

To better understand the dopaminergic defects that give rise to dystonia, Rose et al. generate a knockin mouse model of L-DOPA-responsive dystonia that recapitulates the core features of the human disorder. The timing and extent of dopamine deficiency in combination with abnormal D1 and D2 receptor responses mediates dystonia development.
Keywords: striatum, dopamine, tyrosine hydroxylase, trihexyphenidyl, corticostriatal

To better understand the dopaminergic defects that give rise to dystonia, Rose et al. generate a knockin mouse model of L-DOPA-responsive dystonia that recapitulates the core features of the human disorder. The timing and extent of dopamine deficiency in combination with abnormal D1 and D2 receptor responses mediates dystonia development.
Abstract
Abnormal dopamine neurotransmission is associated with many different genetic and acquired dystonic disorders. For instance, mutations in genes critical for the synthesis of dopamine, including GCH1 and TH cause l-DOPA-responsive dystonia. Despite evidence that implicates abnormal dopamine neurotransmission in dystonia, the precise nature of the pre- and postsynaptic defects that result in dystonia are not known. To better understand these defects, we generated a knock-in mouse model of l-DOPA-responsive dystonia (DRD) mice that recapitulates the human p.381Q>K TH mutation (c.1141C>A). Mice homozygous for this mutation displayed the core features of the human disorder, including reduced TH activity, dystonia that worsened throughout the course of the active phase, and improvement in the dystonia in response to both l-DOPA and trihexyphenidyl. Although the gross anatomy of the nigrostriatal dopaminergic neurons was normal in DRD mice, the microstructure of striatal synapses was affected whereby the ratio of axo-spinous to axo-dendritic corticostriatal synaptic contacts was reduced. Microinjection of l-DOPA directly into the striatum ameliorated the dystonic movements but cerebellar microinjections of l-DOPA had no effect. Surprisingly, the striatal dopamine concentration was reduced to ∼1% of normal, a concentration more typically associated with akinesia, suggesting that (mal)adaptive postsynaptic responses may also play a role in the development of dystonia. Administration of D1- or D2-like dopamine receptor agonists to enhance dopamine signalling reduced the dystonic movements, whereas administration of D1- or D2-like dopamine receptor antagonists to further reduce dopamine signalling worsened the dystonia, suggesting that both receptors mediate the abnormal movements. Further, D1-dopamine receptors were supersensitive; adenylate cyclase activity, locomotor activity and stereotypy were exaggerated in DRD mice in response to the D1-dopamine receptor agonist SKF 81297. D2-dopamine receptors exhibited a change in the valence in DRD mice with an increase in adenylate cyclase activity and blunted behavioural responses after challenge with the D2-dopamine receptor agonist quinpirole. Together, our findings suggest that the development of dystonia may depend on a reduction in dopamine in combination with specific abnormal receptor responses.
Introduction
Dystonia is characterized by involuntary muscle contractions that cause debilitating twisting movements and postures. Although the mechanisms underlying most forms of dystonia are not understood, there are abnormalities shared by many dystonic disorders that provide clues. Abnormal dopamine neurotransmission is associated with many different dystonic disorders (Perlmutter and Mink, 2004; Wichmann, 2008). Mutations in genes critical for the synthesis of dopamine, including GTP cyclohydrolase 1 (GCH1) and tyrosine hydroxylase (TH) cause l-DOPA-responsive dystonia (DRD) (Ichinose et al., 1994; Knappskog et al., 1995; van den Heuvel et al., 1998; Thony and Blau, 2006). Dystonia is also a prominent feature of other inherited disorders associated with disrupted presynaptic dopamine regulation including dopamine transporter (DAT, encoded by SLC6A3) deficiency syndrome (Kurian et al., 2011), amino acid decarboxylase deficiency (Hyland et al., 1992; Brun et al., 2010), vesicular monoamine transporter 2 deficiency (Rilstone et al., 2013), and Lesch-Nyhan disease (Jinnah et al., 2006). Additionally, dystonia sometimes occurs as a complication of chronic 3,4-l-dihydroxyphenylalanine (l-DOPA) treatment in Parkinson’s disease. Similarly, interrupting dopamine neurotransmission with dopamine receptor antagonists can cause drug-induced dystonia (Mehta et al., 2015). Despite strong evidence that abnormal dopamine neurotransmission contributes to both inherited and acquired dystonias, exactly how the dopaminergic defects cause dystonia is poorly understood.
Studies of patients with DRD have provided some insight into the role of abnormal dopamine neurotransmission in dystonia. Classical DRD is characterized by childhood-onset generalized dystonia with diurnal fluctuation whereby symptoms worsen throughout the course of the day (Nygaard et al., 1988; Segawa and Nomura, 1993; Calne, 1994). The distinguishing feature of DRD is the dramatic improvement in symptoms after administration of l-DOPA, the precursor of dopamine, norepinephrine and epinephrine, demonstrating the association between low levels of catecholamines and dystonic movements (Segawa and Nomura, 1993). Mutations in GCH1, which encodes an enzyme necessary for synthesis of the TH cofactor tetrahydrobiopterin (BH4) are the most common causes of autosomal dominant DRD (Clot et al., 2009). The reduced levels of BH4 impair TH activity, resulting in brain dopamine concentrations in DRD patients that are <20% of normal (Ichinose et al., 1994; Rajput et al., 1994). Mutations in TH itself can cause autosomal recessive DRD (Gorke and Bartholome, 1990; Knappskog et al., 1995) or a severe early childhood akinetic-rigidity syndrome (Ludecke et al., 1996; de Rijk-van Andel et al., 2000), depending on the severity of the mutation (Fossbakk et al., 2014). For example, the p.381Q>K mutation, which causes DRD, decreases enzyme activity to ∼15% of normal. In contrast, the p.205 L>P mutation, which causes the more severe phenotype, attenuates enzyme activity to only ∼2% of normal (Ludecke et al., 1996). Combined, the GCH1 and TH mutations suggest that the amount of residual TH activity and brain catecholamine concentrations is a critical determinant in the development of dystonia. However, our understanding of dopaminergic signalling in dystonia remains quite limited. Therefore, we generated a knock-in mouse model of DRD that recapitulates the human p.381Q>K TH mutation and examined both pre- and postsynaptic determinants of dystonia.
Materials and methods
Generation of Thtm1Ehess (DRD) mice
The c.1160C>A (p.382Q>K) mutation in exon 9 of mouse Th, homologous to the DRD-causing c.1141C>A (p.381Q>K) mutation in human TH, was introduced by site-directed mutagenesis into a C57BL/6 bacterial artificial chromosome clone encompassing exons 1–10 of mouse Th (Fig. 1A). After screening C57BL/6-derived embryonic stem cells for homologous recombination by Southern blot, homologous recombination was verified in the genomic DNA from the progeny of the chimeric mice using long PCR (Fig. 1A and B) with primers P1, 5’-GACGTCAGCCTGGCCTTTAAGA-3’, P2, 5’-AGATGGAATGGGAAGGCTCT-3’, and P3, 5’-AGGCCAGAGGCCACTTGTGTAG-3’ to confirm the 5’ end; and primers P4, 5’-GACGAGTTCTTCTGAGGGGATCAA-3’, P5, 5’-ACAGCCTTACCTGTTGTGGG-3’, and P6, 5’-AGTCATGGTAGGCTCTGAAAGTGG-3’ to confirm the 3’ end. Additionally, an amplicon encompassing exon 9 was sequenced to verify that mice testing positive for the long PCR assay also carried the point mutation (Fig. 1C). The Cre-deleter strain C57BL/6-Tg(Zp3-cre)93Knw/J (The Jackson Laboratory) was used to remove the neomycin cassette. Mice were genotyped thereafter with forward and reverse primers 5’-ACACCGAAGCAGAGACTGT-3’ and 5’-CTGATGCTACTTCTCCAGG-3’.
Figure 1.

Generation and molecular characterization of the DRD allele. (A) Schematic of the targeting construct encoding the c.1160C>A mutation in exon 9 of Th, and a neomycin resistance cassette (Neo), flanked by two loxP sites. (B) Results of long PCRs confirming the correct genomic location of the targeting construct. Arrows in A indicate direction and approximate position of primers used to verify homologous recombination; primers P1 and P6 were to genomic DNA outside of the targeting construct. PCR products (4–6 kb) amplified from knock-in (DRD + Neo) and normal mouse genomic DNA correspond to the indicated primers. (C) Sequences obtained from PCR amplicons from genomic DNA verifying the C>A point mutation in Th. (D) Sequence of the reverse transcription-PCR amplicon from brain mRNA illustrating mRNA expression of the c.1160C>A mutation. (E) Quantitative RT-PCR demonstrated no difference in the quantity of Th mRNA between normal (n = 8) and DRD mice (n = 8; P > 0.1, Student’s t-test). Values represent mean ± SEM.
To circumvent the high perinatal lethality exhibited by homozygous C57BL/6J DRD mice, C57BL/6J DRD/+ (heterozygous) mice were crossed to DBA/2J mice to promote hybrid vigour. The F1 heterozygous progeny were crossed to produce DRD mice, heterozygous and wild-type (normal) littermates. Because norepinephrine is essential for cardiovascular development in mice (Thomas et al., 1995), the dams’ drinking water was supplemented with 100 µg/ml of the adrenergic agonist isoproterenol (Sigma-Aldrich), 20 µg/ml phenylephrine (Sigma-Aldrich) and 2.5 mg/ml ascorbic acid (Sigma-Aldrich), at embryonic Day 8.5 through to birth (Portbury et al., 2003). At birth, DRD mice were indistinguishable from normal littermates and born near Mendelian frequency. By the second postnatal week, DRD mice failed to thrive. Therefore, from postnatal Days 9.5 to 16.5, the dams’ drinking water was supplemented with 1.5 mg/ml l-DOPA (Sigma-Aldrich), 0.5 mg/ml benserazide (Sigma-Aldrich), and 2.5 mg/ml ascorbic acid to facilitate feeding, growth and normal movement. Thereafter, mice received a daily injection subcutaneously (s.c.) of 10 mg/kg l-DOPA, 2.5 mg/kg benserazide and 2.5 mg/ml ascorbic acid in saline. TH knockout mice are typically supplemented with 50 mg/kg l-DOPA (Zhou and Palmiter, 1995), however, we found that 10 mg/kg was adequate to restore function. Heterozygous and normal littermates received all treatments in parallel to control for both treatment and handling. l-DOPA supplementation was terminated >24 h prior to all experiments. Housing conditions were in accordance with Emory University’s institutional animal care and use committee. The light period was 7 am to 7 pm. All experiments used 2–4-month-old male and female mice.
Quantitative reverse transcriptase-PCR
Quantitative PCR was performed using an Applied Biosystems Fast 7500 Real-Time PCR system and SYBR® Green Select Mastermix (Life Technologies) in triplicate reactions using brain cDNA. Th primers (5’-GGAACGGTACTGTGGCTACC-3’ and 5’-AACCAGTACACCGTGGAGAG-3’) amplified a 342-bp region containing the c.1160C>A mutation. Other primers included: D1 dopamine receptor (D1DAR) (5’-ATCGTCACTTACACCAGTATCTACAGGA-3’ and 5’-GTGGTCTGGCAGTTCTTGGC-3’), D2DAR (5’-TGGCTGCCCTTCTTCATCACGC -3’ and 5’-TGAAGGCCTTGCGGAACTCAATGT -3’), D3DAR (5’-CCTCTGAGCCAGATAAGCAGC-3’ and 5’- AGACCGTTGCCAAAGATGATG -3’). Data were analysed by the ΔCt method (Schmittgen and Livak, 2008), using 18 s rRNA (5’-TTGACGGAAGGGCACCACCAG-3’ and 5’-GCACCACCACCCACGGAAATCG-3’) as reference. Reverse transcriptase-PCR Th amplicons were sequenced to verify the presence of the c.1160C>A mutation.
Tissue monoamines
Dopamine, 3,4-dihydroxypheynlacetic acid (DOPAC), norepinephrine, l-DOPA, serotonin (5-HT), and 5-hydroxyindoleacetic acid (5-HIAA) were examined by high performance liquid chromatography (HPLC) with electrochemical detection (Song et al., 2012). To assess the effect of l-DOPA administration, mice were injected (s.c.) with l-DOPA, or saline, 45 min before brain collection at 8 pm. The effect of l-3,4-dihydroxyphenylserine (l-DOPS) treatment on tissue monoamines was also assessed at 8 pm (Thomas et al., 1998).
In vivo TH enzyme activity assay
TH enzyme activity was determined in vivo (Carlsson et al., 1972). Mice were injected (s.c.) with 150 mg/kg NSD-1015, an aromatic acid decarboxylase inhibitor, and brains collected 45 min later. Brain regions were dissected and l-DOPA accumulation was assessed by HPLC.
Radioligand binding
Striatal 3H-SCH 23390 and 3H-spiperone saturation binding analyses were performed by adding 0.2–0.4 µg protein to duplicate tubes containing 3H-SCH 23390 (0.125–2 nM; 81.9 Ci/mmol, Perkin Elmer) or 3H-spiperone (0.0625–1 nM; 73.49 Ci/mmol, Perkin Elmer) in a final volume of 0.6 ml and incubated for 30 min at 37°C. Non-specfic binding was determined with 100 µM cis-flupentixol (Sigma).
Adenylate cyclase activity
The effect of SKF 81297 on adenylate cyclase activity in striatal homogenates was determined as described (Lee et al., 2002). Quinpirole-mediated adenylate cyclase activity was assessed as described (Fan and Hess, 2007). Triplicate reactions were initiated with the addition of 10–20 µg membrane protein and incubated at 37°C for 15 min. Reactions were terminated by boiling for 3 min and cAMP accumulation was assessed by enzyme-linked immunosorbent assay (Cell Biolabs).
Immunohistochemistry
Brain tissue for immunohistochemistry was prepared by perfusion fixation with paraformaldehyde solution (Song et al., 2012). Using every sixth coronal section, parallel series of sections were immunostained for TH (1:1000, Pel-Freez) or DAT (1:1000, Millipore) and processed with 3,3’-diaminobenzidine (Egami et al., 2007; Lohr et al., 2014). For DAT, tissue was treated with Citra antigen retrieval reagent (BioGenex) for 1 h at 70°C prior to incubation with the primary antibody.
Stereological assessment
Stereological assessment of TH-positive midbrain neurons was performed as described (Song et al., 2012), with minor adjustments. The TH-positive region of the midbrain was outlined using a ×4 objective using a mouse brain atlas (Paxinos and Franklin, 2001) to estimate the borders of the substantia nigra and ventral tegmental area. TH-positive cells were counted at ×40 using an optical fractionator probe with a 55 185µm2 counting grid and a 10 000 µm2 frame with 30-µm depth. The volume of the dorsal striatum was estimated using the Cavalieri method. The Gundersen coefficient of error was below 0.1.
Electron microscopy
Striatal tissue was immunostained for vesicular glutamate transporters 1 (vGluT1, encoded by SLC17A7; 1:5000, Millipore) or vesicular glutamate transporters 2 (vGluT2, encoded by SLC17A6; 1:5000, Millipore), and processed for electron microscopy (Song et al., 2013). The specificity of these antibodies has been determined previously (Raju and Smith, 2005). Sections from the surface of the blocks were scanned at ×25 000 with a JEOL electron microscope. To estimate synaptic density, immunoreactive boutons were counted from 50 randomly collected images in each animal and expressed as boutons per area scanned. The postsynaptic targets in contact with each labelled terminal were identified based on ultrastructural features (Peters et al., 1991). The ratio of labelled boutons in contact with spines versus dendrites was calculated for each terminal population.
Assessment of abnormal movement
A behavioural inventory was used to define the type of abnormal movement, including tonic flexion (forelimbs, hindlimbs, trunk, head), tonic extension (forelimbs, hindlimbs, trunk, head), clonus (forelimbs and hindlimbs), twisting (trunk, head), and tremor (forelimbs, hindlimbs, trunk, head), as described (Devanagondi et al., 2007; Shirley et al., 2008; Raike et al., 2013). Abnormal movements were scored for 30 s at 10-min intervals for 60 min. Behavioural scores were calculated by summing the scores from all scoring bins. For time of day, l-DOPA, or trihexyphenidyl tests, mice were scored in a novel open field after >24h l-DOPA washout. For dopamine receptor-selective compounds, mice were habituated to test chambers for >24 h before the test. Behavioural raters were blinded to genotype and treatment.
Drug challenges
Compounds were administered (s.c.) in saline, in a volume of 10 ml/kg. Behavioural experiments started 10 min after drug administration, except l-DOPS, which was administered as described (Thomas et al., 1998). For dose response experiments, mice were tested in a repeated measures design with a pseudorandom order of drug doses and vehicle; each mouse received every dose only once within an experiment. Mice were given a 4-day drug washout between challenges. Amphetamine, raclopride, SCH 23390, l-DOPA, and trihexyphenidyl were obtained from Sigma-Aldrich. SKF 81297 and quinpirole were obtained from Tocris Biosciences. l-DOPS was generously provided by Dainippon-Sumitomo Inc. Microinjections were performed as reported (Pizoli et al., 2002; Raike et al., 2013) using 0.5 µl of 400 ng/ml l-DOPA in either midline cerebellum [−6.5 mm anteroposterior (AP) from bregma, 0 mm mediolateral (ML) from midline, −2 mm ventral (V)] or striata, bilaterally (+1 mm AP, ±2 mm ML, −3 mm V). Scoring started 20 min after microinjection. Microinjection sites were histologically verified. Observers were blinded to treatment and genotype.
Locomotor activity
Mice were habituated to photocell activity cages for >24 h during the l-DOPA washout period (Song et al., 2012). For spontaneous locomotor activity, recording started at 1 pm and continued for 24 h. For SKF 81297 and amphetamine challenge, mice were injected at 2 pm, and locomotor activity was recorded for 1 h. For quinpirole, mice were injected at 8 pm and locomotor activity was recorded for 1 h. The time of test was selected to avoid ceiling or floor effects. Mice had access to food and water ad libitum.
Stereotypy
Mice were rated for stereotypy during the locomotor activity tests every 10 min for 30 s. A 0–5 behavioural scale was used: 0 = sleeping; 1 = awake, inactive; 2 = active or exploring; 3 = hyperactive; 4 = hyperactive with bursts of stereotypy; and 5 = continuous stereotypy.
Motor performance tests
Rotarod and pole tests were performed as described (Rose et al., 2014). For rotarod, mice were habituated for 30 s to a 4-cm diameter rod (Columbus Instruments) rotating at 4 rpm. Rotation speed was increased from 4 to 40 rpm over a 6-min period. Mice that did not fall during the 6-min period were recorded as 6 min. For the pole test, mice were placed with their head oriented upwards on a 50-cm tall, 1-cm diameter vertical pole placed inside their home cage. The time for each mouse to descend and have all four paws on the cage floor was recorded. The effect of time of day was assessed on mice trained on the motor task for two consecutive days. Mice remained on their daily l-DOPA supplementation during the training sessions; DRD mice performed comparably to normal mice during training. Mice were tested on the third day after >24 h withdrawal from l-DOPA. The effect of l-DOPA was tested at 2 pm in mice that were naïve to the rotarod or trained on the pole for 2 days before the test.
Statistics
Biochemical and behavioural data were analysed using ANOVA. Significant effects within groups were tested post hoc with the Holm-Sidak test or Tukey’s post hoc test if there was no clear baseline condition (e.g. time of day). Student’s t-test was used for anatomical comparisons between genotypes or for post hoc analyses when the data spanned several orders of magnitude. SigmaStat (Systat Software) was used for all analyses. Detailed statistical analyses are presented in the figure legends.
Results
Generation of a knock-in model of DRD
The DRD-causing p.381Q>K TH mutation (c.1141C>A) was used to create a knock-in mouse because this mutation causes classical DRD in humans and Q381 is conserved across species (Mus musculus Q382). Additionally, although many DRD patients are compound heterozygotes, carrying two different mutant TH alleles, p.381Q>K causes DRD in the homozygous state (Knappskog et al., 1995), so it was possible to genetically recapitulate the disorder with a single knock-in mutation. Homologous recombination (Fig. 1A) was confirmed using genomic DNA from the F1 progeny of the chimeric mice and a series of long PCRs (Fig. 1B). A short sequence encompassing the mutation was amplified and sequenced to independently ascertain the presence of the mutation (Fig. 1C). Expression of the mutation in brain was verified by sequencing the reverse transcriptase PCR product from brain Th (Fig. 1D). Quantitative PCR demonstrated that Th mRNA was expressed at comparable levels in normal mice and DRD mice (Fig. 1E).
Reduced brain TH activity and catecholamines in DRD mice
TH activity was assessed in vivo by measuring l-DOPA accumulation after inhibition of aromatic amino acid decarboxylase by NSD-1015. This indirect assay was selected over more direct in vitro methods to provide a more realistic estimate of actual TH activity in brain. In midbrain, which contains the dopaminergic cell bodies of the substantia nigra and ventral tegmental area, l-DOPA accumulation in DRD mice (homozygous mutants) was significantly reduced to ∼15% of normal (Fig. 2A; P < 0.001). This is consistent with the human p.381Q>K TH activity, which is also ∼15% of normal (Knappskog et al., 1995). Activity was similarly reduced in cortex (P < 0.001) and cerebellum (P < 0.001). However, in striatum, which receives dense innervation of TH-containing axons from the midbrain, l-DOPA accumulation in DRD mice was only ∼1% of normal (P < 0.001). A small but significant reduction in l-DOPA accumulation was also observed in heterozygous mice in midbrain (P < 0.05) and striatum (P < 0.001) demonstrating that the effect is dependent on gene dose. These results imply that the reductions in TH were less severe in the brain regions containing cell soma than in the axon terminal fields, which may be explained by the reported reduction in the stability of the p.381Q>K TH protein (Fossbakk et al., 2014).
Figure 2.

TH dysfunction in DRD mice. (A) TH activity was assessed in vivo in normal (n = 5), heterozygous (n = 7), and DRD mice (n = 8). TH activity was significantly reduced in all brain regions tested in DRD mice, including striatum [F(2,17) = 96.5, P < 0.001], midbrain [F(2,17) = 44.1, P < 0.001], cortex [F(2,17 = 42.3, P < 0.001] and cerebellum [F(2,17) = 8.3, P < 0.01]. (B and C) Regional analysis of dopamine and norepinephrine concentrations in normal (n = 9), heterozygous (n = 6), and DRD mice (n = 6). Dopamine was significantly reduced in striatum [F(2,18) = 137.4, P < 0.001], midbrain [F(2,18) = 17.1, P < 0.001], and cortex [F(2,18) = 15.0, P < 0.001]. Norepinephrine was significantly reduced in striatum [F(2,18) = 10.0, P < 0.001], midbrain [F(2,18) = 136.7, P < 0.001], cortex [F(2,18) = 100.2, P < 0.001] and cerebellum [F(2,18) = 54.5, P < 0.001]. (D) Representative sections immunostained for TH or DAT from striatum or midbrain of normal and DRD mice (Scale bars = 1.5 mm, striatum; 200 µm, midbrain). (E) Stereological cell counts of TH-positive neurons in the substantia nigra (SN) (P > 0.1, Student’s t-test) and ventral tegmental area (VTA) (P > 0.1, Student’s t-test). l-DOPA supplementation was terminated >24 h before sacrificing animals for analysis at 8 pm. Values represent mean ± SEM. Statistical analyses for TH activity and monoamine concentrations were performed for each region using a one-way ANOVA with a Holm-Sidak post hoc analyses where appropriate; *P < 0.05, **P < 0.01, ***P < 0.001 compared to normal.
To determine the neurochemical consequences of the mutation, we measured monoamines and their metabolites in several brain regions. Consistent with the reduction in TH enzyme activity, dopamine was significantly reduced in striatum (P < 0.001), midbrain (P < 0.001) and cortex (P < 0.001) of DRD mice (Fig. 2B). Surprisingly, the reduction in dopamine content was as severe in midbrain (∼99%) as in striatum (∼99%). DOPAC, the primary metabolite of dopamine, was also reduced in DRD mice (P < 0.001). Sex differences were not observed. However, the DOPAC/dopamine ratio in DRD mice was nearly 4-fold higher than normal in midbrain (P < 0.001) and striatum (P < 0.001), suggesting that dopamine turnover occurs at a higher rate in the mutants (Supplementary Table 1), which may explain the low midbrain dopamine concentration despite the partial sparing of TH activity. Males exhibited a higher DOPAC/dopamine ratio than females (P < 0.001) only in the midbrain; the significance of this finding is not clear. Norepinephrine concentrations were also significantly reduced throughout brain (Fig. 2C; P < 0.001). With the exception of brainstem, where 5-HT was significantly increased (P < 0.01), 5-HT concentrations were unaffected (Supplementary Table 1). However, 5-HIAA, the major metabolite of 5-HT, and the 5-HIAA/5-HT ratio were significantly increased in most brain regions (Supplementary Table 1; P < 0.001).
Anatomy of the striatum and midbrain dopamine neurons
To determine if the TH mutation affected the anatomy of midbrain dopamine neurons, we examined the morphology of TH-positive neurons (Fig. 2D). TH immunostaining of normal mice labelled neuronal soma and dendrites in the midbrain and a dense network of axons in the striatum. A similar pattern of immunostaining was observed in DRD mouse brain, although staining appeared weaker, especially in the dendrites and striatum. These results are consistent with the neurochemical studies, implying more severe loss of TH activity in the striatum compared with the midbrain. Despite the relatively weaker TH immunostaining, stereological counts of neurons in the midbrain of DRD mice were not significantly different from normal mice (Fig. 2E). These results imply no loss of TH-positive neurons in the DRD mice.
We next conducted immunostaining for DAT, another marker of dopaminergic neurons. Despite the loss of TH immunostaining in the DRD mouse striatum, DAT staining was normal. Further, stereological measures revealed no difference in striatal volume of DRD mice compared to normal mice (not shown, P > 0.1 Student’s t-test). These results imply that midbrain dopamine neurons and their axonal projections to the striatum are intact, consistent with anatomical studies of DRD patients (Snow et al., 1993; Rajput et al., 1994).
Cortico-striato-pallido-thalamic pathways are implicated in dystonia (Neychev et al., 2008; Lehericy et al., 2013), and glutamatergic synapses in the striatum undergo complex changes in adults with striatal dopamine depletion (Ingham et al., 1998; Villalba et al., 2009; Villalba and Smith, 2013). Therefore, we assessed the distribution and density of corticostriatal and thalamostriatal terminals in DRD mice using vGluT1 as a specific marker of cortical boutons (Fig. 3A) and vGluT2 as a specific marker of thalamic boutons (Fig. 3B; Raju et al., 2006). No significant difference was observed in the density of either population of terminals between normal and DRD mice (Fig. 3C). However, the ratio of axo-spinous to axo-dendritic synaptic contacts was significantly smaller for vGluT1-positive terminals in DRD mice compared to normal mice (Fig. 3D; P < 0.05), and there was a similar trend for vGluT2-positive terminals (P = 0.052). Similar microstructural abnormalities are also observed in DYT1 mutant mice, another model of inherited generalized dystonia (Song et al., 2013).
Figure 3.

Glutamatergic cortico- and thalamo-striatal terminals in DRD mice. (A and B) Examples of vGluT1- (A, corticostriatal) or vGluT2- (B, thalamostriatal) immunoreactive terminals forming an axo-spinous (A) or an axo-dendritic (B) asymmetric synapse in the mouse dorsolateral striatum. Scale bars = 500 nm in A and B. (C) No significant difference was observed in the density of either vGluT1-positive or vGluT2-positive terminals between the genotypes (n = 3/genotype; Student’s t-test). (D) The ratio of vGluT1-positive (n = 3 animals/genotype, 177-180 terminals/genotype) or vGluT2-positive (n = 3/genotype, 133–155 terminals/genotype) axo-spinous to axo-dendritic synaptic contacts was measured. This ratio was significantly smaller (P < 0.05, Student’s t-test) for vGluT1-positive terminals, and approached significance for vGluT2-positive terminals (P = 0.052, Student’s t-test), in the dorsolateral striatum of DRD mice. Values represent mean ± SEM; *P < 0.05. Den = dendrite; Sp = spine; Term = terminal.
DRD mice display the core behavioural features of DRD
DRD mice exhibited overt abnormal involuntary movements. Forelimbs were often tucked tightly into the chest while hindlimbs were lifted off the cage floor in a stereotyped paddling motion with toes spread (Fig. 4A). The movements were comprised largely of flexion of the forelimbs, hindlimbs, and lower trunk. Occasionally, the trunk was pressed down into the cage floor. These movements were dynamic, not fixed postures, and best described as dystonia (Supplementary Videos 1 and 2). No sex bias was observed. Heterozygous mice never exhibited abnormal movements, consistent with the recessive mode of inheritance in humans. Abnormal movements in DRD mice were sometimes observed at the start of the active (dark) period (8 pm) and increased throughout the night with the highest occurrence at the start of the inactive period (8 am) (Fig. 4B and C; P < 0.001). This pattern of activity is reminiscent of DRD patients whose symptoms worsen throughout the day. Consistent with the diurnal fluctuation in dystonic movements, there was no difference in the performance of normal and DRD mice on rotarod or pole test at 8 pm. However, at 8 am, DRD mice were significantly impaired on both tests (Supplementary Fig. 1A, P < 0.01; and Supplementary Fig. 1B, P < 0.001). DRD mice exhibited a normal circadian rhythm with an increase in locomotor activity at the start of the active period but this was followed by a rapid decline to below normal levels throughout the night (Fig. 4D; P < 0.01). The reduction in locomotor activity was most pronounced during the last 6 h of the active period, when dystonic movements were most severe.
Figure 4.

The DRD behavioural phenotype. (A) DRD mice exhibit dystonic movements that occurred most often in the limbs. (B) The abnormal movements were largely composed of transiently sustained flexion in both the inactive (light) and active (dark) period. Data collected in the active are shaded in grey. (C) Dystonic movements were assessed in DRD mice over the course of a day (n = 8). DRD mice expressed significantly fewer dystonic movements at 8 pm than at other times of day [F(3,28) = 6.8, one-way repeated measures ANOVA, P < 0.05 8 pm versus 2 am and 2 pm, P < 0.001 8 pm versus 8 am, Tukey’s post hoc analysis]. (D) Spontaneous locomotor activity was assessed over 24 h in normal and DRD mice (n = 10/genotype). Locomotor activity was significantly reduced in DRD mice compared to normal mice across the entire 24 h period [F(1,18) = 9.6, two-way repeated measures ANOVA; P < 0.01, Holm-Sidak post hoc analysis]. In the active period, there was a significant genotype × time interaction effect [F(1,18) = 5.2, two-way repeated measures ANOVA, P < 0.05, Holm-Sidak post hoc analysis] reflecting the reduction in locomotor activity in DRD mice in the last 6 h of the active period. (E) Peripheral administration of l-DOPA significantly reduced dystonic movements in DRD mice (n = 7/treatment; P < 0.001, paired Student’s t-test) at 2 pm. (F) Administration of trihexyphenidyl (n = 8/dose), a muscarinic acetylcholine receptor antagonist, significantly reduced dystonic movements in DRD mice [F(3,21) = 3.4, one-way repeated measures ANOVA, P < 0.05, Holm-Sidak post hoc analysis] at 2 pm. Values represent mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
The preservation of some locomotor activity in DRD mice suggests that the residual catecholamines are neurochemically and behaviourally relevant. We tested this hypothesis by assessing locomotor activity after challenging DRD and normal mice with 5 mg/kg amphetamine, which causes monoamine release from synaptic terminals. As expected, normal mice exhibited a significant increase in locomotor activity (P < 0.001). DRD mice also exhibited a significant increase in locomotor activity in response to amphetamine (P < 0.01), albeit less than the increase observed in normal mice (Supplementary Fig. 2), suggesting that the low concentrations of catecholamines in DRD mice are adequate to mediate behaviour.
The defining feature of DRD is alleviation of abnormal movements by l-DOPA. Peripheral administration of 10 mg/kg l-DOPA nearly abolished the abnormal movements exhibited by DRD mice (Fig. 4E; P < 0.001). l-DOPA also rescued DRD mouse performance in rotarod and pole test (Supplementary Fig. 1C and D, P < 0.001 and P < 0.001, respectively). Trihexyphenidyl, a non-selective muscarinic acetylcholine receptor antagonist, is also effective in DRD patients (Jarman et al., 1997). DRD mice exhibited a significant dose-dependent reduction in abnormal movements following trihexyphenidyl administration (Fig. 4F, P < 0.05). Because trihexyphenidyl can have sedative effects at high doses, locomotor activity was measured at the same time as the assessment of abnormal movements. Locomotor activity was not affected by any dose of trihexyphenidyl tested [not shown, one-way repeated measures ANOVA, F(3,21) = 0.3, P > 0.1], suggesting that the effect on abnormal movements was not attributable to a global reduction in motor behaviour.
Restoring striatal dopamine alleviates the abnormal movements
To identify the brain region(s) associated with the abnormal movements, we microinjected l-DOPA into regions known to play a prominent role in dystonia. Data from animals and human imaging studies suggest that dystonia is a network disorder involving cortico-striato-pallido-thalamo-cortical and cerebello-thalamo-cortical pathways (Neychev et al., 2008; Carbon et al., 2010; Niethammer et al., 2011; Lehericy et al., 2013). Therefore, the striatum and cerebellum were chosen for microinjection because these regions are often implicated in dystonia and receive abundant catecholaminergic innervation (dopamine in the striatum and norepinephrine in the cerebellum). Bilateral microinjections of l-DOPA into the dorsal striatum significantly reduced the abnormal movements (Fig. 5A, P < 0.01). By contrast, microinjection of l-DOPA into midline cerebellar vermis had no discernible effect on abnormal movements.
Figure 5.

Regional and neurochemical analysis of dystonia in DRD mice (n = 4–5/condition). (A) Microinjection of l-DOPA (200 pg) into the striatum of DRD mice significantly reduced abnormal movements [F(2,8) = 6.8, one way repeated measures ANOVA; P < 0.01 Holm Sidak post hoc analysis], whereas cerebellar microinjections did not (P > 0.1, Holm Sidak post hoc analysis). (B) Peripheral administration of 1 g/kg l-DOPS did not affect abnormal movements in DRD mice (P > 0.4, Student’s t-test). Microinjection experiments were performed at 2 pm and l-DOPS experiments were performed at 8 pm. Values represent mean ± SEM; **P < 0.05.
TH is the rate-limiting enzyme in the synthesis of both dopamine and norepinephrine, and both transmitters are reduced in DRD mice. To better understand the role of each catecholamine in the abnormal movements, we challenged DRD mice with either l-DOPA, which is a precursor for both dopamine and norepinephrine, or l-DOPS, which can be converted to norepinephrine by aromatic acid decarboxylase, but does not directly increase dopamine levels (Thomas et al., 1998). As expected, peripheral administration of 10 mg/kg l-DOPA, a dose that nearly eliminates abnormal movements and restores motor performance, significantly increased dopamine (P < 0.001) and norepinephrine (P < 0.001) concentrations throughout the brain (Supplementary Table 2). Despite ameliorating the motor deficits and abnormal movements (Fig. 4E, Supplementary Fig. 1C and D), striatal dopamine was repleted to only 22% of normal striatal dopamine concentrations. Peripheral administration of l-DOPS (1 g/kg) significantly increased norepinephrine concentrations throughout the brain (Supplementary Table 2, P < 0.05), but had no significant beneficial effect on the abnormal movements (Fig. 5B).
Presynaptic mechanisms associated with the diurnal fluctuation in abnormal movements
Symptoms in DRD patients and mice worsen throughout the course of the active period, but the mechanisms underlying the diurnal variation are not understood. Because TH expression and brain dopamine concentrations have a distinct circadian rhythm (DiRaddo and Kellogg, 1975; Cahill and Ehret, 1981), we assessed striatal TH activity, steady-state dopamine content, and dopamine turnover at 8 am, when dystonia commonly occurs, and 8 pm, when dystonia is infrequent. Striatal TH activity in normal mice was significantly increased at 8 am compared to 8 pm (Fig. 6A; P < 0.01), in agreement with previous studies (Weber et al., 2004; Ferris et al., 2014). There was also a non-significant trend for an increase in TH activity at 8 am in DRD mice. Striatal dopamine concentration was also elevated at 8 am in normal mice (Fig. 6B; P < 0.001), but not in DRD mice. In fact, the trend in DRD mice was toward a reduction in dopamine at 8 am. The diurnal fluctuation in striatal DOPAC concentration supported this trend; DOPAC concentrations were 3-fold higher at 8 pm than at 8 am in DRD mice (Fig. 6C; P < 0.05). By contrast, DOPAC concentrations were significantly lower at 8 pm than 8 am in normal mice (P < 0.01). Most DOPAC is located in the extracellular space (Wallace and Traeger, 2012). Therefore, one explanation for the abnormal increase in DRD mouse DOPAC concentrations at 8 pm is that the residual striatal dopamine in DRD mice is rapidly released and metabolized early in the active period. Additionally, there was a 375% increase in the ratio of striatal DOPAC/dopamine in DRD mice at 8 pm compared to normal mice at 8 pm (P < 0.001) and compared to both DRD and normal mice at 8 am (Fig. 6D; P < 0.001), suggesting that an increase in dopamine turnover is associated with alleviation of the abnormal movements.
Figure 6.
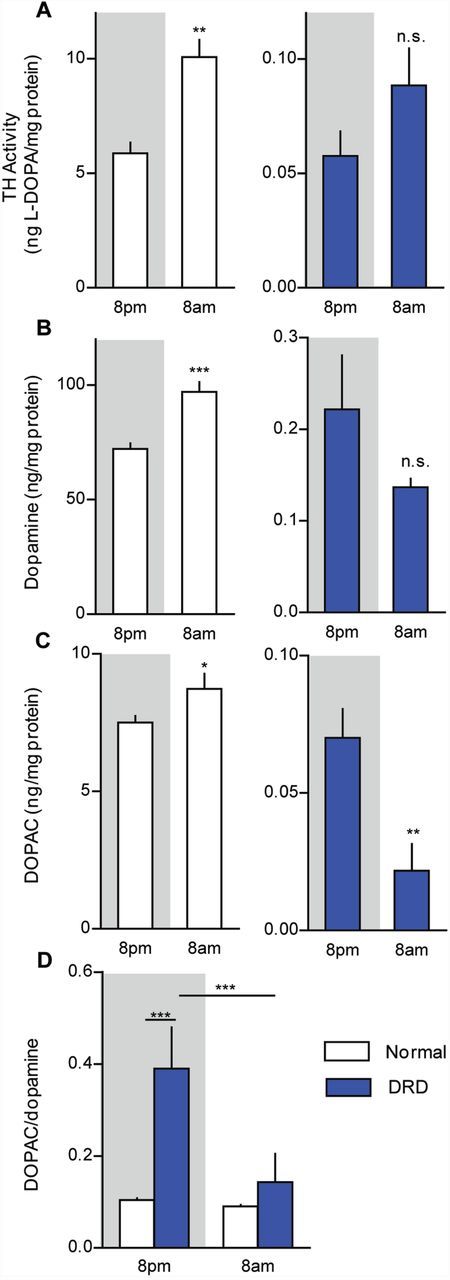
Diurnal fluctuations in dopamine metabolism. (A) Striatal TH activity was assessed in the early active period (8 pm) and early inactive period (8 am) in normal and DRD mice (n = 5–8/condition). There was a significant genotype × time interaction effect [F(1,21) = 25.8, P < 0.001], with normal mice exhibiting significantly more TH activity at 8 am than at 8 pm (P < 0.01) but no significant difference was observed between times of day in DRD mice (P > 0.1). (B) Striatal dopamine and (C) DOPAC concentrations, as well as (D) the DOPAC/dopamine ratio were assessed at 8 am and 8 pm (n = 6–9/condition). (B) For dopamine, there was a significant genotype × time interaction effect [F(1,23) = 23.6, P < 0.001], with the concentration of dopamine in normal mice significantly greater at 8 am than at 8 pm (P < 0.01) but no significant difference observed between times of day in DRD mice. (C) For DOPAC, there was a significant genotype × time interaction effect [F(1,23 = 4.6, P < 0.05], with the concentration of DOPAC in normal mice significantly greater at 8 am than at 8 pm (P < 0.05) but DOPAC was significantly lower at 8 am than at 8 pm in DRD mice (P < 0.01). (D) Similarly, for the DOPAC/dopamine ratio, there was a significant genotype × time interaction effect [F(1,23) = 12.6, P < 0.01] with significantly greater dopamine turnover at 8 pm than at 8 am in DRD mice (P < 0.001), but not normal mice. Statistical analyses were performed using a two-way ANOVA with Student’s t-test post hoc analyses for TH activity, dopamine and DOPAC concentrations, and Holm-Sidak post hoc analysis for turnover values. Values represent mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
D1DAR and D2DARs mediate abnormal movements
If enhanced dopamine turnover at 8 pm underlies the behavioural improvement in DRD mice, then blunting dopamine signalling at this time should worsen the abnormal movements. We tested this hypothesis by challenging DRD mice with D1- or D2-type dopamine receptor antagonists. We observed a significant dose-dependent increase in abnormal movements in response to both the D1DAR antagonist SCH 23390 (Fig. 7A; P < 0.05) and the D2DAR antagonist raclopride (Fig. 7B; P < 0.05). Because antagonists act by blocking the effects of the endogenous agonist, these results suggest that the very low concentrations of dopamine in DRD mice exert a behaviourally relevant effect on dopamine receptors, consistent with the results from amphetamine challenge. Conversely, an increase in dopamine receptor activation should alleviate dystonia. Indeed, low doses of the D1DAR agonist SKF 81297 showed a trend towards reducing the abnormal movements (Fig. 7C; P = 0.051) and the D2DAR agonist quinpirole significantly reduced the abnormal movements (Fig. 7D; P < 0.001).
Figure 7.
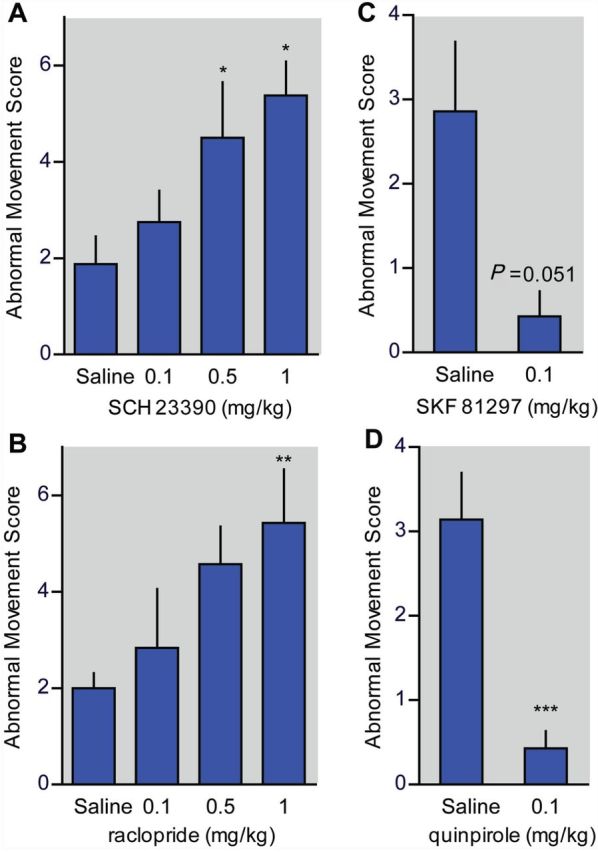
Dopamine receptor-mediated dystonic movements in DRD mice (n = 7–8/dose). (A) The D1DAR antagonist SCH 23390 dose-dependently increased dystonic movements [F(3,21) = 4.64, one-way repeated measures ANOVA, P < 0.05, Holm-Sidak post hoc analysis]. (B) The D2DAR antagonist raclopride dose-dependently increased dystonic movements [F(3,18) = 3.68, one-way repeated measures ANOVA, P < 0.05, Holm-Sidak post hoc analysis]. (C) The D1DAR agonist SKF 81297 reduced dystonic movements (Student’s paired t-test, P = 0.051). (D) The D2DAR agonist quinpirole reduced dystonic movements (Student’s paired t-test, P < 0.001). Values represent mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
Altered dopamine receptor sensitivity in DRD mice
To assess dopamine receptor function, behavioural sensitivity was measured in DRD mice compared to normal mice after challenge with subtype-selective agonists. Locomotor activity and stereotypy were used for these experiments, rather than abnormal movement, because both normal and DRD mice express these behaviours, allowing for direct comparison. The D1DAR agonist SKF 81297 significantly and dose-dependently increased locomotor activity in normal and DRD mice (Fig. 8A; P < 0.001 for each genotype). However, the dose-response curve for DRD mice was shifted to the left; the ED50 for the SKF 81297-induced increase in locomotor activity in normal mice was 1 mg/kg but the ED50 in DRD mice was 0.08 mg/kg, an order of magnitude lower than normal. SKF 81297 also dose-dependently increased stereotypic behaviours in both genotypes (Fig. 8B; P < 0.001 for each genotype), and the dose response curve for DRD mice was again shifted to the left. The D2DAR agonist quinpirole dose-dependently reduced locomotion in normal mice (P < 0.01), in agreement with previous studies (Li et al., 2010; Anzalone et al., 2012). Quinpirole did not suppress locomotor activity in DRD mice (Fig. 8C), although it did alleviate abnormal movements (Fig. 7D). Furthermore, although quinpirole dose-dependently decreased stereotypic behaviours in normal mice (P < 0.01), quinpirole dose-dependently increased stereotypic behaviours in DRD mice (P < 0.05; Fig. 8D). Thus, DRD mice exhibit supersensitivity to D1DAR activation but responses to D2DAR activation are blunted or perhaps altered in valence, indicative of dysregulated D1DAR versus D2DAR signalling.
Figure 8.
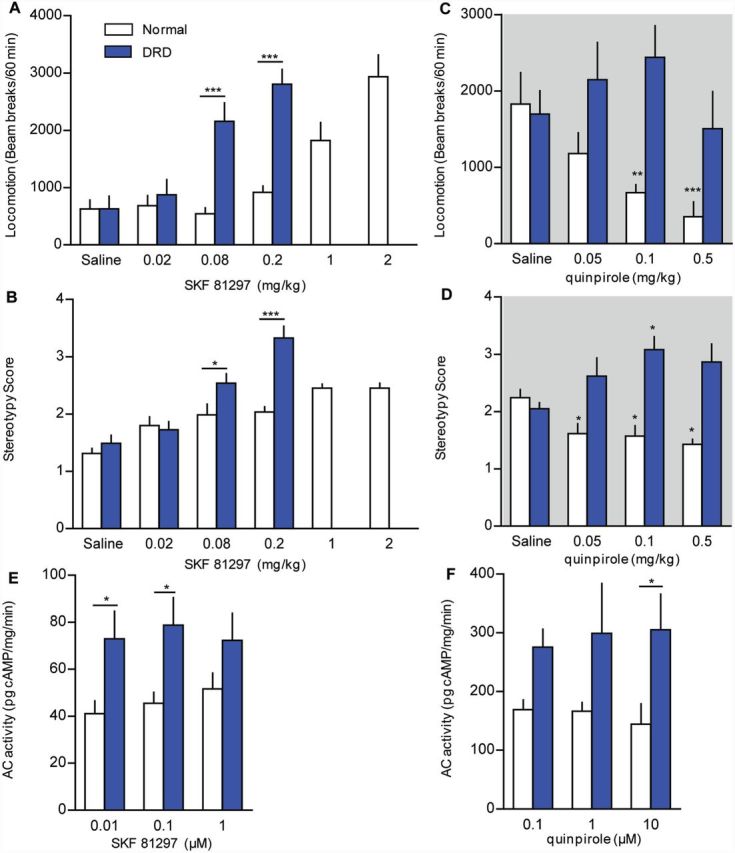
DAR function in DRD mice. (A and C) Locomotor activity and (B and D) stereotypic behaviours were assessed after administration (s.c.) of the D1DAR agonist SKF 81297 at 2 pm or the D2DAR agonist quinpirole at 8 pm (n = 6–8/genotype and condition). (A) For SKF 81297-mediated locomotor activity, there was a significant genotype × dose interaction effect for the lower doses (0.02, 0.08, and 0.2 mg/kg) where both genotypes were tested [F(3,42) = 18.7, two-way repeated measures ANOVA P < 0.001, Holm-Sidak post hoc analysis] with a significant increase in locomotor activity in DRD mice but not normal littermates (P < 0.001). At higher doses, SKF 81297 also induced an increase in locomotor activity in normal mice [F(5,35) = 19.5, one-way repeated measures ANOVA, P < 0.001]. (B) For SKF 81297-mediated stereotypy, there was a significant genotype × dose interaction effect for the lower doses where both genotypes were tested [F(3,42) = 8.9, two-way repeated measures ANOVA, P < 0.001; Holm-Sidak post hoc analysis] with a significant increase in stereotypy in DRD mice but not normal littermates (P < 0.01). At higher doses, SKF 81297 also induced an increase in stereotypy in normal mice [F(5,35) = 17.0, one-way repeated measures ANOVA, P < 0.001]. (C) For quinpirole-mediated locomotor activity, there was a significant main effect of genotype [F(1,33) = 16.4, two-way repeated measures ANOVA, P < 0.01] whereby quinpirole reduced locomotor activity in normal mice [F(3,18) = 5.6, one-way repeated measures ANOVA, P < 0.01], but not in DRD mice. (D) There was a significant genotype × dose interaction effect on stereotypy after administration of quinpirole [F(3,33) = 8.1, two-way repeated measures ANOVA, P < 0.001]. Quinpirole significantly decreased stereotypy in normal mice [F(3,18) = 5.8, one-way repeated measures ANOVA, P < 0.01] but significantly increased stereotypy behaviours in DRD mice [F(3,18) = 5.8, one-way repeated measures ANOVA, P < 0.01]. (E) There was a significant effect of genotype on SKF 81297-induced adenylate cyclase activity [F(1,24) = 14.6, two-way ANOVA, P < 0.001, Holm Sidak post hoc analysis] with increased activity in DRD mice at 0.01 µM (P < 0.05) and 0.1 µM (P < 0.05). (F) There was a significant effect of genotype on quinpirole-induced suppression of adenylate cyclase activity [F(1,23) = 14.6, two-way ANOVA, P < 0.01, Holm Sidak post hoc analysis] with a significant difference between normal and DRD mice in response to 10 µM quinpirole (P < 0.05). Values represent mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
To examine possible mechanisms underlying the behavioural responses, we assessed DAR expression and second messenger responses. Using quantitative reverse transcriptase-PCR to assess mRNA abundance, we observed no changes in the abundance of striatal mRNA encoding D1DARs, D2DARs, or D3DARs or midbrain D2DARs in DRD mice compared to normal mice (Supplementary Table 3). There was no difference in the affinity or density of D1DARs or D2DARs in DRD mice compared to normal mice (Supplementary Table 4), consistent with TH knockout mice, which also do not exhibit changes in dopamine receptor expression (Kim et al., 2000).
To assess functional changes in striatal D1DAR and D2DAR signalling, we measured striatal adenylate cyclase activity. SKF 81297-stimulated adenylate cyclase activity was significantly higher in DRD mice compared to controls (Fig. 8E; P < 0.001). Quinpirole reduced adenylate cyclase activity in normal mice, but adenylate cyclase activity was increased in DRD mice, without a clear dose-dependent effect (Fig. 8F; P < 0.01).
Discussion
DRD mice display the core features of the human disorder, including a causative TH mutation, partial reduction in TH activity, dystonia that worsens throughout the course of the active period, and improvement of the dystonic movements with l-DOPA or trihexyphenidyl. Thus, DRD mice have construct, face and predictive validity. Our results demonstrate that dopamine turnover was about four times higher than normal at the beginning of the active period, but fell to normal levels at the beginning of the inactive period, which may, in part, explain the diurnal fluctuation in dystonia. In addition to the presynaptic defects, both D1DARs and D2DARs appear to mediate dystonia but receptor responses were abnormal. D1DARs were supersensitive while it appears that the valence of D2DARs was reversed, demonstrating a marked differential effect on D1DAR versus D2DAR signalling. Thus, it is likely the presynaptic dopamine deficit in combination with abnormal receptor responses underlie the expression of dystonia.
GCH1 mutations are the most common causes of DRD. However, we elected to create a DRD mouse that carries a TH mutation instead of a GCH1 mutation because BH4 is a cofactor for many enzymes including nitric oxide synthase, phenylalanine hydroxylase and tryptophan hydroxylase, in addition to TH. Disrupting the function of so many neuroactive enzymes with a GCH1 mutation would have obscured the specificity of the effect on catecholamines and added an undesirable level of complexity to the analysis of the mouse mutants. Further, GCH1 mutations may enhance the likelihood of nigrostriatal cell loss (Mencacci et al., 2014), which would have complicated the analysis of a model of DRD. Thus, having a model of DRD with a mutation in TH, instead of GCH1, facilitates mechanistic studies to provide insight specifically into the role of catecholamine dysfunction in dystonia.
Our results provide support for the long-standing hypothesis that abnormal striatal dopamine neurotransmission mediates dystonia in DRD. The attenuated TH activity in DRD affects both dopamine and norepinephrine concentrations throughout the brain. l-DOPA treatment, which repletes dopamine and norepinephrine, abolished dystonia, but l-DOPS, which only increases norepinephrine, did not. Although both striatum and cerebellum are implicated in dystonia, direct delivery of l-DOPA to striatum, where dopamine is abundant but norepinephrine is scarce, ameliorated dystonia. Further, activating dopamine receptors, which are abundant in the striatum but not the cerebellum, also ameliorated the dystonia. In contrast, direct injection of l-DOPA to cerebellum, where norepinephrine is abundant but dopamine is undetectable, did not affect the dystonia. Combined, these results suggest that the abnormal movements are associated with striatal dopamine deficiency, while the contribution of norepinephrine seems to be minor at best. However, these results do not rule out the possibility that early postnatal norepinephrine deprivation contributes to the development of dystonia. This question can be addressed in the future by restoring only norepinephrine and epinephrine synthesis to create mice deficient in dopamine, but not other catecholamines, by rescuing TH expression in non-dopaminergic cells (Zhou and Palmiter, 1995).
The extent and timing of the dopamine deficit may be important for the development of dystonia, as other lines of mice with disrupted TH function do not exhibit dystonia (Kobayashi et al., 1995; Zhou and Palmiter, 1995; Rios et al., 1999). The TH null mutation causes pre- or perinatal death due to norepinephrine deficiency. Restoration of norepinephrine by transgenic rescue results in mice deficient in TH expression only in dopaminergic neurons, dopamine deficient mice, which require daily l-DOPA supplementation starting in the second postnatal week for survival, similar to DRD mice. However, in contrast to DRD mice, after l-DOPA withdrawal, dopamine-deficient mice are akinetic but not dystonic (Szczypka et al., 1999). Unlike the dopamine-deficient mice, TH activity and dopamine levels are low, but not abolished in DRD mice, suggesting that the residual catecholamines in DRD mice may play a role in instigating the abnormal movements. In addition to the degree of dopamine deficiency, the timing of the insult appears to contribute to the behavioural outcome. For example, reserpine treatment or genetic manipulations that acutely and globally deplete dopamine in adults results in profound hypoactivity, but not dystonia, suggesting that the developmental timing of the dopamine depletion and/or the chronic nature of the dopamine deficiency contributes to the induction of dystonia (Carlsson et al., 1957; Sotnikova et al., 2005, 2006). Thus, the phenotype resulting from catecholamine deficiency appears to depend not only on the absolute concentration of dopamine, but also the temporal features of the dopamine depletion. Conditional restoration of dopamine signalling in DRD mice may provide insight into the critical role that timing of the dopamine deficiency plays in the instigation of dystonia.
While presynaptic dopamine dysfunction may be the instigating factor, DAR responses are also implicated. Our findings indicate that both D1DARs and D2DARs mediated the expression of dystonia; D1DAR and D2DAR antagonists exacerbated the dystonia, but activation of either individual receptor subtype ameliorated the dystonia. These findings are consistent with data showing dysregulation of both D1DAR and D2DARs in human dystonia. Reduced striatal D2DAR binding is observed in several forms of dystonia, including blepharospasm (Horie et al., 2009), torticollis (Hierholzer et al., 1994; Naumann et al., 1998), and DYT1 dystonia (Asanuma et al., 2005). Evidence for D1DAR involvement comes from data showing that dystonia is caused by mutations in GNAL (Fuchs et al., 2013; Vemula et al., 2013), the gene encoding the Gα(olf) subunit that transduces D1DAR signalling in spiny projection neurons (Zhuang et al., 2000). Thus, in both humans and mice, a reduction in both D1DAR and D2DAR function is associated with dystonia.
Although restoring DAR signalling by repleting dopamine or agonist administration ameliorated dystonia in DRD mice, dopamine receptor signalling was nonetheless abnormal. DRD mice exhibited behavioural supersensitivity to a D1DAR agonist and a corresponding upregulation in adenylate cyclase stimulation. D1DAR supersensitization is also observed after l-DOPA treatment in adult 6-hydroxydopamine lesioned-rodents, which are used to model the l-DOPA-induced dyskinesias (LIDs) in late-stage Parkinson’s disease (Picconi et al., 2003). Despite the neurochemical parallels between DRD mice and the l-DOPA-induced dyskinesia model, D1DAR agonists induce abnormal movements in the l-DOPA-induced dyskinesia model (Westin et al., 2007), in striking contrast to DRD mice where they reduce dystonia. Understanding D1DAR downstream signalling, such as DARPP-32-ERK1/2-ΔFosB signalling, which has been implicated in LIDs (Andersson et al., 1999; Santini et al., 2007), will be critical for understanding the pathway(s) that distinguish LIDs from dystonias. D2DAR signalling was also abnormal in DRD mice; D2DAR activation, which normally inhibits locomotor activity and adenylate cyclase activity in mice, instead caused subtle increases in both while ameliorating the dystonia. By contrast, D2DARs do not contribute to the expression of LIDs in the adult model (Westin et al., 2007). Thus, the challenge is to understand the interactions between the presynaptic dopamine depletion, (mal)adaptive receptor responses and microstructural synaptic abnormalities to better understand the specific abnormalities that give rise to dystonia. DRD mice provide an unprecedented opportunity to understand the complex dopaminergic signalling defects that cause dystonia.
Supplementary Material
Acknowledgements
We thank Dainippon-Sumitomo Pharmaceuticals Inc. for the l-DOPS and Susan Jenkins for technical assistance.
Glossary
Abbreviations
- l-DOPA
l-3,4-dihydroxyphenylalanine
- DRD
l-DOPA-responsive dystonia
- DOPAC
3,4-dihydroxypheynlacetic acid
- 5-HT
serotonin
- 5-HIAA
5-hydroxyindoleacetic acid
- l-DOPS
l-3,4-dihydroxyphenylserine
- BH4
tetrahydrobiopterin
- DAT
dopamine transporter
- D1DAR
D1-type dopamine receptor
- D2DAR
D2-type dopamine receptor
- vGluT1
vesicular glutamate transporter 1
- vGluT2
vesicular glutamate transporter 2
Funding
This work was supported by the United States National Institute of Health (R21 NS059645, R01 NS088528, T32GM08605), the Bachmann-Strauss Dystonia and Parkinson Foundation, the Pediatric Neurotransmitter Disease Association, and the Yerkes National Primate Center NIH base grant (ORIP/OD P51OD011132).
Supplementary material
Supplementary material is available at Brain online.
References
- Andersson M, Hilbertson A, Cenci MA. Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson's disease. Neurobiol Dis 1999; 6: 461–74. [DOI] [PubMed] [Google Scholar]
- Anzalone A, Lizardi-Ortiz JE, Ramos M, De Mei C, Hopf FW, Iaccarino C, et al. Dual control of dopamine synthesis and release by presynaptic and postsynaptic dopamine D2 receptors. J Neurosci 2012; 32: 9023–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma K, Ma Y, Okulski J, Dhawan V, Chaly T, Carbon M, et al. Decreased striatal D2 receptor binding in non-manifesting carriers of the DYT1 dystonia mutation. Neurology 2005; 64: 347–9. [DOI] [PubMed] [Google Scholar]
- Brun L, Ngu LH, Keng WT, Ch'ng GS, Choy YS, Hwu WL, et al. Clinical and biochemical features of aromatic L-amino acid decarboxylase deficiency. Neurology 2010; 75: 64–71. [DOI] [PubMed] [Google Scholar]
- Cahill AL, Ehret CF. Circadian variations in the activity of tyrosine hydroxylase, tyrosine aminotransferase, and tryptophan hydroxylase: relationship to catecholamine metabolism. J Neurochem 1981; 37: 1109–15. [DOI] [PubMed] [Google Scholar]
- Calne DB. Dopa-responsive dystonia. Ann Neurol 1994; 35: 381–2. [DOI] [PubMed] [Google Scholar]
- Carbon M, Argyelan M, Habeck C, Ghilardi MF, Fitzpatrick T, Dhawan V, et al. Increased sensorimotor network activity in DYT1 dystonia: a functional imaging study. Brain 2010; 133: 690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M, Magnusson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature 1957; 180: 1200. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Atack CV, Lindqvis M, Kehr W, Davis JN. Simultaneous measurement of tyrosine and tyrptophan hydroxylase activities in brain in-vivo using an an inhibitor of aromatic amino-acid decarboxylase. Naunyn Schmiedebergs Arch Pharmacol 1972; 275: 153–68. [DOI] [PubMed] [Google Scholar]
- Clot F, Grabli D, Cazeneuve C, Roze E, Castelnau P, Chabrol B, et al. Exhaustive analysis of BH4 and dopamine biosynthesis genes in patients with Dopa-responsive dystonia. Brain 2009; 132: 1753–63. [DOI] [PubMed] [Google Scholar]
- de Rijk-van Andel JF, Gabreels FJM, Geurtz B, Steenbergen-Spanjers GCH, van den Heuvel L, Smeitink JAM, et al. L-dopa-responsive infantile hypokinetic rigid parkinsonism due to tyrosine hydroxylase deficiency. Neurology 2000; 55: 1926–8. [DOI] [PubMed] [Google Scholar]
- Devanagondi R, Egami K, LeDoux MS, Hess EJ, Jinnah HA. Neuroanatomical substrates for paroxysmal dyskinesia in lethargic mice. Neurobiol Dis 2007; 27: 249–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRaddo J, Kellogg C. In vivo rates of tyrosine and tryptophan hydroxylation in regions of rat brain at four times during the light-dark cycle. Naunyn Schmiedebergs Arch Pharmacol 1975; 286: 389–400. [DOI] [PubMed] [Google Scholar]
- Egami K, Yitta S, Kasim S, Lewers JC, Roberts RC, Lehar M, et al. Basal ganglia dopamine loss due to defect in purine recycling. Neurobiol Dis 2007; 26: 396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan XL, Hess EJ. D2-like dopamine receptors mediate the response to amphetamine in a mouse model of ADHD. Neurobiol Dis 2007; 26: 201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris MJ, Espana RA, Locke JL, Konstantopoulos JK, Rose JH, Chen R, et al. Dopamine transporters govern diurnal variation in extracellular dopamine tone. Proc Natl Acad Sci USA 2014; 111: E2751–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossbakk A, Kleppe R, Knappskog PM, Martinez A, Haavik J. Functional studies of tyrosine hydroxylase missense variants reveal distinct patterns of molecular defects in dopa-responsive dystonia. Hum Mutat 2014; 35: 880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs T, Saunders-Pullman R, Masuho I, Luciano MS, Raymond D, Factor S, et al. Mutations in GNAL cause primary torsion dystonia. Nat Genet 2013; 45: 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorke W, Bartholome K. Biochemical and neurophysiological investigations in two forms of Segawa's disease. Neuropediatrics 1990; 21: 3–8. [DOI] [PubMed] [Google Scholar]
- Hierholzer J, Cordes M, Schelosky L, Richter W, Keske U, Venz S, et al. Dopamine D2 receptor imaging with iodine-123-iodobenzamide SPECT in idiopathic rotational torticollis. J Nucl Med 1994; 35: 1921–7. [PubMed] [Google Scholar]
- Horie C, Suzuki Y, Kiyosawa M, Mochizuki M, Wakakura M, Oda K, et al. Decreased dopamine D receptor binding in essential blepharospasm. Acta Neurol Scand 2009; 119: 49–54. [DOI] [PubMed] [Google Scholar]
- Hyland K, Surtees RA, Rodeck C, Clayton PT. Aromatic L-amino acid decarboxylase deficiency: clinical features, diagnosis, and treatment of a new inborn error of neurotransmitter amine synthesis. Neurology 1992; 42: 1980–8. [DOI] [PubMed] [Google Scholar]
- Ichinose H, Ohye T, Takahashi E, Seki N, Hori T, Segawa M, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet 1994; 8: 236–42. [DOI] [PubMed] [Google Scholar]
- Ingham CA, Hood SH, Taggart P, Arbuthnott GW. Plasticity of synapses in the rat neostriatum after unilateral lesion of the nigrostriatal dopaminergic pathway. J Neurosci 1998; 18: 4732–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarman PR, Bandmann O, Marsden CD, Wood NW. GTP cyclohydrolase I mutations in patients with dystonia responsive to anticholinergic drugs. J Neurol Neurosurg Psychiatry 1997; 63: 304–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnah HA, Visser JE, Harris JC, Verdu A, Larovere L, Ceballos-Picot I, et al. Delineation of the motor disorder of Lesch-Nyhan disease. Brain 2006; 129: 1201–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Szczypka MS, Palmiter RD. Dopamine-deficient mice are hypersensitive to dopamine receptor agonists. J Neurosci 2000; 20: 4405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knappskog PM, Flatmark T, Mallet J, Ludecke B, Bartholome K. Recessively inherited L-DOPA-responsive dystonia caused by a point mutation (Q381K) in the tyrosine hydroxylase gene. Hum Mol Genet 1995; 4: 1209–12. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Morita S, Sawada H, Mizuguchi T, Yamada K, Nagatsu I, et al. Targeted disruption of the Tyrosine-Hydroxylase locus results in severe catecholamine depletion and perinatal lethality in mice. J Biol Chem 1995; 270: 27235–43. [DOI] [PubMed] [Google Scholar]
- Kurian MA, Li Y, Zhen J, Meyer E, Hai N, Christen HJ, et al. Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome: an observational cohort and experimental study. Lancet Neurol 2011; 10: 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Hong JH, Choi IY, Che Y, Lee JK, Yang SD, et al. Impaired D2 dopamine receptor function in mice lacking type 5 adenylyl cyclase. J Neurosci 2002; 22: 7931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehericy S, Tijssen MA, Vidailhet M, Kaji R, Meunier S. The anatomical basis of dystonia: current view using neuroimaging. Mov Disord 2013; 28: 944–57. [DOI] [PubMed] [Google Scholar]
- Li SM, Collins GT, Paul NM, Grundt P, Newman AH, Xu M, et al. Yawning and locomotor behavior induced by dopamine receptor agonists in mice and rats. Behav Pharmacol 2010; 21: 171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr KM, Bernstein AI, Stout KA, Dunn AR, Lazo CR, Alter SP, et al. Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc Natl Acad Sci USA 2014; 111: 9977–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludecke B, Knappskog PM, Clayton PT, Surtees RA, Clelland JD, Heales SJ, et al. Recessively inherited L-DOPA-responsive parkinsonism in infancy caused by a point mutation (L205P) in the tyrosine hydroxylase gene. Hum Mol Genet 1996; 5: 1023–8. [DOI] [PubMed] [Google Scholar]
- Mehta SH, Morgan JC, Sethi KD. Drug-induced movement disorders. Neurol Clin 2015; 33: 153–74. [DOI] [PubMed] [Google Scholar]
- Mencacci NE, Isaias IU, Reich MM, Ganos C, Plagnol V, Polke JM, et al. Parkinson's disease in GTP cyclohydrolase 1 mutation carriers. Brain 2014; 137: 2480–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann M, Pirker W, Reiners K, Lange KW, Becker G, Brucke T. Imaging the pre- and postsynaptic side of striatal dopaminergic synapses in idiopathic cervical dystonia: a SPECT study using [123I] epidepride and [123I] beta-CIT. Mov Disord 1998; 13: 319–23. [DOI] [PubMed] [Google Scholar]
- Neychev VK, Fan XL, Mitev VI, Hess EJ, Jinnah HA. The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain 2008; 131: 2499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer M, Carbon M, Argyelan M, Eidelberg D. Hereditary dystonia as a neurodevelopmental circuit disorder: evidence from neuroimaging. Neurobiol Dis 2011; 42: 202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard TG, Marsden CD, Duvoisin RC. Dopa-responsive dystonia. Adv Neurol 1988; 50: 377–84. [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinattes. San Diego: Academic Press; 2001. [Google Scholar]
- Perlmutter JS, Mink JW. Dysfunction of dopaminergic pathways in dystonia. Adv Neurol 2004; 94: 163–70. [PubMed] [Google Scholar]
- Peters A, Palay S, Webster H. The fine structure of the nervous system: neurons and their supporting cells. New York: Oxford University Press; 1991. [Google Scholar]
- Picconi B, Centonze D, Hakansson K, Bernardi G, Greengard P, Fisone G, et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci 2003; 6: 501–6. [DOI] [PubMed] [Google Scholar]
- Pizoli CE, Jinnah HA, Billingsley ML, Hess EJ. Abnormal cerebellar signaling induces dystonia in mice. J Neurosci 2002; 22: 7825–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portbury AL, Chandra R, Groelle M, McMillian MK, Elias A, Herlong JR, et al. Catecholamines act via a beta-adrenergic receptor to maintain fetal heart rate and survival. Am J of Physiol Heart Circ Physiol 2003; 284: H2069–77. [DOI] [PubMed] [Google Scholar]
- Raike RS, Pizoli CE, Weisz C, van den Maagdenberg A, Jinnah HA, Hess EJ. Limited regional cerebellar dysfunction induces focal dystonia in mice. Neurobiol Dis 2013; 49: 200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajput AH, Gibb WRG, Zhong XH, Shannak KS, Kish S, Chang LG, et al. Dopa-responsive dystonia—pathological and biochemical observation in a case. Ann Neurol 1994; 35: 396–402. [DOI] [PubMed] [Google Scholar]
- Raju DV, Smith Y. Differential localization of the vesicular glutamate transporters 1 and 2 in the rat striatum. In: Bolam JP, Ingham CA, Magill P, editors, IBAGS VIIIth. New York: Plenum Press; 2005. p. 601–10. [Google Scholar]
- Raju DV, Shah DJ, Wright TM, Hall RA, Smith Y. Differential synaptology of vGluT2-containing thalamostriatal afferents between the patch and matrix compartments in rats. J Comp Neurol 2006; 499: 231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rilstone JJ, Alkhater RA, Minassian BA. Brain dopamine-serotonin vesicular transport disease and its treatment. N Engl J Med 2013; 368: 543–50. [DOI] [PubMed] [Google Scholar]
- Rios M, Habecker B, Sasaoka T, Eisenhofer G, Tian H, Landis S, et al. Catecholamine synthesis is mediated by tyrosinase in the absence of tyrosine hydroxylase. J Neurosci 1999; 19: 3519–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose SJ, Kriener LH, Heinzer AK, Fan X, Raike RS, van den Maagdenberg AM, et al. The first knock-in mouse model of episodic ataxia type 2. Exp Neurol 2014; 261: 553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA, et al. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci 2007; 27: 6995–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008; 3: 1101–8. [DOI] [PubMed] [Google Scholar]
- Segawa M, Nomura Y. Hereditary progressive dystonia with marked diurnal fluctuation. Pathophysiological importance of the age of onset. Adv Neurol 1993; 60: 568–76. [PubMed] [Google Scholar]
- Shirley TL, Rao LM, Hess EJ, Jinnah HA. Paroxysmal dyskinesias in mice. Mov Disord 2008; 23: 259–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow BJ, Nygaard TG, Takahashi H, Calne DB. Positron emission tomographic studies of dopa-responsive dystonia and early-onset idiopathic parkinsonism. Ann Neurol 1993; 34: 733–8. [DOI] [PubMed] [Google Scholar]
- Song CH, Bernhard D, Bolarinwa C, Hess EJ, Smith Y, Jinnah HA. Subtle microstructural changes of the striatum in a DYT1 knock-in mouse model of dystonia. Neurobiol Dis 2013; 54: 362–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CH, Fan XL, Exeter CJ, Hess EJ, Jinnah HA. Functional analysis of dopaminergic systems in a DYT1 knock-in mouse model of dystonia. Neurobiol Dis 2012; 48: 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotnikova TD, Beaulieu JM, Barak LS, Wetsel WC, Caron MG, Gainetdinov RR. Dopamine-independent locomotor actions of amphetamines in a novel acute mouse model of Parkinson disease. PLoS Biol 2005; 3: e271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotnikova TD, Caron MG, Gainetdinov RR. DDD mice, a novel acute mouse model of Parkinson's disease. Neurology 2006; 67: S12–17. [DOI] [PubMed] [Google Scholar]
- Szczypka MS, Rainey MA, Kim DS, Alaynick WA, Marck BT, Matsumoto AM, et al. Feeding behavior in dopamine-deficient mice. Proc Natl Acad Sci USA 1999; 96: 12138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SA, Matsumoto AM, Palmiter RD. Noradrenaline is essential for mouse fetal development. Nature 1995; 374: 643–46. [DOI] [PubMed] [Google Scholar]
- Thomas SA, Marck BT, Palmiter RD, Matsumoto AM. Restoration of norepinephrine and reversal of phenotypes in mice lacking dopamine beta-hydroxylase. J Neurochem 1998; 70: 2468–76. [DOI] [PubMed] [Google Scholar]
- Thony B, Blau N. Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase. Hum Mut 2006; 27: 870–8. [DOI] [PubMed] [Google Scholar]
- van den Heuvel L, Luiten B, Smeitink JAM, Rijk-van Andel JF, Hyland K, Steenbergen-Spanjers GCH, et al. A common point mutation in the tyrosine hydroxylase gene in autosomal recessive L-DOPA-responsive dystonia in the Dutch population. Hum Genet 1998; 102: 644–6. [DOI] [PubMed] [Google Scholar]
- Vemula SR, Puschmann A, Xiao J, Zhao Y, Rudzinska M, Frei KP, et al. Role of Galpha(olf) in familial and sporadic adult-onset primary dystonia. Hum Mol Genet 2013; 22: 2510–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba RM, Lee H, Smith Y. Dopaminergic denervation and spine loss in the striatum of MPTP-treated monkeys. Exp Neurol 2009; 215: 220–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba RM, Smith Y. Differential striatal spine pathology in Parkinson's disease and cocaine addiction: a key role of dopamine? Neuroscience 2013; 251: 2–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace LJ, Traeger JS. Dopac distribution and regulation in striatal dopaminergic varicosities and extracellular space. Synapse 2012; 66: 160–73. [DOI] [PubMed] [Google Scholar]
- Weber M, Lauterburg T, Tobler I, Burgunder JM. Circadian patterns of neurotransmitter related gene expression in motor regions of the rat brain. Neurosci Lett 2004; 358: 17–20. [DOI] [PubMed] [Google Scholar]
- Westin JE, Vercammen L, Strome EM, Konradi C, Cenci MA. Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol Psychiatry 2007; 62: 800–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann T. Dopaminergic dysfunction in DYT1 dystonia. Exp Neurol 2008; 212: 242–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou QY, Palmiter RD. Dopamine-deficient mice are severly hypoactive, adipsic, and aphagic. Cell 1995; 83: 1197–209. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Belluscio L, Hen R. G(olf)alpha mediates dopamine D1 receptor signaling. J Neurosci 2000; 20: RC91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.