomatous damage in 129B6F1, but not CASTB6F1 mice that carried a Col4a1 mutation. These data demonstrate that genetic context differences are one factor that may contribute to the variable penetrance and severity of ASD and glaucoma in patients with COL4A1 mutations.
Keywords: type IV collagen, glaucoma anterior segment, basement membrane, genetic context, phenotypic variation
Glaucoma is a group of neurodegenerative diseases where progressive death of retinal ganglion cells leads to vision loss. Over 60 million people suffer from glaucoma-related vision loss worldwide making it a leading cause of blindness.1,2 Developmental glaucoma represents a severe form of the disease that often occurs early in life and responds poorly to current treatments.3 Patients with developmental glaucoma often have IOP elevation that is attributed to developmental defects of the anterior segment of the eye. Anterior segment dysgenesis (ASD) is a general term that describes the failure of normal development of the tissues and structures of the anterior segment of the eye where there are visible morphologic abnormalities of the lens, iris, cornea, and iridocorneal angle. Anterior segment dysgenesis is both clinically and genetically heterogeneous and a number of genes have been identified that cause ASD and glaucoma.4 While there are exceptions and phenotypic overlap among clinical entities, mutations in the genes encoding paired-like homeodomain 2 (PITX2) and forkhead box C1 (FOXC1) are the most common genetic causes of ASD and glaucoma in patients with Axenfeld-Rieger–like diseases and together account for approximately 40% of cases.4 Identification of additional ASD-causing genes and determining their roles in normal ocular development will help to achieve an integrated understanding of the pathogenic mechanisms that underlie ASD and glaucoma.
Collagen type IV alpha 1 (COL4A1) and alpha 2 (COL4A2) are major components of specialized structures of extracellular matrix called basement membranes. COL4A1 and COL4A2 assemble into heterotrimers within the endoplasmic reticulum and are secreted into the extracellular space where they polymerize into higher order molecular structures. COL4A1 and COL4A2 are present in nearly every basement membrane and, accordingly, mutations in either gene cause multisystem disorders.5,6 Although often cited for their involvement in cerebrovascular disease including porencephaly and hemorrhagic stroke, Col4a1 and Col4a2 point mutations were first identified in independent forward genetic screens in mice with cataracts and ocular dysgenesis.7–9 Homozygous mutant mice are rarely viable7,8,10 and complete COL4A1/COL4A2 deficiency is lethal.11 Moreover, the absence of an obvious phenotype in mice that are heterozygous for null mutations of both genes suggests that mutations typically act via a dominant negative mechanism.11 More recently, patients with COL4A1 mutations were reported with a variety of ASD phenotypes including microphthalmia, cataracts, Axenfeld-Rieger malformations and glaucoma.6,12–23
The disease spectrum in patients is broad and severity varies, as does the presence or absence of extraocular findings. For example, while most patients both have ocular and nonocular findings, one large family was reported with only isolated, nonsyndromic congenital cataracts in all 15 affected members.19 It is clear that both allelic heterogeneity and genetic context influence the penetrance and severity of phenotypes caused by Col4a1 and Col4a2 mutations.7–9,24,25 In an earlier study, we described ASD and high IOP in mice with the Col4a1Δex41 mutation (a splice acceptor mutation that skips exon 41) on a C57BL/6J (B6) genetic background.9,10 However, Col4a1+/Δex41 mice on a B6 background also had optic nerve hypoplasia (which is also reported in patients with COL4A1 mutations15), and so we could not conclude whether or not these mice developed glaucomatous optic nerve damage. In Col4a1+/Δex41 mice, both ASD and optic nerve hypoplasia are suppressed when the mice were crossed for a single generation (F1) to 129S6/SvEvTac or to CAST/EiJ (referred to as 129B6F1 and CASTB6F1, respectively).9 In another study of Col4a1 mutant mice ASD and optic nerve head cupping were reported but IOPs were not measured and the genetic context was not stated.7 In the present study, we aged 129B6F1 and CASTB6F1 mice and evaluated them for ASD severity, IOP elevation, and hallmarks of glaucoma. These strains were selected because they are genetically distinct from the original strain (B6) and highly divergent from each other.26 With a few notable exceptions, pathology in CASTB6F1 mice was uniformly and nearly completely suppressed, even in mice aged for over 30 months. In contrast, Col4a1+/Δex41 129B6F1 mice had highly variable ASD and hallmarks of glaucoma including age-related IOP elevation, loss and damage to retinal ganglion cell axons, and optic nerve head cupping. These findings indicate that Col4a1+/Δex41 129B6F1 mice represent a genetic model for age-related glaucoma and will be useful for understanding both anterior segment development and how ASD can progress to glaucoma.
Materials and Methods
Animals
The Col4a1+/Δex41 mice were originally identified in a mutagenesis screen conducted at The Jackson Laboratory (Bar Harbor, ME, USA) and the ocular phenotypes have been previously described.9,10 Col4a1+/Δex41 mice that had been backcrossed to C56BL/6J (B6) mice for at least six generations were mated with CAST/EiJ (CAST) or 129S6/SvEvTac (129) mice to produce CASTB6F1 and 129B6F1 mice, respectively. All animals were maintained in full-barrier facilities free of specific pathogens on a 12-hour light/dark cycle with food and water ad libitum. All experiments were compliant with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco (CA, USA).
Slit-Lamp Biomicroscopy and IOP Measurement
Ocular anterior segment examinations were performed on Col4a1+/+ and Col4a1+/Δex41 mice by observers masked to the genotypes using a slit-lamp biomicroscope (Topcon SL-D7; Topcon Medical Systems, Oakland, NJ, USA) attached to a digital SLR camera (Nikon D200; Nikon, Melville, NY, USA). Intraocular pressures were measured using the microneedle method as previously described.27,28 Briefly, mice were anesthetized with ketamine (99 mg/kg) and xylazine (9 mg/kg) and placed on the measurement platform. Intraocular pressures were measured by inserting a microneedle into the anterior chamber within 3 to 4 minutes after sufficient anesthesia. All measurements were performed during the light cycle of the room and both male and female mice were used for each age group. We detected an effect of sex in 5- to 7-month-old 129B6F1 mice (2-way ANOVA; sex, P = 0.0002; genotype, P = 0.4144; sex and genotype interaction, 0.7316) in both Col4a1+/+ and Col4a1+/Δex41 mice (post hoc Tukey's multiple comparison test; P = 0.011 and 0.018, respectively). We did not detect a sex effect for other groups and so we combined the data from both sexes.
Histologic Analysis
Iridocorneal Angles and Optic Nerve Heads.
Eyes were harvested at appropriate ages and fixed in situ with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (pH 7.2), dehydrated in graded ethanol and embedded in fresh Historesin (Leica, Heidelberg, Germany). Embedded tissues were sectioned and stained with hematoxylin and eosin (H&E). The extent of damage is indicated using a 4-tiered scale as described previously.29 Briefly, three slides, each containing eight sections (n = 24 sections), were analyzed from different regions of the eye and the assigned grades were averaged. Scoring was done without knowledge of the mouse genotype and used a numerical scheme (0, 1, 2, or 3).
Optic Nerves.
Postorbital, intracranial portions of optic nerves were processed and analyzed as previously described.30 Briefly, the top of the skull and most of the brain overlying the optic nerves were removed and the remaining tissue was fixed overnight with 0.8% PFA and 1.2% glutaraldehyde in 0.1 M phosphate buffer. The tissue was embedded in Embed 812 resin (Electron Microscopy Sciences, Ft. Washington, PA, USA). One-micron cross sections of postorbital optic nerve were stained with paraphenylenediamine (PPD). Paraphenylenediamine differentially stains the axoplasm of sick or dying axons darkly, thus permitting detection of axon injury. An optic nerve grading system was used as previously described31 to determine the level of glaucomatous damage. The damage level accounts for several factors including the number of healthy axons remaining, the number of damaged axons, and the amount of scarring associated with gliosis. All nerves were scored by at least two investigators masked for the age and genotype of the mouse. Both investigators were unaware of the grade assigned by the other investigator. When the grades from the two investigators did not agree, a third investigator graded the nerves and the most common grade was used as the final grade for the optic nerve.
Funduscopy and Optical Coherence Tomography
Mice were anesthetized with a steady flow of 1.5% to 3% isofluorane. The eyes were topically anesthetized with one drop of proparacaine, dilated with one drop of a 1:1 mixture of 1% tropicamide and 2.5% phenylephrine. The corneas were kept moist with regular application of 2.5% methylcellulose. Eyes were examined using the Micron III retinal imaging system (Phoenix Research Labs, Pleasanton, CA, USA) and raw images were adjusted using Photoshop CS6 (Adobe, San Jose, CA, USA). Spectral-domain optical coherence tomography (OCT) images were acquired with the Micron Image Guided SD-OCT System (Phoenix Research Labs) by averaging 10 to 20 scans. Levels were adjusted using Photoshop CS6 (Adobe). Optical coherence tomography images were assessed by three investigators masked for the genotypes for cupping of the optic nerve head.
Results
Clinical Phenotypes Are Dependent Upon Genetic Context
Our prior studies showed that both CASTB6F1 and 129B6F1 genetic contexts suppressed severe ASD and optic nerve hypoplasia typically seen in Col4a1+/Δex41 mice on the B6 background.9 However, even mild developmental defects might still be sufficient to contribute to progressive pathology as mice age. To determine if CASTB6F1 and 129B6F1 mutant mice developed glaucoma, we aged and analyzed cohorts of Col4a1+/+ and Col4a1+/Δex41 mice. Slit-lamp biomicroscopy of Col4a1+/+ and Col4a1+/Δex41 CASTB6F1 revealed relatively normal appearance of anterior ocular structures, yet mildly enlarged anterior chambers, in mutant eyes at all ages (Figs. 1A–D). While ASD in this genetic context was generally very mild, a few exceptions were observed. Out of 22 eyes analyzed at 30 months of age, one had severe cataract and enlarged anterior chamber, a second had iris coloboma and a third had corneal neovascularization (Supplementary Fig. S1). In the 129B6F1 genetic context, Col4a1+/Δex41 mice had much more variable and severe ASD at all ages (Figs. 1E–L). The most highly penetrant phenotypes were abnormally tortuous iris vasculature, persistent pupillary membrane, and pigment on the anterior surface of the lens. The frequencies of these phenotypes were similar regardless of age and enlargement of the anterior chamber was the most variable phenotype. Although almost all eyes had enlarged anterior chambers some mice had severely enlarged chambers (compare Figs. 1J and 1L). The frequency of severely enlarged eyes tended to increase with age suggesting high IOP may develop with age.
Figure 1.
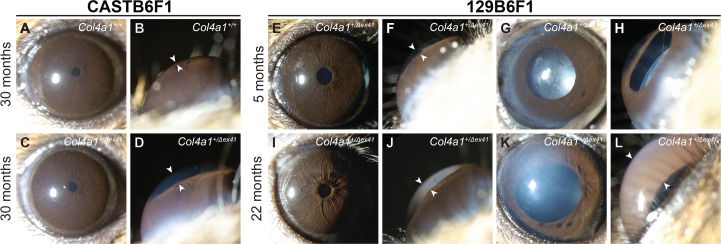
Col4a1 mutation leads to variable ASD depending on genetic context. Col4a1+/Δex41 mice maintained on a C57BL/6J background were crossed for a single generation to CAST/EiJ (called CASTB6F1 and shown in [A–D]) or to 129S6/SvEvTac (called 129B6F1 and shown in [E–L]) and analyzed by slit-lamp biomicroscopy. On the CASTB6F1 background, ASD in Col4a1+/Δex41 mice was uniformly suppressed and the vast majority of Col4a1+/Δex41 eyes had grossly normal appearances ([C] compared with [A]) with the exceptions of mildly enlarged anterior chambers (arrowheads in [D] compared with [B]). Col4a1+/Δex41 mice on the 129B6F1 background had ASD with variable severity (E–L). Among the eyes of 5-month-old mutant mice we observed enlarged and tortuous iris vasculature (21/24 eyes), cataracts (4/27 eyes), enlarged anterior chambers (27/29 eyes; arrowheads in [F, J, L]), persistent pupillary membranes (25/26 eyes) and pigment on the lens (17/27 eyes). Note: sample sizes vary because not all phenotypes could be assessed in all eyes. Similar subphenotypes were also present in the 22-month-old cohort where Col4a1+/Δex41 mice had abnormal iris vasculature (18/18 eyes), cataracts (3/21 eyes), enlarged anterior chambers (20/20 eyes), persistent pupillary membranes (13/21 eyes), and pigment on the lens (11/21 eyes). Frequency of severely enlargement of anterior chamber increases with age suggesting age-related IOP elevation.
Iridocorneal Angles Show Variable Abnormalities of the Anterior Segment That Increase With Age
To better assess the effects of CASTB6F1 and 129B6F1 genetic contexts on the extent of pathology, we performed histologic analyses of the iridocorneal angles in eyes from Col4a1+/+ and Col4a1+/Δex41 mice (Fig. 2). Despite the relatively normal clinical appearance of the eyes from CASTB6F1 mutant mice, histologic analyses revealed pathology including anterior synechia and hypoplastic or absent Schlemm's canal and trabecular meshwork (Figs. 2A–D). Consistent with our observations with slit-lamp biomicroscopy, the morphologic defects in Col4a1+/Δex41 129B6F1 mice were more severe and included hypoplastic or absent Schlemm's canal, hypoplastic trabecular meshwork, and hypoplastic ciliary body (Figs. 2E–H). Iridocorneal angles were also closed to varying extents and anterior synechia were present (Figs. 2F, 2H). The histologic phenotypes found in individual mice and aged cohorts demonstrate that significant variation was observed between individual eyes. Notably, although generally considered to be ‘developmental defects,' by 22 months these pathologies not only persisted but also increased in severity indicating the occurrence of disease-related remodeling that is unrelated to development (Figs. 2I–K).
Figure 2.

Histologic analyses of iridocorneal angle dysgenesis. Histologic analyses revealed morphologic defects of the iridocorneal angle that progressed with age. In control eyes (A, C, E, G) there was a robust, foliated ciliary body, clearly identifiable Schlemm's canal (arrow), and trabecular meshwork. Analyses of Col4a1+/Δex41 CASTB6F1 mice (B, D) showed prevalent, but usually mild, iridocorneal angle defects with hypoplastic or absent Schlemm's canal (13/15 eyes), hypoplastic trabecular meshwork (13/15 eyes), and anterior synechia (10/15 eyes; black arrowhead). Frequencies are indicated for 5-month-old mice. Eyes from Col4a1+/Δex41 mice on the 129B6F1 background between 1.5 and 5 months of age (F) showed hypoplastic Schlemm's canal, trabecular meshwork, and ciliary body as well as anterior synechia (black arrowhead; n = 6). Eyes from mice between 22 and 31 months (H) had severe and extensive iridocorneal adhesions and an unrecognizable Schlemm's canal and trabecular meshwork (n = 6). The pars plana portion of the ciliary body is indicated (white arrowhead). Semiquantitative analyses of iridocorneal dysgenesis for each strain or for individual Col4a1+/Δex41 129B6F1 eyes are shown in ([I–K], respectively). Normal Schlemm's canal (SC), trabecular meshwork (TM), and ciliary body (CB) are indicated with white bars. Mild pathology (yellow) indicates that 50% to 100% of the TM, SC, or CB is present; moderate pathology (orange) indicates that 10% to 49% of these regions are present and severe (red) indicates that less than 10% of these regions are present. The percent angle closure was scored as normal (0%–25% closed), mild (25%–50% closed), moderate (50%–75% closed), or severe (>75% closed). Scale bar: 100 μm.
Aged 129B6F1 Mutant Mice Have Elevated IOP
The CASTB6F1 and 129B6F1 genetic contexts suppress ASD compared with Col4a1+/Δex41 on a B6 background. Despite this, mutant mice from both strains had histologic abnormalities in the iridocorneal angles and enlarged anterior chambers suggesting that they may also have ocular hypertension. To address this possibility we measured IOPs of Col4a1+/+ and Col4a1+/Δex41 mice from both genetic backgrounds over multiple ages (Figs. 3A, 3B). Consistent with the rescuing effect of the CAST background, CASTB6F1 mutant mice had IOP values and distributions that were nearly indistinguishable from control animals even when aged to 22 months (Mann-Whitney U test, P = 0.928) with the exceptions of one Col4a1+/Δex41 eye with very low IOP and one with mildly elevated IOP (22.4 mm Hg). Overall the mean IOPs for both Col4a1+/+ and Col4a1+/Δex41 CASTB6F1 mice significantly decreased with age. In 129B6F1 mice, mean IOP values also decreased with age in Col4a1+/+ mice. In contrast, an age-related IOP decrease did not occur in Col4a1+/Δex41 mice, and the IOPs of Col4a1+/Δex41 mice were higher than Col4a1+/+ mice at 12 to 15 and 18 to 22 months of age (P = 0.032 and 0.039, respectively). In addition, the ranges of IOPs in eyes from Col4a1+/Δex41 mice increased with age (5.5–24.1, 3.0–24.6, and 4.9–36.7 mm Hg for 5–7, 12–15, and 18–23 months of age, respectively). Importantly, we identified a number of Col4a1+/Δex41 eyes that had elevated IOPs. At each age group, respectively, there were 2 (7.1%), 10 (23.8%), and 10 (17.2%) mutant eyes with IOPs that were greater than the highest measured age-matched control IOP (22.5, 20.2, and 19.8 mm Hg for control mice at 5–7, 12–15, and 18–23 months of age, respectively).
Figure 3.
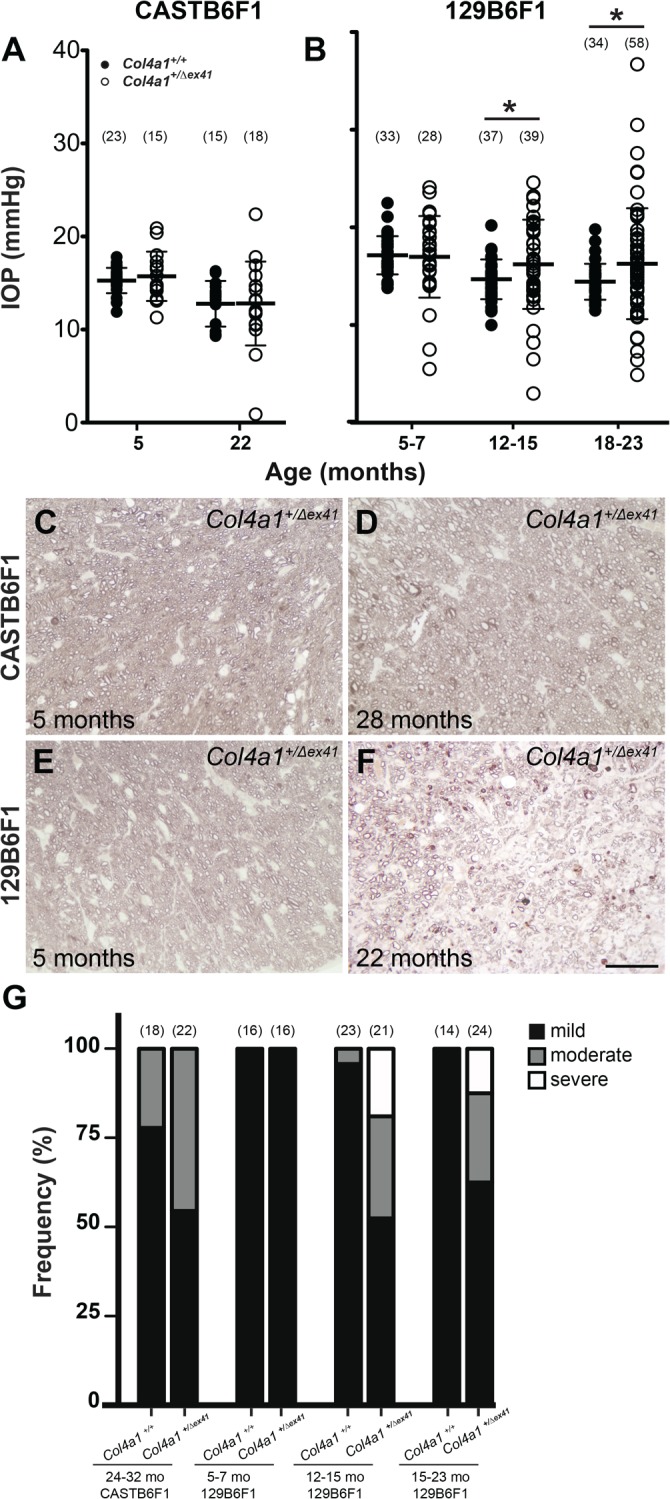
Age-related IOP dysregulation and optic nerve damage in Col4a1+/Δex41 129B6F1 mice. (A) Up to 5 months of age, IOPs of Col4a1+/Δex41 CASTB6F1 mice were comparable with their Col4a1+/+ counterparts (mean ± SD = 15.27 ± 1.37 and 15.72 ± 2.65 respectively; P = 0.929, Mann-Whitney U test). In Col4a1+/Δex41 mice at 22 months, there were rare outliers but the cohort was otherwise indistinguishable from Col4a1+/+ mice (mean ± SD = 12.78 ± 2.45 and 12.80 ± 4.50 respectively; P = 0.928). Overall, the mean IOP for both Col4a1+/+ and Col4a1+/Δex41 mice decreased with age (mean ± SD = 15.27 ± 1.37 and 12.78 ± 2.45 for Col4a1+/+ mice and 15.72 ± 2.65 and 12.80 ± 4.50 for Col4a1+/Δex41 mice at 5 and 22 months of age, respectively). (B). In the 129B6F1 genetic context, the mean IOP decreased with age in Col4a1+/+ (mean ± SD = 17.15 ± 1.95 and 14.44 ± 1.84 for mice at 5–7 and 18–23 months of age, respectively) but retained in old Col4a1+/Δex41 mice (mean ± SD = 16.99 ± 4.17 and 16.28 ± 5.68 for mice at 5–7 and 18–23 months of age, respectively). Intraocular pressures covered a greater range for Col4a1+/Δex41 compared with Col4a1+/+ mice at all ages, and the mean IOP for Col4a1+/Δex41 mice were higher than Col4a1+/+ mice at both 12 to 15 months and 18 to 22 months of age (mean ± SD = 14.69 ± 2.04 and 16.22 ± 4.57, for Col4a1+/+ and Col4a1+/Δex41 mice at 12–15 months of age, respectively, P = 0.032; Mean ± SD = 14.44 ± 1.84 and 16.28 ± 5.68, for Col4a1+/+ and Col4a1+/Δex41 mice at 18–22 months of age, respectively, P = 0.039). Numbers in parentheses indicate sample sizes for each genotype on each genetic background. Black bars indicate mean and SD for each group. *P < 0.05. (C–F). Histologic analysis and PPD staining of retinal ganglion cell axons in retro-orbital optic nerve sections demonstrates gliosis and sick or dying retinal ganglion cells in old Col4a1+/Δex41 129B6F1 (F) but not in young Col4a1+/Δex41 129B6F1 (E) nor in optic nerves from old and young Col4a1+/Δex41 CASTB6F1 (C, D) mice. Healthy axons have a clear axoplasm that is surrounded by darkly stained myelin while diseased axons stain darkly throughout. Scale bar: 20 μm. (G) Using a grading system to represent damage, we found severely damaged optic nerves in 4 of 21 and 3 of 24 Col4a1+/Δex41 129B6F1 mice at 12 to 15 and 15 to 23 months of age, respectively. The proportions of damaged nerves were significantly different between Col4a1+/+ and Col4a1+/Δex41 mice (Fisher's exact test, P = 0.002, and P = 0.032 for 12–15 and 15–23 months of age, respectively). None of the Col4a1+/Δex41 CASTB6F1 optic nerves were severely damaged; even for mice aged for more than 2 years.
Glaucoma Hallmarks in Col4a1+/Δex41 Mice on the 129B6F1 Background
Loss of, and damage to, retinal ganglion cell axons in the optic nerve and subsequent excavation of the optic nerve head are hallmarks of glaucoma. Col4a1+/Δex41 on a B6 background had optic nerve hypoplasia that precluded assessment of glaucomatous optic nerve damage in these animals.9 However, optic nerves appear to develop normally in Col4a1+/Δex41 CASTB6F1 and 129B6F1 mice9 and so we tested whether these mice had hallmarks of glaucoma. To do this we sectioned optic nerves from CASTB6F1 and 129B6F1 mice and stained them with PPD, which differentially stains the axoplasm of sick or dying axons. Stained optic nerve sections were then graded for the extent of damage by at least two independent investigators that were masked to the genotypes (Figs. 3C–G).31 Although we observed moderately damaged optic nerves in both Col4a1+/+ and Col4a1+/Δex41 mice aged for over 2 years we did not detect severely damaged nerves from CASTB6F1 mice of either genotype (Figs. 3C, 3D, 3G). In 129B6F1 mice we did not detect differences between optic nerves from Col4a1+/+ and Col4a1+/Δex41 animals up to 5 months of age; however, we identified a portion of severely damaged optic nerves in older Col4a1+/Δex41 mice. The numbers of damaged optic nerves were significantly different between Col4a1+/+ and Col4a1+/Δex41 mice at 12 to 15 months and 18 to 23 months of age (Fisher's exact test, P = 0.002 and P = 0.032, respectively; Figs. 3E–G).
Next, we tested whether optic nerve damage in Col4a1+/Δex41 129B6F1 mice was accompanied by optic nerve head excavation. While the optic nerve heads of Col4a1+/+ and Col4a1+/Δex41 CASTB6F1 mice (Figs. 4A–F) and Col4a1+/+ 129B6F1 mice (Figs. 4G–I) appeared normal, eyes from aged Col4a1+/Δex41 129B6F1 mice revealed optic nerve head excavation (Figs. 4J–L). Hematoxylin and eosin–stained sections showed clearly visible loss of both the retinal ganglion cells and the nerve fiber layer and the optic nerve head was cupped and disorganized in 3/5 mutant 129B6F1 eyes. Finally, we used OCT to screen additional mice from both backgrounds for evidence of optic nerve head excavation. The optic nerve heads of Col4a1+/Δex41 CASTB6F1 mice were indistinguishable from those from their Col4a1+/+ littermate controls (Fig. 4B vs. 4E); however, Col4a1+/Δex41 129B6F1 mice were deemed to have optic nerve head excavation by OCT (Figs. 4K vs. 4H).
Figure 4.

Optic nerve head excavation in Col4a1+/Δex41 129B6F1 mice. Analyses of optic nerve heads from both Col4a1+/+ (A–C) and Col4a1+/Δex41 CASTB6F1 (D–F) revealed well-defined ganglion cells (arrow) and robust nerve fiber layers (arrowheads). In contrast, while control 129B6F1 eyes (G–I) had morphologically normal optic nerve heads in three of five Col4a1+/Δex41 129B6F1 eyes at 22 months (J–L), both the ganglion cell and nerve fiber layers were absent and the optic nerve head was cupped, a sign of severe glaucoma. Optical coherence tomography imaging is able to detect optic nerve cupping in vivo and, using OCT, we identified cupping in a portion of Col4a1+/Δex41 129B6F1 eyes. Among the 10 Col4a1+/Δex41 129B6F1 eyes independently examined by three investigators, six were unanimously considered as having optic nerve head cupping. In contrast, all Col4a1+/Δex41 CASTB6F1 eyes (n = 8) and eyes from Col4a1+/+ mice on either background (n = 8 and 6 for CASTB6F1 and 129B6F1, respectively) had normal optic nerve head morphology. Horizontal bars in fundus images (A, D, G, J) indicate the position of OCT scanning beams.
Discussion
Here, we show that a Col4a1 mutation can cause ASD, age-related IOP elevation and glaucomatous neurodegeneration with characteristic retinal ganglion cell axon loss and optic nerve head cupping in a subset of eyes. Genetic context modifies these age-related phenotypes, whereby 129B6F1 mutant mice had highly variable pathology including some eyes with relatively severe ASD. Although the majority of these mice had some level of ASD, only a portion of the129B6F1 mice develops glaucoma. In the CASTB6F1 genetic context, ASD was strongly suppressed in the vast majority of Col4a1+/Δex41 mice. Notably however, at least three eyes had very severe phenotypes suggesting that even in genetically identical animals other factors can determine the phenotypic outcome. Our data also suggest that despite the obvious presence of developmental defects, iridocorneal angle malformations, and IOP elevation both progress with age. If this holds true in patients, then there may indeed be a therapeutic window for intervention to prevent or delay vision loss in patients with COL4A1 mutations.
To date, there are over 70 COL4A1 mutations identified in patients and approximately one-third of these are reported to have ASD or cataracts.5,6,15,17,20,23,32 In contrast, all 13 mutations in mice were identified primarily because of this phenotype.5 Both observations could reflect ascertainment biases. COL4A1 is often considered for its role in cerebrovascular disease and is only recently being considered as an ASD candidate gene. However, in mice, ASD is readily identified in forward genetic mutagenesis screens and a role in cerebrovascular disease could have more easily gone undetected. Given that COL4A1 and COL4A2 mutations are very likely to contribute to multisystemic disorders that include ASD (e.g., Muscle-Eye-Brain disease32) and not just isolated ASD, it is likely that patients with COL4A1 and COL4A2 mutations may be selected against by exclusion criteria in studies focused solely on identifying genes underlying more typical Axenfeld-Rieger types of dysgenesis. With the ongoing shift toward analyzing all genes through exome or genome sequencing, rather than selecting candidates, we expect that the number of ASD patients with COL4A1 or COL4A2 mutations will continue to increase. Our data predict that patients with COL4A1 and COL4A2 mutations may range from having severe ASD and optic nerve hypoplasia to age-related ocular hypertension with or without RGC loss, or perhaps to no ocular pathology at all. However, both genes should be considered as candidate genes in patients with ASD, especially those where ASD is part of a multisystem disorder.
PITX2 and FOXC1 mutations are the most common genetic causes of ASD identified. While proteins with which PITX2 and FOXC1 interact have been identified33–36 and cellular pathways37–42 have been implicated, the mechanisms underlying ASD are largely unknown. Thus, it remains unknown if COL4A1 mutations interact with Pitx2 and Foxc1 or if they represent a mechanistically distinct form of ASD. Like COL4A1 and COL4A2, the roles of PITX2 and FOXC1 in normal ocular development and function are still being fully understood, as are the mechanisms by which mutations in either gene cause ASD and glaucoma. PITX2 and FOXC1 are transcription factors that are expressed in overlapping subsets of periocular mesenchyme cells that differentiate into the ocular anterior segment structures.43–45 In contrast, COL4A1 and COL4A2 are extracellular matrix proteins that are present in ocular basement membranes through development.46 Collagen processing enzymes, procollagen lysyl hydroxylases, which hydroxylate lysines in collagens, and thus creating carbohydrate binding sites and promoting the stability of the collagen network, have been demonstrated to be PITX2 transcriptional targets,40 and it is possible that the pathogenic pathways are shared and that the pathogenic effects of COL4A1 and COL4A2 mutations are genetically downstream of PITX2 and FOXC1. Alternatively, extracellular matrix molecules can act as instructive signals or regulators for many developmental processes and it is possible that COL4A1 and COL4A2 are genetically upstream of PITX2 and FOXC1. Furthermore, multiple additional extracellular matrix proteins and their modifying enzymes also contribute to ASD in mice and patients, including FBN2,47 LAMB2,48 heparin sulfate proteoglycans COL18,49 AGRN,50 and heparan sulfate synthesize enzymes.50,51 Perhaps the most relevant is the recent identification that mutations in peroxidasin, a key enzyme in extracellular organization of COL4A1 and COL4A2, cause congenital cataracts, ASD and developmental glaucoma in patients52,53 and in mice.54 The extents to which the pathogenic mechanisms involved in these cases are distinct or overlapping need to be explored.
The partial and nearly complete suppression of ASD and glaucoma in 129B6F1 and CASTB6F1 mice, respectively, demonstrates reproducible differential effects of the two backgrounds. One possibility is that both inbred strains carry the same genetic modifier but that CAST has additional properties that confer more profound suppression. A second possibility is that each strain has a different modifier locus (or loci). Interestingly, we have recently shown that the CASTB6F1 background suppresses intracerebral hemorrhages yet 129B6F1 had no effect,25 indicating that the capacity for genetic modification between the two strains differs for this particular phenotype. Current efforts are underway to identify the pathway(s) by which the genetic modifier(s) suppress pathology. Once identified, we can test the relevance of this pathway to ASD in other matrix and matrix-associated genes. Moreover, pharmacologic manipulation of this same pathway may represent a therapeutic alternative to prevent vision loss in ASD patients that do not respond to current treatments.
Supplementary Material
Acknowledgments
The authors thank Gould lab members for helpful discussions with the manuscript. The authors also thank Kendall Hoff and Tanav Popli for maintaining the mouse colonies. The authors also thank other members of the John lab for technical assistance especially with mouse colonies and physiology.
Supported by National Institute of Health (Bethesda, MD, USA) Grants EY019887 (DBG), EY11721 (SWMJ), P30EY002162, Research to Prevent Blindness (DBG and RTL; New York, NY, USA), Karl Kirchgessner Foundation (DBG; Redondo Beach, CA, USA), That Man May See (MM and DBG; San Francisco, CA, USA), and Knights Templar Eye Foundation (MM; Flower Mound, TX, USA).
Disclosure: M. Mao, None; R.S. Smith, None; M.V. Alavi, None; J.K. Marchant, None; M. Cosma, None; R.T. Libby, None; S.W.M. John, None; D.B. Gould, None
References
- 1. Quigley HA,, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006; 90: 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Quigley HA. Glaucoma. Lancet. 2011; 377: 1367–1377. [DOI] [PubMed] [Google Scholar]
- 3. Strungaru MH,, Dinu I,, Walter MA. Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Invest Ophthalmol Vis Sci. 2007; 48: 228–237. [DOI] [PubMed] [Google Scholar]
- 4. Reis LM,, Semina EV. Genetics of anterior segment dysgenesis disorders. Curr Opin Ophthalmol. 2011; 22: 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuo DS,, Labelle-Dumais C,, Gould DB. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum Mol Genet. 2012; 21: R97 –R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meuwissen ME,, Halley DJ,, Smit LS,, et al. The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature [published online ahead of print February 26, 2015]. Genet Med. doi:http://dx.doi.org/10.1038/gim.2014.210. [DOI] [PubMed]
- 7. Van Agtmael T,, Schlotzer-Schrehardt U,, McKie L,, et al. Dominant mutations of Col4a1 result in basement membrane defects which lead to anterior segment dysgenesis and glomerulopathy. Hum Mol Genet. 2005; 14: 3161–3168. [DOI] [PubMed] [Google Scholar]
- 8. Favor J,, Gloeckner CJ,, Janik D,, et al. Type IV procollagen missense mutations associated with defects of the eye, vascular stability, the brain, kidney function and embryonic or postnatal viability in the mouse, Mus musculus: an extension of the Col4a1 allelic series and the identification of the first two Col4a2 mutant alleles. Genetics. 2007; 175: 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gould DB,, Marchant JK,, Savinova OV,, Smith RS,, John SW. Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum Mol Genet. 2007; 16: 798–807. [DOI] [PubMed] [Google Scholar]
- 10. Gould DB,, Phalan FC,, Breedveld GJ,, et al. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005; 308: 1167–1171. [DOI] [PubMed] [Google Scholar]
- 11. Poschl E,, Schlotzer-Schrehardt U,, Brachvogel B,, Saito K,, Ninomiya Y,, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004; 131: 1619–1628. [DOI] [PubMed] [Google Scholar]
- 12. Coupry I,, Sibon I,, Mortemousque B,, Rouanet F,, Mine M,, Goizet C. Ophthalmological features associated with COL4A1 mutations. Arch Ophthalmol. 2010; 128: 483–489. [DOI] [PubMed] [Google Scholar]
- 13. Shah S,, Kumar Y,, McLean B,, et al. A dominantly inherited mutation in collagen IV A1 (COL4A1) causing childhood onset stroke without porencephaly. Eur J Paediatr Neurol. 2010; 14: 182–187. [DOI] [PubMed] [Google Scholar]
- 14. Rouaud T,, Labauge P,, Tournier Lasserve E,, et al. Acute urinary retention due to a novel collagen COL4A1 mutation. Neurology. 2010; 75: 747–749. [DOI] [PubMed] [Google Scholar]
- 15. Yoneda Y,, Haginoya K,, Kato M,, et al. Phenotypic spectrum of COL4A1 mutations: porencephaly to schizencephaly. Ann Neurol. 2013; 73: 48–57. [DOI] [PubMed] [Google Scholar]
- 16. Shah S,, Ellard S,, Kneen R,, et al. Childhood presentation of COL4A1 mutations. Dev Med Child Neurol. 2012; 54: 569–574. [DOI] [PubMed] [Google Scholar]
- 17. Rodahl E,, Knappskog PM,, Majewski J,, et al. Variants of anterior segment dysgenesis and cerebral involvement in a large family with a novel COL4A1 mutation. Am J Ophthalmol. 2013; 155: 946–953. [DOI] [PubMed] [Google Scholar]
- 18. Decio A,, Tonduti D,, Pichiecchio A,, et al. A novel mutation in COL4A1 gene: a possible cause of early postnatal cerebrovascular events. Am J Med Genet A. 2015; 167: 810–815. [DOI] [PubMed] [Google Scholar]
- 19. Xia XY,, Li N,, Cao X,, et al. A novel COL4A1 gene mutation results in autosomal dominant non-syndromic congenital cataract in a Chinese family. BMC Med Genet. 2014; 15: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deml B,, Reis LM,, Maheshwari M,, Griffis C,, Bick D,, Semina EV. Whole exome analysis identifies dominant COL4A1 mutations in patients with complex ocular phenotypes involving microphthalmia. Clin Genet. 2014; 86: 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Colin E,, Sentilhes L,, Sarfati A,, et al. Fetal intracerebral hemorrhage and cataract: think COL4A1. J Perinatol. 2014; 34: 75–77. [DOI] [PubMed] [Google Scholar]
- 22. Slavotinek AM,, Garcia ST,, Chandratillake G,, et al. Exome sequencing in 32 patients with anophthalmia/microphthalmia and developmental eye defects [published online ahead of print December 2, 2014]. Clin Genet. doi:http://dx.doi.org/10.1111/cge.12543. [DOI] [PMC free article] [PubMed]
- 23. Livingston J,, Doherty D,, Orcesi S,, et al. COL4A1 mutations associated with a characteristic pattern of intracranial calcification. Neuropediatrics. 2011; 42: 227–233. [DOI] [PubMed] [Google Scholar]
- 24. Kuo DS,, Labelle-Dumais C,, Mao M,, et al. Allelic heterogeneity contributes to variability in ocular dysgenesis, myopathy and brain malformations caused by Col4a1 and Col4a2 mutations. Hum Mol Genet. 2014; 23: 1709–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jeanne M,, Jorgensen J,, Gould DB. Molecular and genetic analysis of collagen type IV mutant mouse models of spontaneous intracerebral hemorrhage identify mechanisms for stroke prevention [published online ahead of print March 9 2015]. Circulation. doi:http://dx.doi.org/10.1161/CIRCULATIONAHA.114.013395. [DOI] [PMC free article] [PubMed]
- 26. Keane TM,, Goodstadt L,, Danecek P,, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011; 477: 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. John SW,, Hagaman JR,, MacTaggart TE,, Peng L,, Smithes O. Intraocular pressure in inbred mouse strains. Invest Ophthalmol Vis Sci. 1997; 38: 249–253. [PubMed] [Google Scholar]
- 28. Savinova OV,, Sugiyama F,, Martin JE,, et al. Intraocular pressure in genetically distinct mice: an update and strain survey. BMC Genet. 2001; 2: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Libby RT,, Smith RS,, Savinova OV,, et al. Modification of ocular defects in mouse developmental glaucoma models by tyrosinase. Science. 2003; 299: 1578–1581. [DOI] [PubMed] [Google Scholar]
- 30. Anderson MG,, Libby RT,, Mao M,, et al. Genetic context determines susceptibility to intraocular pressure elevation in a mouse pigmentary glaucoma. BMC Biol. 2006; 4: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anderson MG,, Libby RT,, Gould DB,, Smith RS,, John SW. High-dose radiation with bone marrow transfer prevents neurodegeneration in an inherited glaucoma. Proc Natl Acad Sci U S A. 2005; 102: 4566–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Labelle-Dumais C,, Dilworth DJ,, Harrington EP,, et al. COL4A1 mutations cause ocular dysgenesis, neuronal localization defects, and myopathy in mice and Walker-Warburg syndrome in humans. PLoS Genet. 2011; 7: e1002062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Acharya M,, Huang L,, Fleisch VC,, Allison WT,, Walter MA. A complex regulatory network of transcription factors critical for ocular development and disease. Hum Mol Genet. 2011; 20: 1610–1624. [DOI] [PubMed] [Google Scholar]
- 34. Acharya M,, Sharp MW,, Mirzayans F,, et al. Yeast two-hybrid analysis of a human trabecular meshwork cDNA library identified EFEMP2 as a novel PITX2 interacting protein. Mol Vis. 2012; 18: 2182–2189. [PMC free article] [PubMed] [Google Scholar]
- 35. Huang L,, Chi J,, Berry FB,, Footz TK,, Sharp MW,, Walter MA. Human p32 is a novel FOXC1-interacting protein that regulates FOXC1 transcriptional activity in ocular cells. Invest Ophthalmol Vis Sci. 2008; 49: 5243–5249. [DOI] [PubMed] [Google Scholar]
- 36. Huang Y,, Huang K,, Boskovic G,, et al. Proteomic and genomic analysis of PITX2 interacting and regulating networks. FEBS Lett. 2009; 583: 638–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tamimi Y,, Lines M,, Coca-Prados M,, Walter MA. Identification of target genes regulated by FOXC1 using nickel agarose-based chromatin enrichment. Invest Ophthalmol Vis Sci. 2004; 45: 3904–3913. [DOI] [PubMed] [Google Scholar]
- 38. Berry FB,, Skarie JM,, Mirzayans F,, et al. FOXC1 is required for cell viability and resistance to oxidative stress in the eye through the transcriptional regulation of FOXO1A. Hum Mol Genet. 2008; 17: 490–505. [DOI] [PubMed] [Google Scholar]
- 39. Paylakhi SH,, Moazzeni H,, Yazdani S,, et al. FOXC1 in human trabecular meshwork cells is involved in regulatory pathway that includes miR-204, MEIS2, and ITGbeta1. Exp Eye Res. 2013; 111: 112–121. [DOI] [PubMed] [Google Scholar]
- 40. Hjalt TA,, Amendt BA,, Murray JC. PITX2 regulates procollagen lysyl hydroxylase (PLOD) gene expression: implications for the pathology of Rieger syndrome. J Cell Biol. 2001; 152: 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zacharias AL,, Gage PJ. Canonical Wnt/beta-catenin signaling is required for maintenance but not activation of Pitx2 expression in neural crest during eye development. Dev Dyn. 2010; 239: 3215–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Strungaru MH,, Footz T,, Liu Y,, et al. PITX2 is involved in stress response in cultured human trabecular meshwork cells through regulation of SLC13A3. Invest Ophthalmol Vis Sci. 2011; 52: 7625–7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hjalt TA,, Semina EV,, Amendt BA,, Murray JC. The Pitx2 protein in mouse development. Dev Dyn. 2000; 218: 195–200. [DOI] [PubMed] [Google Scholar]
- 44. Gage PJ,, Rhoades W,, Prucka SK,, Hjalt T. Fate maps of neural crest and mesoderm in the mammalian eye. Invest Ophthalmol Vis Sci. 2005; 46: 4200–4208. [DOI] [PubMed] [Google Scholar]
- 45. Berry FB,, Lines MA,, Oas JM,, et al. Functional interactions between FOXC1 and PITX2 underlie the sensitivity to FOXC1 gene dose in Axenfeld-Rieger syndrome and anterior segment dysgenesis. Hum Mol Genet. 2006; 15: 905–919. [DOI] [PubMed] [Google Scholar]
- 46. Bai X,, Dilworth DJ,, Weng YC,, Gould DB. Developmental distribution of collagen IV isoforms and relevance to ocular diseases. Matrix Biol. 2009; 28: 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shi Y,, Tu Y,, Mecham RP,, Bassnett S. Ocular phenotype of Fbn2-null mice. Invest Ophthalmol Vis Sci. 2013; 54: 7163–7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zenker M,, Aigner T,, Wendler O,, et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet. 2004; 13: 2625–2632. [DOI] [PubMed] [Google Scholar]
- 49. Ylikarppa R,, Eklund L,, Sormunen R,, et al. Lack of type XVIII collagen results in anterior ocular defects. FASEB J. 2003; 17: 2257–2259. [DOI] [PubMed] [Google Scholar]
- 50. Fuerst PG,, Rauch SM,, Burgess RW. Defects in eye development in transgenic mice overexpressing the heparan sulfate proteoglycan agrin. Dev Biol. 2007; 303: 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Iwao K,, Inatani M,, Matsumoto Y,, et al. Heparan sulfate deficiency leads to Peters anomaly in mice by disturbing neural crest TGF-beta2 signaling. J Clin Invest. 2009; 119: 1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khan K,, Rudkin A,, Parry DA,, et al. Homozygous mutations in PXDN cause congenital cataract, corneal opacity, and developmental glaucoma. Am J Hum Genet. 2011; 89: 464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Choi A,, Lao R,, Ling-Fung Tang P,, et al. Novel mutations in PXDN cause microphthalmia and anterior segment dysgenesis. Eur J Hum Genet. 2015; 23: 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yan X,, Sabrautzki S,, Horsch M,, et al. Peroxidasin is essential for eye development in the mouse. Hum Mol Genet. 2014; 23: 5597–5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.