

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is expected to become the second leading cause of cancer death by 2030. Current therapeutic options are limited, warranting an urgent need to explore innovative treatment strategies. Due to specific microenvironment constraints including an extensive desmoplastic stroma reaction, PDAC faces major metabolic challenges, principally hypoxia and nutrient deprivation. Their connection with oncogenic alterations such as KRAS mutations has brought metabolic reprogramming to the forefront of PDAC therapeutic research. The Warburg effect, glutamine addiction, and autophagy stand as the most important adaptive metabolic mechanisms of cancer cells themselves, however metabolic reprogramming is also an important feature of the tumor microenvironment, having a major impact on epigenetic reprogramming and tumor cell interactions with its complex stroma. We present a comprehensive overview of the main metabolic adaptations contributing to PDAC development and progression. A review of current and future therapies targeting this range of metabolic pathways is provided.
Keywords: glycolysis, warburg effect, metformin, glutamine, hypoxia
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is currently the fifth leading cause of cancer death and the second leading digestive cancer in incidence in Western countries [1]. By 2030, it is expected to be the second leading cause of cancer death [2]. PDAC is considered to be the tumor with the worst prognosis among all digestive malignancies, with a 5-year survival rate of less than 5% [1, 3].
PDAC are highly invasive tumors with early metastatic potential, for which therapeutic options are limited [4]. Gemcitabine has been the reference chemotherapy regimen since 1997. In 2011, the FOLFIRINOX regimen combining 5-fluorouracil, leucovorin, oxaliplatin, and irinotecan was shown to be superior to gemcitabine (median overall survival [OS]: 11.1 versus 6.8 months, p < 0.001) in selected patients; those with a performance status 0–1 and absence of cholestasis [5]. In 2013, the combination of gemcitabine with nanoparticles of albumin-bound paclitaxel (nab-paclitaxel) demonstrated a statistically significant increase in OS compared with gemcitabine alone (median OS: 8.5 versus 6.7 months, p < 0.001) [6]. Nonetheless, despite these encouraging improvements, overall prognosis in this patient population remains dismal and new therapeutic approaches are urgently needed.
Cancer cells need large amounts of both energy (adenosine triphosphate [ATP]) and macromolecules to sustain their proliferation. As a hallmark of cancer, metabolism reprogramming highlights the fact that changes in cell metabolism are necessary for tumor initiation and progression. Both oncogenes and the tumor microenvironment are involved in this process [7–11]. PDAC displays one of the most extensive and poorly vascularized desmoplastic stromal reactions of all carcinomas, leading to tumor hypoxia and nutrient deprivation, yet without evidence of major cell death. Taken together, this suggests that pancreatic tumor cells adapt to metabolically challenging survival conditions in their microenvironment [12]. Targeting PDAC-specific metabolic pathways thus represents a novel strategy to explore for the development of innovative therapies.
In this review, we provide a comprehensive overview of the metabolic deregulations in PDAC and their supportive role in tumor development and progression, and then focus on crucial metabolic nodes that could be leveraged in future therapeutic strategies.
METABOLIC ADAPTIVE MECHANISMS
PDACs are characterized by a prominent desmoplastic stromal reaction, and the extent of the stroma is often greater than the epithelial component of the tumor (up to 80% of tumor volume) [13–15]. Activated pancreatic stellate cells (PSC) are responsible for the excessive production of extracellular matrix [16–18]. The resulting dense and fibrotic stroma compresses vessels and generates high interstitial pressure thereby limiting tumor vascularization. As a consequence, tumor cells are confronted with hypoxia and nutrient deprivation [19, 20].
Hypoxia is a typical feature of PDAC and is associated with poor prognosis [19, 21–27]. Preclinical studies in PDAC models showed that hypoxia increases cancer cell proliferation, survival, epithelial-to-mesenchymal transition (EMT), invasiveness, and metastasis, as well as resistance to chemotherapy and radiotherapy, through hypoxia-inducible factor (HIF)-1α -dependent and -independent mechanisms [25, 26, 28–36].
Cells in hypovascularized PDAC have to adapt to their metabolically challenging environment early in tumor development. Several changes occur in response to oxygen deprivation: increased glycolysis as well as increased amino acid (AA) production derived from protein degradation, protein glycosylation, and fatty acid synthesis. In addition recycling and scavenging of cellular components has been shown to be applicable in PDAC. This early adaptive mechanism is known as the “metabolic switch” and is described in detail below [Figure 1].
Figure 1. Overview of PDAC cell metabolism in response to microenvironment constraints and oncogenic signals.
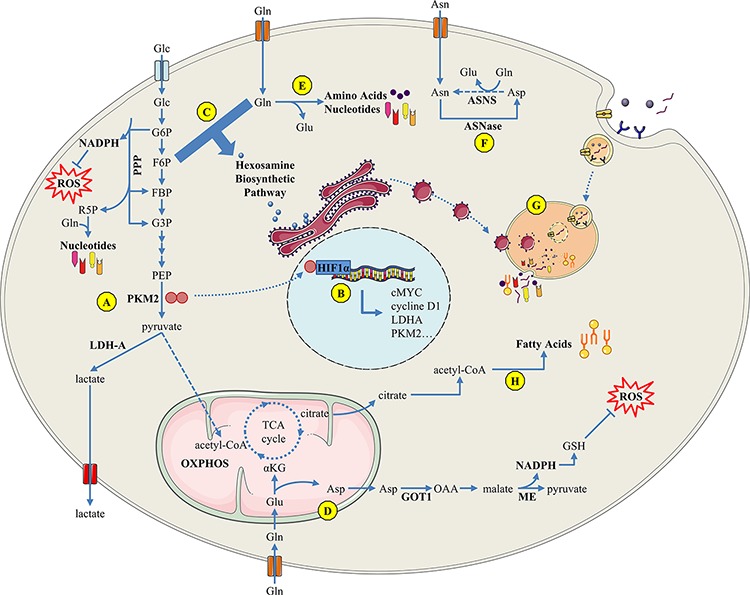
A. The Warburg effect sustains metabolic needs of PDAC proliferative cells; B. The PKM2 tyrosine kinase enhances transcriptional activity of several factors such as hypoxia-inducible factor HIF1-α, inducing the Warburg effect through a positive feedback loop; C. the hexosamine biosynthetic pathway uses glucose and glutamine influx for protein O-GlcNAc glycosylation and its inhibition induces an unfolded-protein response-dependent cell death; D. PDAC-specific glutamine metabolism: glutamine-derived aspartate is converted into oxaloacetate, then into malate, and finally into pyruvate, resulting in an increased NADPH/NADP+ ratio that provides the reducing power to maintain reduced glutathione pools; E. glutamine is a nitrogen donor for amino acid and nucleotide biosynthesis; F. ASNase may be a promising therapy since a majority of PDAC express no or low ASNS; G. macropinocytosis and autophagy support the metabolic needs of PDAC cells; H. PDAC overexpresses enzymes involved in fatty acid synthesis. Glc : glucose; Gln: glutamine; Glu : glutamate; Asn : asparagine; ASNase : asparaginase; ASNS : asparagine synthetase; GSH : glutathion; LDH-A : lactate dehydrogenase-A; ME : malic enzyme; NADP : nicotinamide adenine dinucleotide phosphate; OXPHOS : oxidative phosphorylation; PKM : pyruvate kinase muscle-isozyme.
Glycolysis and the Warburg effect
In the presence of oxygen, normal cells produce ATP from glucose-derived pyruvate by oxidative phosphorylation (OXPHOS) via the mitochondrial tricarboxylic acid (TCA) cycle. In the 1920s, Otto Warburg observed that some proliferative tissues, notably tumor cells, display increased glucose uptake and preferentially metabolize glucose-derived pyruvate to lactate even in the presence of oxygen [37–39]. This phenomenon of aerobic glycolysis is also known as the “Warburg effect”.
The glycolytic switch is an early phenomenon characterized by increased expression of lactate dehydrogenase (LDH, that converts pyruvate into lactate) and inactivation of pyruvate dehydrogenase (PDH, that converts pyruvate into acetyl-CoA for the TCA cycle) [40–46]. The glycolytic switch is thought to be driven by the hypoxic tumor microenvironment through HIF-1α activation, aberrant signaling due to oncogene activation (e.g., Ras, PI3K/mTOR, c-Myc), tumor suppressor gene inactivation (e.g., p53), or by mutations in the OXPHOS pathway [47, 48].
In preclinical models, hypoxic PDAC cells overexpress glycolytic markers. Constitutively activated K-Ras, present in more than 90% of PDAC, has a key role in metabolic reprogramming and particularly in the glycolytic switch [49–52]. Gene expression and metabolic flux analyses showed oncogenic KRAS upregulates expression of glucose transporter (GLUT)-1 (increasing glucose influx) and of the hexokinase (HK) 1–2 and phosphofructokinase enzymes, which speed up glycolytic activity. Oncogenic KRAS also supports biomass synthesis (i.e. proteins, nucleic acids etc.) required for cancer cell proliferation by rewiring glucose toward anabolic pathways, such as the pentose phosphate pathway (PPP), while maintaining a low level of reactive oxygen species (ROS) by limiting ROS production and ROS-related apoptosis [53]. TP53 loss-of-function (50% of PDAC) also contributes to the glycolytic switch through deregulation of GLUT1 and GLUT4 transcription and loss of expression of TIGAR (TP53-inductible glycolytic and apoptotic regulator) which acts as a fructose-2, 6-biphosphatase (FBP-ase) [54, 55]. Although the physiological substrate of TIGAR remains controversial, when silenced, FBP levels increase enhancing pyruvate kinase (PKM) glycolytic activity [56, 57]. Interestingly, genetic mutations may be a consequence of metabolic stress, such as glucose deprivation, dynamically interconnecting oncogenic and metabolic alterations [58].
The glycolytic switch also mediates interconnections between tumor compartments [45]. Far from being a waste product of the Warburg effect, lactate may be an important vector for tumor-stroma interactions and symbiotic spatial energy fuel exchange between cell compartments within the tumor [59]. Lactate produced by hypoxic cancer cells can diffuse to the extracellular environment through lactate transporter MCT-4 and be taken up by normoxic cancer cells through MCT-1 to be used for oxidative metabolism, thereby sparing glucose for hypoxic cancer cells [34, 40]. Lactate also “feeds” stromal cells providing a fuel source for OXPHOS [60].
Moreover, acidification of the microenvironment by lactic acid contributes to pro-tumor immunologic remodeling by promoting chronic inflammation, while suppressing T-cell mediated adaptive immune response [61–63]. Lactate-dependent interleukin-17 and interleukin-23 production can induce an inflammatory tumor environment that will result in the attraction of pro-tumoral immune cells [64–68]. Thus, the end products of the Warburg effect participate in the inter-compartment dialogue and symbiosis within PDAC and generate a favorable immunologic microenvironment for cancer cells. Not surprisingly, high lactate concentrations and acidic pH, representative of “glycolytic tumors”, has been associated with poor prognosis and a more aggressive phenotype [69, 70].
Responding to amino acid deprivation
PDAC cells also face AA shortage, which can have a critical impact on cell survival especially for essential AA. It has been suggested that the increased AA requirement for cancer cells is a very early phenomenon in tumor development and that metabolic reprogramming to provide cancer cells with branched-chain AA (BCAA) precedes PDAC diagnosis by about 5 years [71]. Mayers et al. showed that elevated plasma levels of all three proteinogenic, essential, BCAA (isoleucine, leucine and valine) are associated with future diagnosis of PDAC. BCAA elevations are derived from a long-term pool of AA of muscular origin. This study reveals that protein breakdown clearly predates PDAC diagnosis and clinical cachexia. The mechanisms underlying this protein breakdown are still under investigation.
Although glutamine is a non-essential AA, most cancer cells exhibit glutamine addiction [72, 73]. The metabolic fate of glutamine is multifaceted; it can be used for lipid biosynthesis, as a nitrogen donor for AA and nucleotide biosynthesis, as a carbonic substrate for the re-feeding of the mitochondrial TCA cycle through a phenomenon called anaplerosis, and even as fuel for cell energy production [74–76]. PDAC cells metabolize glutamine through a non-canonical pathway in which transaminases play a crucial role. Whereas most cells use glutamate dehydrogenase (GDH-1) to convert glutamine-derived glutamate into α-ketoglutarate in the mitochondria to fuel the TCA cycle, PDAC relies on a distinct pathway in which glutamine-derived aspartate is transported into the cytoplasm where it can be converted into oxaloacetate by aspartate transaminase (i.e. glutamic-oxaloacetic transaminase [GOT-1]), then into malate, and finally into pyruvate. Conversion of malate to pyruvate by malic enzyme results in an increased NADPH/NADP+ ratio (nicotinamide adenine dinucleotide phosphate), providing the reducing power to maintain reduced glutathione pools to protect cells against oxidative damage [77]. Low expression of GDH-1 and overexpression of glutaminase, GOT-1, and enzymes using glutamine as a nitrogen donor (cytidine triphosphate synthase, guanine monophosphate synthetase, asparagine synthetase) are characteristic features of PDAC [42, 77]. In these tumors, transcriptional reprogramming of key metabolic enzymes in the glutamine pathway (e.g. GDH-1, GOT1) is driven by KRAS or MYC oncogenes [77–79]. Thus, more than an anaplerotic precursor for the TCA cycle, glutamine is necessary to sustain PDAC cell growth required for biomass synthesis and maintenance of the redox balance.
Glucose deprivation has been shown to induce the expression of asparagine synthetase (ASNS) probably through the unfolded-protein response (UPR) pathway as a means to protect cells from apoptosis [80, 81]. However, in contrast to normal pancreatic tissue that expresses high levels of ASNS, approximately half of PDAC cells express no or low ASNS levels [82]. These tumors may thus harbor an intrinsic fragility to asparagine deprivation that may be exploited therapeutically by L-asparaginase therapy [83].
Upregulation of the hexosamine biosynthetic pathway
The hexosamine biosynthetic pathway (HBP) is responsible for N-acetylglucosamine (GlcNAc) production for protein O-GlcNAc glycosylation. Glucosamine-fructose-6-phosphate aminotransferase (GFPT) uses glutamine as a substrate to convert fructose-6-phosphate into glucosamine-6-phosphate, which is one of the precursors for UDP-GlcNAc synthesis and O-GlcNAc glycosylation. HBP activity thus depends on both glutamine as well as glucose (which is converted into fructose-6-phosphate). PDAC cells exhibit high levels of O-GlcNAc glycosylated proteins due to upregulation of GFPT1, GFPT2, and O-GlcNAc-transferase, and low levels of O-GlcNAcase, the enzyme catalyzing deglycosylation [84, 85]. Increased glucose and glutamine uptake and KRAS-dependent upregulation of GFPT, the rate-limiting enzyme in this process, result in increased HBP activity in PDAC, which has been associated with tumor invasion and metastasis [34, 53, 86].
O-GlcNAc glycosylation can redirect glucose to the PPP by inhibiting phosphofructokinase-1 and stabilizes key transcription factors such as p53, c-Myc or β-catenin [87–89]. It also promotes aneuploidy and participates in cancer cell phenotype by enhancing insulin, TGF-β, and FGF pathway activity through transcriptional and epigenetic mechanisms [90, 91]. In addition, HBP can modulate tyrosine kinase receptor (TKR) signaling [92]. HBP inhibition using tunicamycin (a nucleoside antibiotic that blocks GlcNAc-1-phosphotransferase) in PDAC, resulted in decreased protein levels and membrane expression of several TKR such as EGFR (epidermal growth factor receptor), ErbB2, ErbB3, and IGFR (insulin-like growth factor receptor) [93]. Of note, glucose deprivation reduces HBP activity, which decreases protein glycosylation and induces UPR-dependent cell death [94]. The metabolic switch induced by HBP is thus at the crossroads between growth factor survival and microenvironment signaling and may represent an innovative approach in cancer therapy.
Activation of lipid metabolism
Fatty acid (FA) synthesis occurs at a low level in most normal tissues, with the exception of liver and adipose tissues. However in cancer cells, FA are synthesized at high levels and undergo esterification, mainly providing phospholipids for membrane formation. PDAC cells overexpress enzymes involved in FA and cholesterol synthesis such as FA synthase (FAS) and ATP citrate lyase, while levels of several enzymes involved in FA β-oxidation in mitochondria are reduced [42]. FA synthesis requires NADPH that is produced in PDAC cells either by the KRAS-activated PPP or by malic enzyme during glutaminolysis. Overexpression of FAS in PDAC is associated with poor prognosis [95]. As reviewed by Swierczinski et al. [96], the oncogenic potential of FAS exploits several mechanisms; FAS expression is strongly induced by hypoxia, the PI3K/AKT/mTOR pathway through activation of SREBP1c transcription factor, and by microenvironment acidification through epigenetic modifications of the FAS promoter [97–99].
In cancer cells, activation of de novo lipogenesis induces an excess of monounsaturated lipids (which are less susceptible to lipid peroxidation than polyunsaturated) in cell membranes, increasing the resistance of cancer cells to oxidative stress [100]. Besides, plasma membranes exhibit specific subdomains, named lipid rafts, which are enriched in sphingolipids and cholesterol. Caveolae, a type of lipid raft, are principally composed of caveolin-1, which is deregulated in several human malignancies including PDAC [101, 102]. Interestingly, co-expression of caveolin-1 and FAS correlates significantly with poor clinical features and reduced survival in PDAC patients suggesting that these proteins are potential therapeutic targets in this indication [103]. Moreover, these lipid rafts are essential in cancer cell signaling processes, forming platforms for growth-factor receptors [104].
Recent work of Guillaumond et al. revealed cholesterol uptake and more specifically low-density lipoprotein receptor (LDLR) as a highly attractive target for PDAC metabolic therapy. They showed that lipoprotein catabolism and cholesterol synthesis pathways are enriched in PDAC, compared with nonmalignant pancreas [105]. This increase in tumor cell cholesterol content is consistent with the increased of lipid raft levels observed in cancer cells. Interestingly, cholesterol level of lipid rafts has been shown to modulate EGFR-dependent survival pathway [106]. Cholesterol uptake disruption through shRNA silencing of LDLR inhibit proliferation and ERK1/2 pathway activation of PDAC cells [105].
Autophagy and pinocytosis
Recycling and scavenging are often necessary for cancer cells to sustain their biomass needs. Macroautophagy is a catabolic process that consists of degrading macromolecular complexes and cytoplasmic organelles into AA, lipids, and nucleosides that are then recycled. Autophagy is triggered by nutrient shortage, protein damage, or by oxidative stress occurring through inhibition of the AMP kinase (AMPK) and mTOR pathways, and by activation of UPR [107–109].
The role of autophagy in cancer progression has been controversial, and both pro- and anti-tumorigenic effects have been described [110, 111]. In most cases, PDACs exhibit basal autophagy activity [112]. Rosenfeldt et al. recently provided new insight into this complex issue, bringing to light the role of p53 in the process [113]. In mouse models of PDAC, inhibition of autophagy blocked KRAS tumorigenicity in a wild type TP53 background, but favored pancreatic intraepithelial neoplastic (PanIN) transformation into invasive PDAC in the context of a coexisting oncogenic KRAS mutation and TP53 deletion. In tumors with intact p53, autophagy inhibition resulted in decreased metabolism activity, whereas in tumors with loss of p53 function (embryonic homozygous TP53 deletion), it induced an increase in glucose consumption for anabolic pathway activity, fueling cancer cell proliferation. PDAC cell dependence on autophagy may thus vary according to the genetic background of the tumor. However, more recently, using an alternative mouse model with stochastic loss of heterozygosity of TP53, tumor cell lines, and genetically-characterized patient-derived xenografts, Yang A. et al. [114] showed that p53 status does not seem to affect response to autophagy inhibition. These findings have important implications on ongoing clinical trials.
Cancer cells are also able to absorb and degrade extracellular components through an endocytic process called macropinocytosis. KRAS-dependent upregulation of macropinocytosis contributes to the metabolic needs of PDAC cell lines, with macropinocytosis inhibition shown to reduce KRAS-transformed cell growth [115, 116].
TARGETING METABOLISM IN PANCREATIC CANCER
Activating KRAS mutations in PDAC are acknowledged to be a major driver of carcinogenesis; however, to date they have proven to be poorly druggable targets. Addressing downstream metabolic alterations may circumvent this allowing inhibition of tumor growth in PDAC, as suggested by preliminary data [117, 118].
Blocking the heart of the glycolytic switch via PKM2
Pyruvate kinase controls the penultimate step of glycolysis, catalyzing the production of pyruvate and ATP from phosphoenopyruvate (PEP) and adenosine 5′-diphosphate (ADP), putting PKM2 at the core of the glycolytic switch in cancer cells [Text Box 1]. This enzyme has several isoforms (M1, M2, L, R), with PKM1 and PKM2 resulting from an alternative splicing of the same pre-mRNA. PKM2 is found in several tissues (liver, lung, pancreatic islets, and retina) and is preferentially expressed over PKM1 in cancer cells through cMyc-dependent splicing modulation [119].
Box 1. PKM2 at the core of the glycolytic switch in cancer cells.
PKM2 glycolytic activity is regulated by different mechanisms, including allosteric and post-translational modifications [163–166]. PKM2 is present as either active tetramers or inactive dimers. In cancer cells, it is predominantly found in dimers with low activity. Active tetramers induce OXPHOS whereas inactive dimers favor cytoplasmic conversion of pyruvate into lactate by LDH-A [122]. The low glycolytic activity of PKM2 dimers allows upstream glycolytic metabolite accumulation and their redirection towards anabolic pathways (for review, see [167]).
Furthermore, monomeric PKM2 can translocate into the nucleus and acts as a co-transcription factor. Activation of the EGFR pathway promotes PKM2 nuclear translocation via EGFR-activated ERK1/2 which directly binds and phosphorylates PKM2 on Ser37, resulting in its nuclear translocation and activation, without any effect on PKM1 [121, 168]. Through a positive feedback loop, PKM2 binding to succinyl-5-aminoimidazole-4-carboxamide-1-ribose-5′-phosphate (SAICAR), an intermediate of the de novo purine nucleotide biosynthesis that is abundant in proliferative cells, leads to phosphorylation and activation of ERK1/2 [169]. In the nucleus, PKM2 interacts with nuclear HIF1-α and p300 to induce transcription of hypoxia-responsive genes (e.g. anaerobic glycolysis genes). PKM2 also binds to β-catenin and promotes expression of pro-proliferative MYC and CCDN1 genes. In addition, PKM2 interacts with STAT3 and histone H3 whose phosphorylation on threonine 11 depends on EGFR activation and is required for the dissociation of HDAC3 from the CCND1 and MYC promoter regions [170, 171]. As PKM gene expression is modulated by c-Myc, STAT3, β-catenin, and HIF1-α, and PKM alternative splicing is under c-Myc control, the kinase activity of PKM2 induces a positive feedback loop that globally enhances the glycolytic phenotype of cancer cells and plays a crucial role in cancer cell metabolism reprogramming [172].
As the dimer/tetramer status of PKM2 drives pyruvate fate towards OXPHOS or lactate production, targeting PKM2 by constraining its conformation may have therapeutic potential. The inactive dimer being the main PKM2 form in tumors, allosteric activators maintaining PKM2 in its highly active tetrameric form could inhibit cancer cell growth without toxicity since active tetramers are the form present in normal tissues [120]. These activators may in fact prevent the accumulation of glycolytic intermediates and their rewiring into anabolic pathways that are crucial for biomass synthesis of highly proliferative cells. Moreover, these compounds might prevent PKM2 nuclear translocation and the positive feedback loop with ERK proteins that enhance the Warburg effect [121]. This might be particularly relevant in PDAC, which are characterized by activation of the MAP kinase pathway downstream of constitutively activated oncogenic KRAS. Several PKM2 inhibitors that effectively inhibit cancer cell growth in vitro have already been identified, a number of which merit evaluation in PDAC [122].
Addressing glycolysis via LDH-A
LDH controls the rate-limiting final step of glycolysis, converting pyruvate into lactate in the cytoplasm. LDH activity is not required in normal tissues under normoxic conditions. The two LDH isoforms (LDH-A and -B) can be combined as five different tetramers (LDH-1–5). LDH-A is predominantly expressed in the liver and muscles and LDH-B in the myocardia. LDH-5 is composed of four LDH-A units which is overexpressed in many cancers including PDAC as a result of post-translational or transcriptional c-Myc, K-Ras, HIF-1α, and FOXM1 (forkhead box protein M1) dependent regulation, and is associated with poor prognosis [43, 123–126].
In vitro and in vivo constitutive expression of LDH-A enhances cell growth while its silencing decreases tumorigenicity of PDAC cells [43]. Several LDH-5 inhibitors are in preclinical development but their efficacy in vivo is limited by their pharmacokinetic profile (short half-life) warranting optimization of their structure/stability and/or administration modalities. Interestingly, an LDH-A genetic deficiency causes myopathy only after major physical effort and individuals carrying this anomaly are healthy, suggesting that LDH-5 inhibition would present limited toxicities.
Blocking lactate transport
Lactate efflux plays a critical role in intracellular pH regulation and in tumor-stroma interactions contributing to cancer cell invasiveness and immune escape. Lactate transport occurs via monocarbonate transporters (MCT): MCT-4 for lactate efflux of highly glycolytic cells, and MCT-1 for lactate import into cells that use lactate as an oxidative combustible (e.g. heat, skeletal muscle, normoxic PDAC cells) [34]. High levels of both MCT-1 and MCT-4 are associated with poor prognosis, and MCT-1 inhibition reduces growth and tumorigenicity of RAS-mutated fibroblasts [59, 127]. AZD3965, a MCT-1 inhibitor, is currently being evaluated in a Phase I trial (NCT01791595).
Both MCT-1 and MCT-4 are associated with CD147 (also known as EMMPRIN or basigin), an immunoglobulin-family chaperone. CD147, MCT-1, and MCT-4 expressions at the cell surface are mutually dependent [128–130]. Proof-of-principle that targeting CD147 is an attractive approach has been established by knockdown studies and anti-CD147 antibodies showing that loss of CD147 function markedly reduced the levels of both MCT-1 and MCT-4 proteins and impaired the growth of tumor xenografts in mice [131, 132]. CD147 being ubiquitously expressed and not specific to MCT-1 and MCT-4, more selective inhibitors are required.
Targeting glutamine addiction
Glutamine analog inhibitors have been developed, from in vitro studies to clinical trials, but all studied analogs showed considerable off-target effects. More recently, targeting specific nodes of glutamine metabolism raised some interest. Notably, glutaminase inhibitors, such as bis-2-(5-phenylacetamido-1, 2, 4-thiadiazol-2-yl)ethyl sulfide and compound 968, demonstrated antiproliferative effects in vitro and in xenografts models. Similarly, aminooxyacetate, a non-specific aminotransferase inhibitor, has demonstrated efficacy in xenograft models. However, it has not been established whether target effects or off-target effects of these compounds were responsible for their antitumor activity, and their clinical development has been suspended [133].
Enhancing asparagine deprivation with L-asparaginase
L-asparaginase catalyzes the hydrolysis of asparagine into aspartic acid and ammonia, inducing asparagine deprivation. L-asparaginase is one of the most efficient agents against acute lymphoblastic leukemia, being used in the clinic for almost 50 years [134]. Leukemic lymphoblasts exhibit very low levels of ASNS - as do PDAC cells - meaning they are unable to produce de novo asparagine and these cells thus rely on exogenous supplementation. L-asparaginase-induced asparagine deprivation triggers cell apoptosis, and in vitro and in vivo experiments show that PDAC cells expressing low ASNS are very sensitive to L-asparaginase [82]. Interestingly, asparagine depletion may be rescued by glutamine through a transamidation reaction catalyzed by ASNS; asparaginase anti-leukemic activity correlated strongly with asparaginase-induced glutamine reduction, eventually resulting in protein synthesis inhibition and initiation of autophagy [135, 136].
The classically formulated L-asparaginase is limited by toxicity and development of an immune response. A new formulation encapsulating L-asparaginase in erythrocytes has increased bioavailability and a better toxicity profile while retaining strong antitumor activity in leukemia [137]. Based on preclinical data showing activity, a phase II clinical trial in PDAC with this formulation as second-line therapy is ongoing in the PDAC metastatic setting (NCT02195180).
Regulating fatty acid synthesis
Two widely used drugs, metformin and statins, provide evidence that targeting lipid metabolism in cancer may have therapeutic efficacy. Metformin has antitumor effects in preclinical PDAC models, notably by inhibiting de novo FA synthesis via downregulation of Sp transcription factors that reduce FAS expression (see below) [138, 139]. Statins are inhibitors of the HMG-CoA reductase, which is involved in the synthesis of cholesterol precursors. Some data suggest that statins might prevent PDAC and enhance survival of PDAC patients [96]. However, a recent randomized phase 2 trial failed to show a survival benefit for simvastatine in patients treated with gemcitabine [140].
Plant-derived compounds such as green tea polyphenols or flavonoids can inhibit FAS and have shown cytotoxic effects in vitro in human PDAC cell lines, but further studies are warranted [141]. Several FAS inhibitors are currently in preclinical development such as the cerulenin analog C75 that showed antitumor activity in breast and prostate cancers as well as in lymphoma [96].
Since cholesterol synthesis inhibition appears to be ineffective for PDAC treatment, blocking cholesterol uptake through LDLR blockade may be a more appropriate strategy. Moreover, this strategy sensitize PDAC cells to chemotherapeutic drugs such as gemcitabine [105]. LDLR inactivating compounds are warranted to develop this novel approach of PDAC metabolic targeting.
Pinocytosis and autophagy inhibitors
The finding that survival of RAS-transformed cells depends on autophagy offers a potential approach for inhibition. Hydroxychloroquine is a compound approved for malaria and several rheumatologic diseases that prevents lysosome acidification, thus inhibiting autophagy and macropinocytosis. Several trials testing hydroxychloroquine in patients with PDAC are ongoing (NCT01978184; NCT01128296; NCT01506973; NCT01494155; NCT01273805). Wolpin et al. [142] reported the results of a phase II study evaluating hydroxychloroquine monotherapy in 20 patients with previously treated metastatic PDAC. Median progression-free survival and OS were limited (46.5 and 69.0 days, respectively). In addition, recent data showed that autophagy inhibition in PDAC cells lacking p53 may result in increased anabolism and tumor progression [113]. This raises the question of the biological relevance of this strategy and the identification of the right tumor context in which these inhibitors can be used safely. The outcomes of patients recruited in above clinical trials should shed light on this strategy.
The mTOR pathway is constitutively activated in 25%-75% of human PDAC tumors and mTOR inhibition can lead to proliferation arrest of PDAC cells [143]. Rapamycin induces autophagy in rapamycin-sensitive pancreatic cell lines only, which suggests that autophagy induction may be a downstream consequence of the antitumor effects of mTOR inhibitors [144, 145]. Recent results from phase II clinical trials of mTOR inhibitors in PDAC patients failed to demonstrate clinical benefit [146, 147].
Non-specific OXPHOS inhibitors: metformin
Metformin is an antidiabetic drug that belongs to the biguanide family. Retrospective observational studies showed that metformin might reduce the risk of PDAC in diabetic patients and have antitumor properties, following the observation that it was associated with increased survival in diabetic patients with PDAC [148–150]. In in vitro and in vivo models, metformin was shown to impair proliferation and tumorigenicity of PDAC and cancer stem cells [151–155].
Metformin inhibits OXPHOS (mitochondrial complex I), TCA cycle anaplerosis, and de novo FA palmitate synthesis from glucose-derived acetyl-CoA [152, 156]. Thus, metformin may contribute to limit cell membrane synthesis. With cholesterol and FA de novo synthesis inhibited, glucose metabolism is channeled towards lactate production, which is consistent with one of the observed side effects, lactic acidosis. In a stem cell-enriching culture model, metformin exposure significantly decreased mitochondrial transmembrane potential and increased mitochondrial ROS production [151]. However, its effects on ROS production are controversial [157].
Metformin may also exert an antitumor effect by inhibiting the mTOR pathway [155], as suggested by its association with reduced phospho-mTOR and phospho-p70S6K levels, independently of AKT inhibition [151, 158]. Metformin activates AMPK, which negatively regulates mTORC1. In addition, metformin-induced activation of AMPK disrupts crosstalk between the insulin/IGF-1 receptor and G protein-coupled receptor signaling in PDAC models in vitro and in vivo [159, 160]. However, neither AMPK activators nor mTOR inhibitors (e.g. rapamycin) were able to mimic these cellular effects in PDAC cells. Clinical trials evaluating PI3K/AKT/mTOR pathway inhibitors in PDAC also failed to demonstrate a survival improvement, suggesting that AMPK-dependent inhibition of mTOR is not the driving mechanism for metformin activity in PDAC [151]. This suggests pathways, such as Sonic Hedgehog, contribute to metformin's antitumor effect [161].
In preclinical models, metformin was used at concentrations ranging from 5 to 20 mM, whereas its serum concentration in patients at therapeutic antidiabetic doses is 2 000–10 000 times less concentrated (around 2 μM)—a potentially major pitfall in the translation of these results to the clinic [162]. Clinical trials testing antitumor activity of metformin at antidiabetic doses are ongoing and will contribute to resolve these issues.
DISCUSSION AND CONCLUSION
To survive under severe metabolic constraints, PDAC cells rely on specific metabolic adaptations, offering a source of innovative strategies to treat PDAC patients in the coming years. Only few of them have reached the clinical development stage [Box 2]. Promising novel targets have been highlighted in this review, including PKM2 as a master regulator of tumor metabolism and the potential use of allosteric regulators, and LDH-A inhibitors. Strategies to metabolically starve tumors are also very appealing and interestingly enough, many compounds targeting tumor metabolism are expected to have low toxicities. Given the proven metabolic plasticity associated with tumors, intratumor heterogeneity, and the multiplicity of cell types involved in symbiotic metabolic interactions, targeting tumor metabolism will almost certainly benefit from combination with other targeted agents or cytotoxic compounds.
Box 2. Metabolism-modulating agents in clinical development for pancreatic ductal adenocarcinoma therapy.
| Name (mechanism of action) | Trial identifier | Phase | Current status |
|---|---|---|---|
| AZD3965 (MCT-1 inhibitor) | |||
| NCT01791595 | 1 | Recruiting | |
| ERY001 (L-asparaginase encapsulated in red blood cells) | |||
| - Metastatic PDAC | |||
| NCT02195180 | 2 | Recruiting | |
| Hydroxychloroquine (autophagy inhibitor) | |||
| - Neo-adjuvant setting | |||
| NCT01978184 | 2 | Recruiting (combined with Gem and nab-P) | |
| NCT01494155 | 2 | Recruiting (combined with capecitabine and radiotherapy) | |
| NCT01128296 | 1/2 | Active, not recruiting (combined with gemcitabine) | |
| - Locally advanced or metastatic PDAC | |||
| NCT01506973 | 1/2 | Status unknown (combined with Gem and nab-P) | |
| NCT01273805 | 2 | Active, not recruiting (monotherapy) | |
| Metformin (non-specific inhibitor) | |||
| - Neo-adjuvant setting | |||
| NCT02153450 | 2 | Recruiting (combined with stereotactic radiosurgery) | |
| - Adjuvant setting | |||
| NCT02005419 | 2 | Recruiting (combined with Gem) | |
| - Locally advanced or metastatic PDAC | |||
| NCT01210911 | 2 | Completed (combined with Gem and erlotinib) | |
| NCT01167738 | 2 | Terminated (concern of detrimental effect) | |
| NCT01666730 | 2 | Recruiting (combined with FOLFOX 6) | |
| NCT02336087 | 1 | Not yet recruiting (combined with Gem, nab-P, and a standardized dietary supplement) | |
| NCT01488552 | 1/2 | Recruiting (combined with Gem + nab-P or FOLFIRINOX) | |
| NCT02048384 | 1/2 | Recruiting (combined with rapamycin) | |
| NCT01971034 | 2 | Completed (combined with paclitaxel) | |
Gem: gemcitabine; nab-P: nab-paclitaxel; PDAC: pancreatic ductal adenocarcinoma
Acknowledgments
None.
Abbreviations
- ADP
adenosine 5′-diphosphate
- ATP
adenosine 5′-triphosphate
- AA
amino acid
- ASNS
asparagine synthetase
- BCAA
branched-chain amino acid
- CA
carbonic anhydrase
- EGFR
epidermal growth factor receptor
- EMT
epithelio-mesenchymal transition
- ERK
extracellular signal-regulated kinase
- FA
fatty acid
- FAS
fatty acid synthase
- FBP
fructose-2, 6-biphosphate
- FGF
fibroblast growth factor
- FOXM1
forkhead box protein M1
- GFPT
glucosamine-fructose-6-phosphate aminotransferase
- GLUT
glucose transporter
- GDH
glutamate dehydrogenase
- GOT
glutamic oxaloacetic transaminase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HK
hexokinase
- HBP
hexosamine biosynthetic pathway
- HIF
hypoxia-inducible factor
- IGFR
insulin-like growth factor receptor
- LDH
lactate dehydrogenase
- LDLR
low-density lipoprotein receptor
- MCT
monocarbonate transporter
- NADP
nicotinamide adenine dinucleotide phosphate
- GlcNAc
N-acetylglucosamine
- OS
overall survival
- OXPHOS
oxidative phosphorylation
- PDAC
pancreatic ductal adenocarcinoma
- PanIN
pancreatic intraepithelial neoplasia
- PSC
pancreatic stellate cell
- PPP
pentose phosphate pathway
- PEP
phosphoenopyruvate
- PDH
pyruvate dehydrogenase
- PDHK
pyruvate dehydrogenase kinase
- PKM
pyruvate kinase muscle-isozyme
- ROS
reactive oxygen species
- TIGAR
TP53-inductible glycolytic and apoptotic regulator
- TCA
tricarboxylic acid
- TGF
transforming growth factor
- TKR
tyrosine kinase receptor
- UPR
unfolded-protein response
Footnotes
CONFLICTS OF INTEREST
All authors declare no potential conflicts of interest.
GRANT SUPPORT
None.
REFERENCES
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics, 2010. CA: A Cancer Journal for Clinicians. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Research. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 3.Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. [DOI] [PubMed]
- 4.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, Leach SD, Stanger BZ. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul J-L, Gourgou-Bourgade S, de la Fouchardière C, Bennouna J, Bachet J-B, Khemissa-Akouz F, et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. New England Journal of Medicine. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 6.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. New England Journal of Medicine. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Chen J-Q, Russo J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochimica et Biophysica Acta. 2012;1826:370–384. doi: 10.1016/j.bbcan.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Hsu PP, Sabatini DM. Cancer Cell Metabolism: Warburg and Beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 10.Galluzzi L, Kepp O, Heiden MGV, Kroemer G. Metabolic targets for cancer therapy. Nature Reviews Drug Discovery. 2013;12:829–846. doi: 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- 11.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nature Reviews Drug Discovery. 2011;10:671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 12.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nature Reviews Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hidalgo M. Pancreatic cancer. New England Journal of Medicine. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 14.Chu GC, Kimmelman AC, Hezel AF, DePinho RA. Stromal biology of pancreatic cancer. Journal of Cellular Biochemistry. 2007;101:887–907. doi: 10.1002/jcb.21209. [DOI] [PubMed] [Google Scholar]
- 15.Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Molecular Cancer Therapeutics. 2007;6:1186–1197. doi: 10.1158/1535-7163.MCT-06-0686. [DOI] [PubMed] [Google Scholar]
- 16.Masamune A, Shimosegawa T. Signal transduction in pancreatic stellate cells. Journal of Gastroenterology. 2009;44:249–260. doi: 10.1007/s00535-009-0013-2. [DOI] [PubMed] [Google Scholar]
- 17.Eguchi D, Ikenaga N, Ohuchida K, Kozono S, Cui L, Fujiwara K, Fujino M, Ohtsuka T, Mizumoto K, Tanaka M. Hypoxia enhances the interaction between pancreatic stellate cells and cancer cells via increased secretion of connective tissue growth factor. Journal of Surgical Research. 2013;181:225–233. doi: 10.1016/j.jss.2012.06.051. [DOI] [PubMed] [Google Scholar]
- 18.Erkan M, Reiser-Erkan C, Michalski CW, Deucker S, Sauliunaite D, Streit S, Esposito I, Friess H, Kleeff J. Cancer-Stellate Cell Interactions Perpetuate the Hypoxia-Fibrosis Cycle in Pancreatic Ductal Adenocarcinoma. Neoplasia. 2009;11:497–508. doi: 10.1593/neo.81618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmann A-C, Mori R, Vallbohmer D, Brabender J, Klein E, Drebber U, Baldus SE, Cooc J, Azuma M, Metzger R, Hoelscher AH, Danenberg KD, Prenzel KL, et al. High Expression of HIF1a Is a Predictor of Clinical Outcome in Patients with Pancreatic Ductal Adenocarcinomas and Correlated to PDGFA, VEGF, and bFGF. Neoplasia. 2008;10:674–679. doi: 10.1593/neo.08292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masamune A, Kikuta K, Watanabe T, Satoh K, Hirota M, Shimosegawa T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2008;295:709–717. doi: 10.1152/ajpgi.90356.2008. [DOI] [PubMed] [Google Scholar]
- 21.Miyake K, Yoshizumi T, Imura S, Sugimoto K, Batmunkh E, Kanemura H, Morine Y, Shimada M. Expression of Hypoxia-Inducible Factor-1α, Histone Deacetylase 1, and Metastasis-Associated Protein 1 in Pancreatic Carcinoma: Correlation With Poor Prognosis With Possible Regulation. Pancreas. 2008;36:e1–9. doi: 10.1097/MPA.0b013e31815f2c2a. [DOI] [PubMed] [Google Scholar]
- 22.Sun H-C, Qiu Z-J, Liu J, Sun J, Jiang T, Huang K-J, Yao M, Huang C. Expression of hypoxia-inducible factor-1 alpha and associated proteins in pancreatic ductal adenocarcinoma and their impact on prognosis. International Journal of Oncology. 2007;30:1359–1367. [PubMed] [Google Scholar]
- 23.Couvelard A, O'Toole D, Leek R, Turley H, Sauvanet A, Degott C, Ruszniewski P, Belghiti J, Harris AL, Gatter K, Pezzella F. Expression of hypoxia-inducible factors is correlated with the presence of a fibrotic focus and angiogenesis in pancreatic ductal adenocarcinomas. Histopathology. 2005;46:668–676. doi: 10.1111/j.1365-2559.2005.02160.x. [DOI] [PubMed] [Google Scholar]
- 24.Kitada T, Seki S, Sakaguchi H, Sawada T, Hirakawa K, Wakasa K. Clinicopathological significance of hypoxia-inducible factor-1α expression in human pancreatic carcinoma. Histopathology. 2003;43:550–555. doi: 10.1111/j.1365-2559.2003.01733.x. [DOI] [PubMed] [Google Scholar]
- 25.Büchler P, Reber HA, Lavey RS, Tomlinson J, Büchler MW, Friess H, Hines OJ. Tumor hypoxia correlates with metastatic tumor growth of pancreatic cancer in an orthotopic murine model1. Journal of Surgical Research. 2004;120:295–303. doi: 10.1016/j.jss.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 26.Chang Q, Jurisica I, Do T, Hedley DW. Hypoxia Predicts Aggressive Growth and Spontaneous Metastasis Formation from Orthotopically Grown Primary Xenografts of Human Pancreatic Cancer. Cancer Research. 2011;71:3110–3120. doi: 10.1158/0008-5472.CAN-10-4049. [DOI] [PubMed] [Google Scholar]
- 27.Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, Bastidas AJ, Vierra M. Pancreatic tumors show high levels of hypoxia. International Journal of Radiation Oncology*Biology*Physics. 2000;48:919–922. doi: 10.1016/s0360-3016(00)00803-8. [DOI] [PubMed] [Google Scholar]
- 28.Niizeki H, Kobayashi M, Horiuchi I, Akakura N, Chen J, Wang J, Hamada J, Seth P, Katoh H, Watanabe H, Raz A, Hosokawa M. Hypoxia enhances the expression of autocrine motility factor and the motility of human pancreatic cancer cells. British Journal of Cancer. 2002;86:1914–1919. doi: 10.1038/sj.bjc.6600331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang W, Reiser-Erkan C, Michalski CW, Raggi MC, Quan L, Yupei Z, Friess H, Erkan M, Kleeff J. Hypoxia inducible BHLHB2 is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Biochemical and Biophysical Research Communications. 2010;401:422–428. doi: 10.1016/j.bbrc.2010.09.070. [DOI] [PubMed] [Google Scholar]
- 30.Zhu S, Zhou Y, Wang L, Zhang J, Wu H, Xiong J, Zhang J, Tian Y, Wang C, Wu H. Transcriptional upregulation of MT2-MMP in response to hypoxia is promoted by HIF-1α in cancer cells. Molecular Carcinogenesis. 2011;50:770–780. doi: 10.1002/mc.20678. [DOI] [PubMed] [Google Scholar]
- 31.Yokoi K, Fidler IJ. Hypoxia Increases Resistance of Human Pancreatic Cancer Cells to Apoptosis Induced by Gemcitabine. Clinical Cancer Research. 2004;10:2299–2306. doi: 10.1158/1078-0432.ccr-03-0488. [DOI] [PubMed] [Google Scholar]
- 32.Chen C, Yu Z. siRNA Targeting HIF-1α Induces Apoptosis of Pancreatic Cancer Cells through NF-κB-independent and -dependent Pathways under Hypoxic Conditions. Anticancer Research. 2009;29:1367–1372. [PubMed] [Google Scholar]
- 33.Chen J, Zhao S, Nakada K, Kuge Y, Tamaki N, Okada F, Wang J, Shindo M, Higashino F, Takeda K, Asaka M, Katoh H, Sugiyama T, et al. Dominant-Negative Hypoxia-Inducible Factor-1α Reduces Tumorigenicity of Pancreatic Cancer Cells through the Suppression of Glucose Metabolism. The American Journal of Pathology. 2003;162:1283–1291. doi: 10.1016/s0002-9440(10)63924-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guillaumond F, Leca J, Olivares O, Lavaut M-N, Vidal N, Berthezène P, Dusetti NJ, Loncle C, Calvo E, Turrini O, Iovanna JL, Tomasini R, Vasseur S. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proceedings of the National Academy of Sciences. 2013;110:3919–3924. doi: 10.1073/pnas.1219555110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guillaumond F, Iovanna JL, Vasseur S. Pancreatic tumor cell metabolism: focus on glycolysis and its connected metabolic pathways. Archives of Biochemistry and Biophysics. 2014;545:69–73. doi: 10.1016/j.abb.2013.12.019. [DOI] [PubMed] [Google Scholar]
- 36.Izuishi K, Kato K, Ogura T, Kinoshita Taira, Esumi H. Remarkable Tolerance of Tumor Cells to Nutrient Deprivation: Possible New Biochemical Target for Cancer Therapy. Cancer Research. 2000;60:6201–6207. [PubMed] [Google Scholar]
- 37.Warburg O, Posener K, Negelein E. Über der Stoffwechsel der Carcinomzelle. Biochemische Zeitschrift. 1924;152:319–344. [Google Scholar]
- 38.Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. The Journal of general physiology. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 40.Feron O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiotherapy and Oncology. 2009;92:329–233. doi: 10.1016/j.radonc.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 41.Zhou W, Capello M, Fredolini C, Piemonti L, Liotta LA, Novelli F, Petricoin EF. Proteomic Analysis of Pancreatic Ductal Adenocarcinoma Cells Reveals Metabolic Alterations. Journal of Proteome Research. 2011;10:1944–1952. doi: 10.1021/pr101179t. [DOI] [PubMed] [Google Scholar]
- 42.Zhou W, Capello M, Fredolini C, Racanicchi L, Piemonti L, Liotta LA, Novelli F, Petricoin EF. Proteomic Analysis Reveals Warburg Effect and Anomalous Metabolism of Glutamine in Pancreatic Cancer Cells. Journal of Proteome Research. 2012;11:554–563. doi: 10.1021/pr2009274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rong Y, Wu W, Ni X, Kuang T, Jin D, Wang D, Lou W. Lactate dehydrogenase A is overexpressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumor Biology. 2013;34:1523–1530. doi: 10.1007/s13277-013-0679-1. [DOI] [PubMed] [Google Scholar]
- 44.Mikuriya K, Kuramitsu Y, Ryozawa S, Fujimoto M, Mori S, Oka M, Hamano K, Okita K, Sakaida I, Nakamura K. Expression of glycolytic enzymes is increased in pancreatic cancerous tissues as evidenced by proteomic profiling by two-dimensional electrophoresis and liquid chromatography-mass spectrometry/mass spectrometry. International Journal of Oncology. 2007;30:849–855. [PubMed] [Google Scholar]
- 45.Bensinger SJ, Christofk HR. New aspects of the Warburg effect in cancer cell biology. Seminars in Cell & Developmental Biology. 2012;23:352–361. doi: 10.1016/j.semcdb.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 46.Sanders E, Diehl S. Analysis and interpretation of transcriptomic data obtained from extended Warburg effect genes in patients with clear cell renal cell carcinoma. Oncoscience. 2015;2:151–186. doi: 10.18632/oncoscience.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth Factors Can Influence Cell Growth and Survival through Effects on Glucose Metabolism. Molecular and Cellular Biology. 2001;21:5899–5912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nature Reviews Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 49.Feldmann G, Beaty R, Hruban RH, Maitra A. Molecular genetics of pancreatic intraepithelial neoplasia. Journal of Hepato-Biliary-Pancreatic Surgery. 2007;14:224–232. doi: 10.1007/s00534-006-1166-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends in Biochemical Sciences. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Molecular Systems Biology. 2011;7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dell' Antone P. Energy metabolism in cancer cells: How to explain the Warburg and Crabtree effects? Medical Hypotheses. 2012;79:388–392. doi: 10.1016/j.mehy.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 53.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shen L, Sun X, Fu Z, Yang G, Li J, Yao L. The Fundamental Role of the p53 Pathway in Tumor Metabolism and Its Implication in Tumor Therapy. Clinical Cancer Research. 2012;18:1561–1567. doi: 10.1158/1078-0432.CCR-11-3040. [DOI] [PubMed] [Google Scholar]
- 55.Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The Tumor Suppressor p53 Down-Regulates Glucose Transporters GLUT1 and GLUT4 Gene Expression. Cancer Research. 2004;64:2627–2633. doi: 10.1158/0008-5472.can-03-0846. [DOI] [PubMed] [Google Scholar]
- 56.Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 57.Gerin I, Noël G, Bolsée J, Haumont O, Van Schaftingen E, Bommer GT. Identification of TP53-induced glycolysis and apoptosis regulator (TIGAR) as the phosphoglycolate-independent 2, 3-bisphosphoglycerate phosphatase. Biochemical Journal. 2014;458:439–448. doi: 10.1042/BJ20130841. [DOI] [PubMed] [Google Scholar]
- 58.Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JKV, Markowitz S, Zhou S, Diaz LA, Velculescu VE, Lengauer C, et al. Glucose Deprivation Contributes to the Development of KRAS Pathway Mutations in Tumor Cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. Journal of Clinical Investigation. 2013;123:3685–3692. doi: 10.1172/JCI69741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364–373. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- 61.Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, Renner K, Timischl B, Mackensen A, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 62.Gottfried E. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. 2006;107:2013–2021. doi: 10.1182/blood-2005-05-1795. [DOI] [PubMed] [Google Scholar]
- 63.Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. International Journal of Cancer. 2012;131:633–640. doi: 10.1002/ijc.26410. [DOI] [PubMed] [Google Scholar]
- 64.Shime H, Yabu M, Akazawa T, Kodama K, Matsumoto M, Seya T, Inoue N. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. The Journal of Immunology. 2008;180:7175–7183. doi: 10.4049/jimmunol.180.11.7175. [DOI] [PubMed] [Google Scholar]
- 65.Yabu M, Shime H, Hara H, Saito T, Matsumoto M, Seya T, Akazawa T, Inoue N. IL-23-dependent and -independent enhancement pathways of IL-17A production by lactic acid. International Immunology. 2011;23:29–41. doi: 10.1093/intimm/dxq455. [DOI] [PubMed] [Google Scholar]
- 66.Liu C-Y, Xu J-Y, Shi X-Y, Huang W, Ruan T-Y, Xie P, Ding J-L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Laboratory Investigation. 2013;93:844–854. doi: 10.1038/labinvest.2013.69. [DOI] [PubMed] [Google Scholar]
- 67.Protti MP, De Monte L. Immune infiltrates as predictive markers of survival in pancreatic cancer patients. Frontiers in Physiology. 2013;4:210. doi: 10.3389/fphys.2013.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, Sakoda M, Ueno S, Natsugoe S, Takao S. Significance of M2-Polarized Tumor-Associated Macrophage in Pancreatic Cancer. Journal of Surgical Research. 2011;167:e211–219. doi: 10.1016/j.jss.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 69.Hirschhaeuser F, Sattler UGA, Mueller-Klieser W. Lactate: A Metabolic Key Player in Cancer. Cancer Research. 2011;71:6921–6925. doi: 10.1158/0008-5472.CAN-11-1457. [DOI] [PubMed] [Google Scholar]
- 70.Alfarouk KO, Verduzco D, Rauch C, Muddathir AK, Adil HB, Elhassan GO, Ibrahim ME, Orozco JDP, Cardone RA, Reshkin SJ others. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience. 2014;1:777. doi: 10.18632/oncoscience.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, Yuan C, Bao Y, Townsend MK, Tworoger SS, Davidson SM, Papagiannakopoulos T, Yang A, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nature Medicine. 2014;20:1193–1198. doi: 10.1038/nm.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends in Biochemical Sciences. 2010;35:427–433. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eagle H. Nutrition Needs of Mammali Cells in Tissue Culture. Science. 1955;122:501–504. doi: 10.1126/science.122.3168.501. [DOI] [PubMed] [Google Scholar]
- 74.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proceedings of the National Academy of Sciences. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kovasevic Z. The pathway of glutamine and glutamate oxidation in isolated mitochondria from mammalian cells. Biochemical Journal. 1971;3:757–763. doi: 10.1042/bj1250757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X-Y, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. The Journal of Cell Biology. 2007;178:93–105. doi: 10.1083/jcb.200703099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cui H, Darmanin S, Natsuisaka M, Kondo T, Asaka M, Shindoh M, Higashino F, Hamuro J, Okada F, Kobayashi M. Enhanced expression of asparagine synthetase under glucose-deprived conditions protects pancreatic cancer cells from apoptosis induced by glucose deprivation and cisplatin. Cancer research. 2007;67:3345–3355. doi: 10.1158/0008-5472.CAN-06-2519. [DOI] [PubMed] [Google Scholar]
- 81.Barbosa-Tessmann IP, Chen C, Zhong C, Schuster SM, Nick HS, Kilberg MS. Activation of the Unfolded Protein Response Pathway Induces Human Asparagine Synthetase Gene Expression. Journal of Biological Chemistry. 1999;274:31139–31144. doi: 10.1074/jbc.274.44.31139. [DOI] [PubMed] [Google Scholar]
- 82.Dufour E, Gay F, Aguera K, Scoazec J-Y, Horand F, Lorenzi PL, Godfrin Y. Pancreatic tumor sensitivity to plasma L-asparagine starvation. Pancreas. 2012;41:940–948. doi: 10.1097/MPA.0b013e318247d903. [DOI] [PubMed] [Google Scholar]
- 83.Balasubramanian MN, Butterworth EA, Kilberg MS. Asparagine synthetase: regulation by cell stress and involvement in tumor biology. AJP: Endocrinology and Metabolism. 2013;304:E789–799. doi: 10.1152/ajpendo.00015.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pan S, Chen R, Tamura Y, Crispin DA, Lai LA, May DH, McIntosh MW, Goodlett DR, Brentnall TA. Quantitative Glycoproteomics Analysis Reveals Changes in N-Glycosylation Level Associated with Pancreatic Ductal Adenocarcinoma. Journal of Proteome Research. 2014;13:1293–1306. doi: 10.1021/pr4010184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ma Z, Vocadlo DJ, Vosseller K. Hyper-O-GlcNAcylation Is Anti-apoptotic and Maintains Constitutive NF- B Activity in Pancreatic Cancer Cells. Journal of Biological Chemistry. 2013;288:15121–15130. doi: 10.1074/jbc.M113.470047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Remmers N, Anderson JM, Linde EM, DiMaio DJ, Lazenby AJ, Wandall HH, Mandel U, Clausen H, Yu F, Hollingsworth MA. Aberrant Expression of Mucin Core Proteins and O-Linked Glycans Associated with Progression of Pancreatic Cancer. Clinical Cancer Research. 2013;19:1981–1993. doi: 10.1158/1078-0432.CCR-12-2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Slawson C, Copeland RJ, Hart GW. O-GlcNAc signaling: a metabolic link between diabetes and cancer? Trends in Biochemical Sciences. 2010;35:547–555. doi: 10.1016/j.tibs.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Olivier-Van Stichelen S, Guinez C, Mir A-M, Perez-Cervera Y, Liu C, Michalski J-C, Lefebvre T. The hexosamine biosynthetic pathway and O-GlcNAcylation drive the expression of -catenin and cell proliferation. AJP: Endocrinology and Metabolism. 2012;302:E417–424. doi: 10.1152/ajpendo.00390.2011. [DOI] [PubMed] [Google Scholar]
- 89.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, Peters EC, Driggers EM, Hsieh-Wilson LC. Phosphofructokinase 1 Glycosylation Regulates Cell Growth and Metabolism. Science. 2012;337:975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hanover JA, Krause MW, Love DC. Post-translational modifications: Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nature Reviews Molecular Cell Biology. 2012;13:312–321. doi: 10.1038/nrm3334. [DOI] [PubMed] [Google Scholar]
- 91.Tian E, Hoffman MP, Ten Hagen KG. O-glycosylation modulates integrin and FGF signalling by influencing the secretion of basement membrane components. Nature Communications. 2012;3:869. doi: 10.1038/ncomms1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wellen KE, Lu C, Mancuso A, Lemons JMS, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, Thompson CB. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes & Development. 2010;24:2784–2799. doi: 10.1101/gad.1985910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Contessa JN, Bhojani MS, Freeze HH, Rehemtulla A, Lawrence TS. Inhibition of N-Linked Glycosylation Disrupts Receptor Tyrosine Kinase Signaling in Tumor Cells. Cancer Research. 2008;68:3803–3809. doi: 10.1158/0008-5472.CAN-07-6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Palorini R, Cammarata F, Balestrieri C, Monestiroli A, Vasso M, Gelfi C, Alberghina L, Chiaradonna F. Glucose starvation induces cell death in K-ras-transformed cells by interfering with the hexosamine biosynthesis pathway and activating the unfolded protein response. Cell Death and Disease. 2013;4:e732. doi: 10.1038/cddis.2013.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alo PL, Amini M, Piro F, Pizzuti L, Sebastiani V, Botti C, Murari R, Zotti G, Di Tondo U. Immnunohistochemical expression and prognostic significance of fatty acid synthase in pancreatic carcinoma. Anticancer research. 2007;27:2523–2527. [PubMed] [Google Scholar]
- 96.Swierczynski J. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World Journal of Gastroenterology. 2014;20:2279–2303. doi: 10.3748/wjg.v20.i9.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Choi W-I, Jeon B-N, Park H, Yoo J-Y, Kim Y-S, Koh D-I, Kim M-H, Kim Y-R, Lee C-E, Kim K-S, Osborne TF, Hur M-W. Proto-oncogene FBI-1 (Pokemon) and SREBP-1 Synergistically Activate Transcription of Fatty-acid Synthase Gene (FASN) Journal of Biological Chemistry. 2008;283:29341–29354. doi: 10.1074/jbc.M802477200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bandyopadhyay S, Pai SK, Watabe M, Gross SC, Hirota S, Hosobe S, Tsukada T, Miura K, Saito K, Markwell SJ. FAS expression inversely correlates with PTEN level in prostate cancer and a PI 3-kinase inhibitor synergizes with FAS siRNA to induce apoptosis. Oncogene. 2005;24:5389–5395. doi: 10.1038/sj.onc.1208555. [DOI] [PubMed] [Google Scholar]
- 99.Menendez JA, Decker JP, Lupu R. In support of fatty acid synthase (FAS) as a metabolic oncogene: Extracellular acidosis acts in an epigenetic fashion activating FAS gene expression in cancer cells. Journal of Cellular Biochemistry. 2005;94:1–4. doi: 10.1002/jcb.20310. [DOI] [PubMed] [Google Scholar]
- 100.Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D, Daniels VW, Machiels J, Vanderhoydonc F, Smans K, Waelkens E, et al. De novo Lipogenesis Protects Cancer Cells from Free Radicals and Chemotherapeutics by Promoting Membrane Lipid Saturation. Cancer Research. 2010;70:8117–8126. doi: 10.1158/0008-5472.CAN-09-3871. [DOI] [PubMed] [Google Scholar]
- 101.Williams TM. Caveolin-1 in oncogenic transformation, cancer, and metastasis. AJP: Cell Physiology. 2004;288:C494–506. doi: 10.1152/ajpcell.00458.2004. [DOI] [PubMed] [Google Scholar]
- 102.Suzuoki M, Miyamoto M, Kato K, Hiraoka K, Oshikiri T, Nakakubo Y, Fukunaga A, Shichinohe T, Shinohara T, Itoh T, Kondo S, Katoh H. Impact of caveolin-1 expression on prognosis of pancreatic ductal adenocarcinoma. British Journal of Cancer. 2002;87:1140–1144. doi: 10.1038/sj.bjc.6600619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Witkiewicz AK, Nguyen KH, Dasgupta A, Kennedy EP, Yeo CJ, Lisanti MP, Brody JR. Co-expression of fatty acid synthase and caveolin-1 in pancreatic ductal adenocarcinoma. Cell Cycle. 2008;7:3021–3025. doi: 10.4161/cc.7.19.6719. [DOI] [PubMed] [Google Scholar]
- 104.Staubach S, Hanisch F-G. Lipid rafts: signaling and sorting platforms of cells and their roles in cancer. Expert Review of Proteomics. 2011;8:263–277. doi: 10.1586/epr.11.2. [DOI] [PubMed] [Google Scholar]
- 105.Guillaumond F, Bidaut G, Ouaissi M, Servais S, Gouirand V, Olivares O, Lac S, Borge L, Roques J, Gayet O, Pinault M, Guimaraes C, Nigri J, et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proceedings of the National Academy of Sciences. 2015;112:2473–2478. doi: 10.1073/pnas.1421601112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Oh HY, Lee EJ, Yoon S, Chung BH, Cho KS, Hong SJ. Cholesterol level of lipid raft microdomains regulates apoptotic cell death in prostate cancer cells through EGFR-mediated Akt and ERK signal transduction. The Prostate. 2007;67:1061–1069. doi: 10.1002/pros.20593. [DOI] [PubMed] [Google Scholar]
- 107.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 108.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Molecular biology of the cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mazure NM, Pouysségur J. Hypoxia-induced autophagy: cell death or cell survival? Current Opinion in Cell Biology. 2010;22:177–180. doi: 10.1016/j.ceb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 110.Iacobuzio-Donahue CA, Herman JM. Autophagy, p53, and Pancreatic Cancer. New England Journal of Medicine. 2014;370:1352–1353. doi: 10.1056/NEJMcibr1400189. [DOI] [PubMed] [Google Scholar]
- 111.Sousa CM, Kimmelman AC. The Complex Landscape of Pancreatic Cancer Metabolism. Carcinogenesis. 2014;35:1441–1450. doi: 10.1093/carcin/bgu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'Antonio G, Mautner J, Tonon G, Haigis M, et al. Pancreatic cancers require autophagy for tumor growth. Genes & Development. 2011;25:717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, Adams PD, Anderson KI, Gottlieb E, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 114.Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, Von Hoff DD, Maitra A, Kimmelman AC. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discovery. 2014;4:905–913. doi: 10.1158/2159-8290.CD-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, Rabinowitz JD. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proceedings of the National Academy of Sciences. 2013;110:8882–8887. doi: 10.1073/pnas.1307237110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, Rabinowitz JD, Metallo CM, Vander Heiden MG, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–637. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.White E. Exploiting the bad eating habits of Ras-driven cancers. Genes & Development. 2013;27:2065–2071. doi: 10.1101/gad.228122.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bhardwaj V, Rizvi N, Lai MB, Lai JCK, Bhushan A. Glycolytic Enzyme Inhibitors Affect Pancreatic Cancer Survival by Modulating Its Signaling and Energetics. Anticancer Research. 2010;30:743–749. [PubMed] [Google Scholar]
- 119.Yang W, Lu Z. Regulation and function of pyruvate kinase M2 in cancer. Cancer Letters. 2013;339:153–158. doi: 10.1016/j.canlet.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Seminars in Cancer Biology. 2005;15:300–308. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 121.Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nature Cell Biology. 2012;14:1295–1304. doi: 10.1038/ncb2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tamada M, Suematsu M, Saya H. Pyruvate Kinase M2: Multiple Faces for Conferring Benefits on Cancer Cells. Clinical Cancer Research. 2012;18:5554–5561. doi: 10.1158/1078-0432.CCR-12-0859. [DOI] [PubMed] [Google Scholar]
- 123.Zhao D, Zou S-W, Liu Y, Zhou X, Mo Y, Wang P, Xu Y-H, Dong B, Xiong Y, Lei Q-Y, Guan K-L. Lysine-5 Acetylation Negatively Regulates Lactate Dehydrogenase A and Is Decreased in Pancreatic Cancer. Cancer Cell. 2013;23:464–476. doi: 10.1016/j.ccr.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhou W, Capello M, Fredolini C, Racanicchi L, Piemonti L, Liotta LA, Novelli F, Petricoin EF. MS analysis reveals O-methylation of L-lactate dehydrogenase from pancreatic ductal adenocarcinoma cells: General. Electrophoresis. 2012;33:1850–1854. doi: 10.1002/elps.201200017. [DOI] [PubMed] [Google Scholar]
- 125.Cui J, Shi M, Xie D, Wei D, Jia Z, Zheng S, Gao Y, Huang S, Xie K. FOXM1 Promotes the Warburg Effect and Pancreatic Cancer Progression via Transactivation of LDHA Expression. Clinical Cancer Research. 2014;20:2595–2606. doi: 10.1158/1078-0432.CCR-13-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Martín-Bernabé A, Cortés R, Lehmann SG, Seve M, Cascante M, Bourgoin-Voillard S. Quantitative Proteomic Approach to Understand Metabolic Adaptation in Non-Small Cell Lung Cancer. Journal of Proteome Research. 2014;13:4695–4704. doi: 10.1021/pr500327v. [DOI] [PubMed] [Google Scholar]
- 127.Floch RL, Chiche J, Marchiq I, Naiken T, Ilc K, Murray CM, Critchlow SE, Roux D, Simon M-P, Pouysségur J. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proceedings of the National Academy of Sciences. 2011;108:16663–16668. doi: 10.1073/pnas.1106123108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ovens MJ, Manoharan C, Wilson MC, Murray CM, Halestrap AP. The inhibition of monocarboxylate transporter 2 (MCT2) by AR-C155858 is modulated by the associated ancillary protein. Biochemical Journal. 2010;431:217–225. doi: 10.1042/BJ20100890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kirk P, Wilson MC, Heddle C, Brown MH, Barclay AN, Halestrap AP. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. The EMBO journal. 2000;19:3896–3904. doi: 10.1093/emboj/19.15.3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gallagher SM, Castorino JJ, Wang D, Philp NJ. Monocarboxylate transporter 4 regulates maturation and trafficking of CD147 to the plasma membrane in the metastatic breast cancer cell line MDA-MB-231. Cancer research. 2007;67:4182–4189. doi: 10.1158/0008-5472.CAN-06-3184. [DOI] [PubMed] [Google Scholar]
- 131.Schneiderhan W, Scheler M, Holzmann K-H, Marx M, Gschwend JE, Bucholz M, Gress TM, Seufferlein T, Adler G, Oswald F. CD147 silencing inhibits lactate transport and reduces malignant potential of pancreatic cancer cells in in vivo and in vitro models. Gut. 2009;58:1391–1398. doi: 10.1136/gut.2009.181412. [DOI] [PubMed] [Google Scholar]
- 132.Wang S. CD147 silencing via RNA interference reduces tumor cell invasion, metastasis and increases chemosensitivity in pancreatic cancer cells. Oncology Reports. 2012;27:2003–2009. doi: 10.3892/or.2012.1729. [DOI] [PubMed] [Google Scholar]
- 133.Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. Journal of Clinical Investigation. 2013;123:3678–84. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dolowy WC, Henson D, Cornet J, Sellin H. Toxic and antineoplastic effects of L-asparaginase. Study of mice with lymphoma and normal monkeys and report on a child with leukemia. Cancer. 1966;19:1813–1819. doi: 10.1002/1097-0142(196612)19:12<1813::aid-cncr2820191208>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 135.Emadi A, Zokaee H, Sausville EA. Asparaginase in the treatment of non-ALL hematologic malignancies. Cancer Chemotherapy and Pharmacology. 2014;73:875–883. doi: 10.1007/s00280-014-2402-3. [DOI] [PubMed] [Google Scholar]
- 136.Avramis VI, Sencer S, Periclou AP, Sather H, Bostrom BC, Cohen LJ, Ettinger AG, Ettinger LJ, Franklin J, Gaynon PS, Hilden JM, Lange B, Majlessipour F, et al. A randomized comparison of nativeEscherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: a Children's Cancer Group study. Blood. 2002;99:1986–1994. doi: 10.1182/blood.v99.6.1986. [DOI] [PubMed] [Google Scholar]
- 137.Domenech C, Thomas X, Chabaud S, Baruchel A, Gueyffier F, Mazingue F, Auvrignon A, Corm S, Dombret H, Chevallier P, Galambrun C, Huguet F, Legrand F, et al. l-asparaginase loaded red blood cells in refractory or relapsing acute lymphoblastic leukaemia in children and adults: results of the GRASPALL 2005–01 randomized trial: GRASPA® in Relapsing ALL. British Journal of Haematology. 2011;153:58–65. doi: 10.1111/j.1365-2141.2011.08588.x. [DOI] [PubMed] [Google Scholar]
- 138.Nair V, Pathi S, Jutooru I, Sreevalsan S, Basha R, Abdelrahim M, Samudio I, Safe S. Metformin inhibits pancreatic cancer cell and tumor growth and downregulates Sp transcription factors. Carcinogenesis. 2013;34:2870–2879. doi: 10.1093/carcin/bgt231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lu S, Archer MC. Sp1 coordinately regulates de novo lipogenesis and proliferation in cancer cells. International Journal of Cancer. 2010;126:416–425. doi: 10.1002/ijc.24761. [DOI] [PubMed] [Google Scholar]
- 140.Hong JY, Nam EM, Lee J, Park JO, Lee S-C, Song S-Y, Choi SH, Heo JS, Park SH, Lim HY, Kang WK, Park YS. Randomized double-blinded, placebo-controlled phase II trial of simvastatin and gemcitabine in advanced pancreatic cancer patients. Cancer Chemotherapy and Pharmacology. 2014;73:125–130. doi: 10.1007/s00280-013-2328-1. [DOI] [PubMed] [Google Scholar]
- 141.Harris DM, Li L, Chen M, Lagunero FT, Go VLW, Boros LG. Diverse mechanisms of growth inhibition by luteolin, resveratrol, and quercetin in MIA PaCa-2 cells: a comparative glucose tracer study with the fatty acid synthase inhibitor C75. Metabolomics. 2012;8:201–210. doi: 10.1007/s11306-011-0300-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wolpin BM, Rubinson DA, Wang X, Chan JA, Cleary JM, Enzinger PC, Fuchs CS, McCleary NJ, Meyerhardt JA, Ng K, Schrag D, Sikora AL, Spicer BA, et al. Phase II and Pharmacodynamic Study of Autophagy Inhibition Using Hydroxychloroquine in Patients With Metastatic Pancreatic Adenocarcinoma. The Oncologist. 2014;19:637–638. doi: 10.1634/theoncologist.2014-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Morran DC, Wu J, Jamieson NB, Mrowinska A, Kalna G, Karim SA, Au AYM, Scarlett CJ, Chang DK, Pajak MZ. Australian Pancreatic Cancer Genome Initiative (APGI) Oien KA, McKay CJ, et al. Targeting mTOR dependency in pancreatic cancer. Gut. 2014;63:1481–1489. doi: 10.1136/gutjnl-2013-306202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Utomo WK, Narayanan V, Biermann K, van Eijck CHJ, Bruno MJ, Peppelenbosch MP, Braat H. mTOR is a promising therapeutical target in a subpopulation of pancreatic adenocarcinoma. Cancer Letters. 2014;346:309–317. doi: 10.1016/j.canlet.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 145.Mirzoeva OK, Hann B, Hom YK, Debnath J, Aftab D, Shokat K, Korn WM. Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3K—mTOR pathway in pancreatic adenocarcinoma. Journal of Molecular Medicine. 2011;89:877–889. doi: 10.1007/s00109-011-0774-y. [DOI] [PubMed] [Google Scholar]
- 146.Wolpin BM, Hezel AF, Abrams T, Blaszkowsky LS, Meyerhardt JA, Chan JA, Enzinger PC, Allen B, Clark JW, Ryan DP, Fuchs CS. Oral mTOR Inhibitor Everolimus in Patients With Gemcitabine-Refractory Metastatic Pancreatic Cancer. Journal of Clinical Oncology. 2009;27:193–198. doi: 10.1200/JCO.2008.18.9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Javle MM, Shroff RT, Xiong H, Varadhachary GA, Fogelman D, Reddy SA, Davis D, Zhang Y, Wolff RA, Abbruzzese JL. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC cancer. 2010;10:368. doi: 10.1186/1471-2407-10-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Currie CJ, Poole CD, Gale EAM. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009;52:1766–1777. doi: 10.1007/s00125-009-1440-6. [DOI] [PubMed] [Google Scholar]
- 149.Li D, Yeung SJ, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic Therapies Affect Risk of Pancreatic Cancer. Gastroenterology. 2009;137:482–488. doi: 10.1053/j.gastro.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sadeghi N, Abbruzzese JL, Yeung S-CJ, Hassan M, Li D. Metformin Use Is Associated with Better Survival of Diabetic Patients with Pancreatic Cancer. Clinical Cancer Research. 2012;18:2905–2912. doi: 10.1158/1078-0432.CCR-11-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lonardo E, Cioffi M, Sancho P, Sanchez-Ripoll Y, Trabulo SM, Dorado J, Balic A, Hidalgo M, Heeschen C. Metformin Targets the Metabolic Achilles Heel of Human Pancreatic Cancer Stem Cells. PLoS ONE. 2013;8:e76518. doi: 10.1371/journal.pone.0076518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cantoria MJ, Boros LG, Meuillet EJ. Contextual inhibition of fatty acid synthesis by metformin involves glucose-derived acetyl-CoA and cholesterol in pancreatic tumor cells. Metabolomics. 2014;10:91–104. doi: 10.1007/s11306-013-0555-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife Sciences. 2014;3:e02242. doi: 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Karnevi E, Said K, Andersson R, Rosendah AH. Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer. 2013;13 doi: 10.1186/1471-2407-13-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith J, Rozengurt E. Different Patterns of Akt and ERK Feedback Activation in Response to Rapamycin, Active-Site mTOR Inhibitors and Metformin in Pancreatic Cancer Cells. PLoS ONE. 2013;8:e57289. doi: 10.1371/journal.pone.0057289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Andrzejewski S, Gravel S-P, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer & metabolism. 2014;2:12. doi: 10.1186/2049-3002-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bridges HR, Jones AJY, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochemical Journal. 2014;462:475–487. doi: 10.1042/BJ20140620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gou S, Cui P, Li X, Shi P, Liu T, Wang C. Low Concentrations of Metformin Selectively Inhibit CD133+ Cell Proliferation in Pancreatic Cancer and Have Anticancer Action. PLoS ONE. 2013;8:e63969. doi: 10.1371/journal.pone.0063969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin Disrupts Crosstalk between G Protein-Coupled Receptor and Insulin Receptor Signaling Systems and Inhibits Pancreatic Cancer Growth. Cancer Research. 2009;69:6539–6545. doi: 10.1158/0008-5472.CAN-09-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rozengurt E, Sinnett-Smith J, Kisfalvi K. Crosstalk between Insulin/Insulin-like Growth Factor-1 Receptors and G Protein-Coupled Receptor Signaling Systems: A Novel Target for the Antidiabetic Drug Metformin in Pancreatic Cancer. Clinical Cancer Research. 2010;16:2505–2511. doi: 10.1158/1078-0432.CCR-09-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nakamura M, Ogo A, Yamura M, Yamagushi Y, Nakashima H. Metformin Suppresses Sonic Hedgehog Expression in Pancreatic Cancer Cells. Anticancer research. 2014;34:1765–1769. [PubMed] [Google Scholar]
- 162.Wood AJ, Bailey CJ, Turner RC. Metformin. New England Journal of Medicine. 1996;334:574–579. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 163.Gui DY, Lewis CA, Vander Heiden MG. Allosteric Regulation of PKM2 Allows Cellular Adaptation to Different Physiological States. Science signaling. 2013;6:pe7. doi: 10.1126/scisignal.2003925. [DOI] [PubMed] [Google Scholar]
- 164.Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J-k, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, Thomas CJ, Vander Heiden MG, Cantley LC. Inhibition of Pyruvate Kinase M2 by Reactive Oxygen Species Contributes to Cellular Antioxidant Responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hitosugi T, Kang S, Vander Heiden MG, Chung T-W, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Science signaling. 2009;2:ra73. doi: 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lv L, Xu Y-P, Zhao D, Li F-L, Wang W, Sasaki N, Jiang Y, Zhou X, Li T-T, Guan K-L, Lei Q-Y, Xiong Y. Mitogenic and Oncogenic Stimulation of K433 Acetylation Promotes PKM2 Protein Kinase Activity and Nuclear Localization. Molecular Cell. 2013;52:340–352. doi: 10.1016/j.molcel.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Li Z, Yang P, Li Z. The multifaceted regulation and functions of PKM2 in tumor progression. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2014;1846:285–296. doi: 10.1016/j.bbcan.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 168.Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature. 2011;478:118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Keller KE, Doctor ZM, Dwyer ZW, Lee Y-S. SAICAR Induces Protein Kinase Activity of PKM2 that Is Necessary for Sustained Proliferative Signaling of Cancer Cells. Molecular Cell. 2014;53:700–709. doi: 10.1016/j.molcel.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z. PKM2 Phosphorylates Histone H3 and Promotes Gene Transcription and Tumorigenesis. Cell. 2012;150:685–696. doi: 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gao X, Wang H, Yang JJ, Liu X, Liu Z-R. Pyruvate Kinase M2 Regulates Gene Transcription by Acting as a Protein Kinase. Molecular Cell. 2012;45:598–609. doi: 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2009;463:364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]