

Abstract
Inconsistency in the reported associations between the A66G polymorphism in the methionine synthase reductase (MTRR) gene and colorectal cancer (CRC) prompted a meta-analysis, so that we could obtain a more precise estimate. Databases searches of the published literature yielded 20 case–control studies from 17 articles (8,371 cases and 12,574 controls). We calculated pooled odds ratios (ORs) and 95% confidence intervals in three genetic comparisons (A allele, G allele, and A/G genotype). We found no evidence of overall associations between MTRR A66G and CRC risk (OR 0.96–1.05, P = 0.12–0.44). This was materially unchanged when reanalyzed without the Hardy–Weinberg equilibrium (HWE)-deviating studies (OR 0.97–1.06, P = 0.11–0.65). In the A allele comparison, however, outlier treatment generated significant protection (OR 0.91, P = 0.01). Combined removal of the outliers and HWE-deviating studies reflected this summary effect (OR 0.90, P = 0.01) as did the pooled OR from high-quality studies (OR 0.90, P = 0.01). Only the Asian subgroup showed significant (both at P = 0.05) A allele (OR 1.13) and A/G genotype (OR 0.88) associations. In conclusion, post-outlier A allele effects were protective. Our study also suggests ethnic-specific associations with Asian susceptibility and protection in the A allele and A/G genotype comparisons, respectively. Folate status showed no association of this polymorphism with CRC.
Keywords: colorectal cancer, A66G, methionine synthase reductase, polymorphism
Introduction
Colorectal cancer (CRC) is one of the most common cancer types in the world.1 The majority of CRC cases are sporadic, that is, absence of genetic predisposition or family history.2 This indicates that modifiable risk factors (lifestyle and nutrition) are strongly related to disease development. Contributing factors for CRC progression include inflammation3,4 and inflammatory bowel disease.5 These are thought to have genetic and acquired factors. However, genetic predisposition is crucial to CRC susceptibility.6 Over the past decade, the role of folate and genetic polymorphisms of enzymes involved in its metabolism has attracted considerable interest in epidemiological research on this cancer type.7 Folate and methionine metabolisms are essential in DNA synthesis, repair, and methylation, and abnormalities in these processes (due to alterations in enzyme functions) are implicated in colorectal carcinogenesis.7,8 The role of genetic polymorphisms in the folate metabolic pathway has not yet been fully evaluated for association with the risk of CRC. Methionine synthase reductase (MTRR) is essential for providing methyl groups, and it is likely that enzymatic variants due to functional polymorphisms may alter DNA methylation, with subsequent impact on carcinogenesis.9 A genetic polymorphism at nucleotide 66 (A–G) of the MTRR gene (rs1801394), located in chromosome 5p15.2–15.3,10 results in the substitution of isoleucine with methionine at codon 22 (I22M).11 MTRR restores the activity of methionine synthase (MTR) enzyme and plays an essential role in the folate and vitamin B12-dependent remethylation of homocysteine to methionine. Under conditions of adequate methionine, approximately 40% of homocysteine is remethylated to methionine through the activity of these enzymes.12
The role of folate spans a spectrum of effects on the etiology of CRC.13 Although studies have shown that high folate levels elicit reduced the risk of CRC,14,15 others have suggested that high folate intake might increase the risk of CRC in persons harboring premalignant lesions.16,17 On the other end of the spectrum, low folate levels have also been reported to be associated with both increased18 and decreased risks19 of CRC.
Association data for the MTRR A66G polymorphism and its effect on the risk of CRC have remained inconsistent.20–26 Two recent meta-analyses have not exactly concurred in their findings; one failed to find any significant association27 between MTRR A66G and CRC and the other28 found the G allele might increase Caucasian risk. This prompted us to perform a meta-analysis to obtain more precise estimates.
Materials and Methods
Selection of studies
We searched MEDLINE using PubMed and ScienceDirect for association studies as of July 11, 2015. The terms used were “methionine synthase reductase,” “MTRR,” “polymorphism,” “colorectal,” “colon,” and “rectal” as medical subject heading and text, unrestricted by language. References cited in the retrieved articles were also screened manually to identify additional eligible studies. Inclusion criteria were (1) case–control study evaluating the association between MTRR polymorphisms and CRC risk and (2) sufficient genotype frequency data presented to calculate the odds ratios (ORs) and 95% confidence intervals (CIs).
Data extraction and calculations
Two investigators independently extracted data and reached consensus on all the items. The following information were obtained from each publication: first author’s name, published year, country of origin, ethnicity, sources of controls, sample sizes, used matching, addressed the Hardy–Weinberg equilibrium (HWE), genotyping platform, number of cases and controls, and genotype frequencies. We performed two calculations: (1) to determine the statistical power of the each study assuming an OR of 1.5 at a genotypic risk level of = 0.05 (two sided), power was considered adequate at ≥80% (Table 1); (2) to determine deviations from HWE and found them in two studies (Supplementary Table 1).23,29
Table 1.
Characteristics of studies of the A66G polymorphism in the MTRR gene and its association with colorectal cancer.
| FIRST AUTHOR YEAR [REFERENCES] | COUNTRY | ETHNIC GROUP | SOURCE OF CONTROLS | POWER (α = 0.05 OR 1.5) | SAMPLE SIZE | USED MATCH | USED HWE | GENOTYPING | NOS |
|---|---|---|---|---|---|---|---|---|---|
| 1 Matsuo 200223 | Japan | Asian | HB | 47.0 | 383 | Yes | Yes | RFLP | 4 |
| 2 Yoshimitsu 201234 | Japan | Asian | HB | 96.1 | 1,569 | No | Yes | RFLP | 5 |
| 3 Morita 201333 | Japan | Asian | HB | 96.8 | 1,463 | No | Yes | RFLP | 5 |
| 4 Otani 200524 | Japan | Asian | HB | 39.6 | 331 | Yes | No | Taqman | 6 |
| 5 LeMarchand 200222 | USA, Japan* | Asian | PB | 75.1 | 707 | Yes | Yes | RFLP | 8 |
| 6 Curtin 201131 | USA | NHC** (81–83%) | PB | 89.2 | 1,026 | Yes | Yes | GG bead-based | 8 |
| 7 Hazra 200720 | USA | NHC | PB | 90.4 | 1,066 | Yes | Yes | Taqman | 9 |
| 8 Burcos 201035 | Romania | NHC | HB | 24.2 | 180 | No | Yes | RFLP | 3 |
| 9 de Vogel 200929 | Netherlands | NHC | PB | 99.4 | 2,496 | No | Yes | PCR | 4 |
| 10 Hubner 200636 | UK | NHC | PB | 52.5 | 546 | No | Yes | Taqman | 6 |
| 11 Liu 201338 | USA | NHC** (91–93%) | PB | 100.0 | 3,195 | Yes | Yes | GG bead-based | 9 |
| 12 Theodoratou 200826 | UK | NHC | HB | 99.4 | 2,004 | Yes | Yes | Array-based | 7 |
| 13 Steck 200825 | USA | NHC | PB | 79.6 | 840 | Yes | Yes | Taqman | 9 |
| 14 Pardini 201139 | Czechoslovakia | NHC | HB | 98.8 | 2,033 | Yes | Yes | RFLP | 7 |
| 15 Koushik 201121 | USA | NHC | PB | 88.1 | 1,164 | Yes | Yes | Taqman | 8 |
| 16 Jokic 201137 | Croatia | NHC | PB | 68.6 | 600 | Yes | Yes | Taqman | 7 |
| 17 LeMarchand 200222 | USA | NHC | PB | 42.5 | 317 | Yes | Yes | RFLP | 8 |
| 18 Steck 200825 | USA | AA | PB | 64.7 | 561 | Yes | Yes | Taqman | 9 |
| 19 LeMarchand 200222 | USA | Hawaiian | PB | 11.3 | 163 | Yes | Yes | RFLP | 8 |
| 20 Guimaraes 201132 | Brazil | South American | HB | 38.8 | 301 | Yes | Yes | PCR | 7 |
Note: Japanese subjects residing in the USA;
Admixture.
Abbreviations: NHC, non-Hispanic Caucasian; AA, African-American; HB, hospital-based; PB, population-based; OR, odds ratio; HWE, Hardy–Weinberg Equilibrium; RFLP, restriction fragment length polymorphism; PCR, polymerase chain reaction; NOS, Newcastle-Ottawa Score.
Quality assessment of the studies
The Newcastle–Ottawa Scale (NOS)30 was used to assess the methodological quality of the included studies. These studies were judged based on three broad perspectives: selection, comparability, and exposure in case–control studies. The star rating system has scores ranging from zero (worst) to 9 (best). Scores of 5–6 and ≥7 stars indicate moderate and high quality, respectively.
Meta-analysis
Risks (OR) of CRC with the A66G MTRR polymorphisms were estimated for each study. Frequency of the G allele is minor in 1022–25,31–34 of the 19 studies but not in nine21,25,29,35–39 of them where the A allele is minor. Given non-uniformity of the minor allele frequency across the studies, we thus compared the following for A66G: (i) G allele with A/G-A/A genotype, (ii) A allele with A/G-G/G genotype, and (iii) A/G genotype with homozygous A/A and G/G genotypes (heterozygote comparison). To compare the effects on the same baseline, we used raw data for genotype frequencies to calculate pooled ORs, obtained using either fixed40 (in absence of heterogeneity) or random41 (in its presence) effects model. Heterogeneity between studies was (i) estimated using the chi-square (χ2)-based Q test,42 (ii) explored using subgroup analysis,42 and (iii) quantified with the I2 statistic that measures degree of inconsistency among studies43 and its sources detected using the Galbraith plot.44 Using this plot, we identified three studies as sources of heterogeneity.33,34,36 Outlier treatment consisted of eliminating these studies in the overall analysis and subgroups followed by reanalysis. Data were analyzed using Review Manager 5.3 (Cochrane Collaboration), SIGMASTAT 2.03, and SIGMAPLOT 11.0 (Systat Software). Two-sided P values of ≤0.05 were considered significant except in estimations of heterogeneity and publication bias. Given the low power of the χ2-based Q test for heterogeneity, P value was set at ≤0.10,45 as was for publication bias,46 assessed with Egger’s test47 and the Begg–Mazumdar diagnosis.48
Subgroup analyses
We stratified our analysis into four subgroups where, first, Caucasians (non-Hispanic Caucasian (NHC); 6,177 cases/9,290 controls) were compared with Asians (1,766 cases/2,687 controls). Population admixtures were found in two US studies.31,38 The second subgroup is composed of folate intake, where we compared effects when consumption was low at <400 μg/d (313 cases/422 controls) and when it was high at >400 μg/d (356 cases/699 controls). Finally, we also considered a subgroup in confining the analysis to studies with NOS scores of 7–9 (5,963 cases/8,014 controls).
Results
Included studies
Figure 1 outlines our study selection process in a flowchart following Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines.49 A total of 57 citations were identified with the initial search, from which 30 were excluded after title and abstract review. From the remaining 27, eight were excluded for not conforming to the inclusion criteria. Full-text articles of the remaining 19 articles were examined, two of which were excluded for non-availability of genotype data. Thus, the total number of articles (8,371 cases and 12,574 controls) included in the meta-analysis was 17.20–26,29,31–39 Features of the included studies with epidemiological and clinical features are outlined in Table 1. Of the 17 articles, 15 were single studies.20,21,23,24,26,29,31–39 Steck et al25 and LeMarchand et al22 had separate data for more than one ethnic group and were considered as two and three studies, respectively. Thus, the total number of studies was 20. Subjects in 12 studies were NHC,20–22,25,26,29,31,35–39 five Asian,22–24,33,34 one each of African-American,25 Hawaiian,22 and South American.32 Statistical power in 10 studies was adequate20,21,25,26,29,31,33,34,38,39 and the other 10 was not.22–25,32,35–37 Four studies provided separate data on folate intake.24,25,36,38 Methodological quality of the studies, determined by NOS, was moderate to high with a mean of 6.85 ± 1.84 and a median of 7.0. In addition, 13 studies from 10 articles20–22,25,26,31,32,37–39 had high NOS (7–9). Supplementary Table 1 shows the quantitative traits of the included studies. Of the 20 studies, two23,29 had control frequencies that deviated from the HWE. The PRISMA checklist was generated to provide detailed description of this meta-analysis (Supplementary Table 2).
Figure 1.
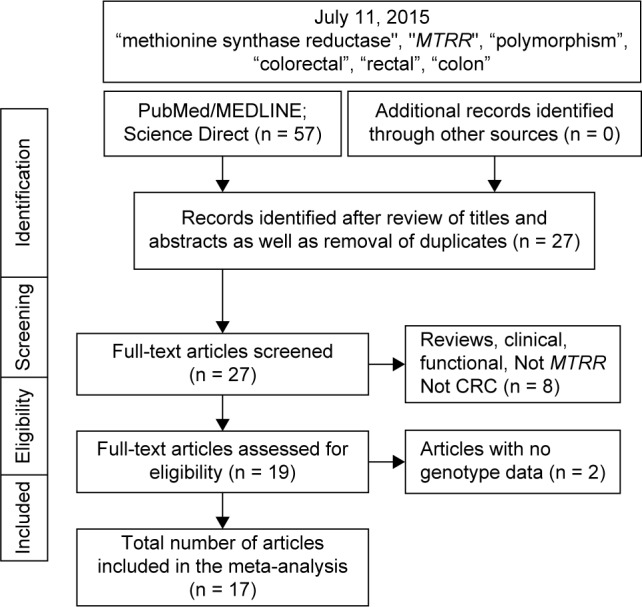
Flowchart of selection of studies for inclusion in the meta-analysis.
Overall and subgroup effects
Table 2 shows the overall pooled effect in the three genetic comparisons indicating absence of significant associations (OR 0.96–1.05, P = 0.12–0.44). These effects were confirmed with removal of the HWE-deviating studies (OR 0.97–1.06, P = 0.11–0.65) and subgroup analysis by NHC ethnicity (OR 0.93–1.05, P = 0.21–0.87). Of the three genetic comparisons, only the A allele comparison was heterogeneous (I2 = 44%) as shown in Figure 2. Subjecting the A allele pooled effect (OR 0.96, P = 0.44) to outlier treatment (Fig. 3) resulted in the following two outcomes: (i) zero heterogeneity (I2 = 0%) and (ii) gain in significance (OR 0.91, P = 0.01; Fig. 4). This post-outlier pooled value was unaltered with the combined removal of outliers plus HWE-deviating studies23,29,33,34,36 (OR 0.90, P = 0.01) and reflected in the high NOS effect (OR 0.90, P = 0.01). Stratified by ethnicity, the Asian homozygous (AA and GG) effects showed increased risk, significant in the A allele comparison (OR 1.13, P = 0.04). In contrast, the heterozygous A/G effect was significantly protective (OR 0.88, P = 0.05). The overall modifier and subgroup effects in all comparisons were deemed robust as they were unaltered by sensitivity treatment except the Caucasian summary effects in the A/G genotype comparison in account of serial omission of two studies.29,38 Of the three genetic comparisons of the overall effects shown in Table 3, only the G allele effects showed evidence of publication bias (Egger’s test: P = 0.04; Begg-Mazumdar test: P < 0.01).
Table 2.
Summary effects in the overall and subgroup analyses.
| TEST OF ASSOCIATION
|
TEST OF HETEROGENEITY
|
||||||
|---|---|---|---|---|---|---|---|
| N | OR | 95% CI | Pa | Pb | I2 (%) | ANALYSIS MODEL | |
| A allele | |||||||
| Overall | 20 | 0.96 | 0.87–1.06 | 0.44 | 0.02 | 44 | R |
| HWE studies only | 18 | 0.98 | 0.92–1.06 | 0.65 | 0.01 | 49 | R |
| Outliers off | 17 | 0.91 | 0.84–0.98 | 0.01 | 0.64 | 0 | F |
| Outliers + HWE off | 15 | 0.90 | 0.83–0.98 | 0.01 | 0.50 | 0 | F |
| Caucasian | 12 | 0.93 | 0.83–1.05 | 0.25 | 0.07 | 40 | R |
| Asian | 5 | 1.13 | 1.00–1.28 | 0.05 | 0.17 | 38 | F |
| 7–9 NOS | 13 | 0.90 | 0.83–0.98 | 0.01 | 0.35 | 9 | F |
| G allele | |||||||
| Overall | 20 | 1.05 | 0.99–1.13 | 0.12 | 0.25 | 16 | F |
| HWE studies only | 18 | 1.06 | 0.99–1.14 | 0.11 | 0.72 | 0 | F |
| Caucasian | 12 | 1.05 | 0.97–1.12 | 0.21 | 0.95 | 0 | F |
| Asian | 5 | 1.09 | 0.71–1.66 | 0.69 | 0.01 | 68 | R |
| 7–9 NOS | 13 | 1.07 | 0.99–1.15 | 0.08 | 0.82 | 0 | F |
| A/G genotype | |||||||
| Overall | 20 | 0.98 | 0.92–1.03 | 0.42 | 0.20 | 20 | F |
| HWE studies only | 18 | 0.97 | 0.91–1.03 | 0.35 | 0.27 | 16 | F |
| Caucasian | 12 | 1.01 | 0.94–1.07 | 0.87 | 0.21 | 24 | F |
| Asian | 5 | 0.88 | 0.78–1.00 | 0.05 | 0.31 | 16 | F |
| 7–9 NOS | 13 | 1.01 | 0.95–1.08 | 0.70 | 0.77 | 0 | F |
Notes: Pa: P value for test of association; Pb: P value for heterogeneity; I2 is a measure of heterogeneity expressed in %. Values in bold indicate significant associations. R: random-effects model, F: fixed-effects model.
Abbreviations: HWE, Hardy-Weinberg Equilibrium; N, number of studies; OR, odds ratio; CI, confidence interval; NOS, Newcastle-Ottawa Score.
Figure 2.

Summary effects in the A allele comparison. The diamond denotes the pooled odds ratio. Squares indicate the odds ratio in each study, with square sizes directly proportional to the weight contribution (%) of the study. Horizontal lines on each side of the squares represent 95% confidence intervals (CI). The chi-square test P value is <0.10 indicating heterogeneity, necessitating use of the random-effects model.
Abbreviations: J, Japanese; C, Caucasian; H, Hawaiian; AA, African-American; M-H, Mantel-Haenszel.
Figure 3.

Galbraith plot analysis to detect sources of heterogeneity in the A allele comparison. The three outliers (indicated by the last name of the first author) are the studies found outside (above) the +2 confidence limit.
Figure 4.

Summary effects in the A allele comparison without the outliers. The diamond denotes the pooled odds ratio. Squares indicate the odds ratio in each study, with square sizes directly proportional to the weight contribution (%) of the study. Horizontal lines on each side of the squares represent 95% confidence intervals (CI). The chi-square test P value is >0.10 and I2 of 0% indicating absence of heterogeneity, necessitating use of the fixed-effects model.
Abbreviations: J, Japanese; C, Caucasian; H, Hawaiian; AA, African-American; M-H, Mantel-Haenszel.
Table 3.
Results of tests for publication bias in the overall analysis.
| GENETIC COMPARISON | EGGER REGRESSION
|
BEGG-MAZUMDAR CORRELATION
|
||
|---|---|---|---|---|
| INTERCEPT | P VALUE | KENDALL’S τ | P VALUE | |
| A allele | −1.11 | 0.17 | −0.18 | 0.27 |
| G allele | 1.04 | 0.04 | 0.38 | <0.01 |
| A/G genotype | −0.50 | 0.47 | −0.07 | 0.65 |
Note: Values in bold indicate significance interpreted as evidence of publication bias.
Folate intake
Table 4 summarizes findings of the folate analysis. The four folate intake studies (669 cases/1,121 controls)24,25,36,38 showed non-significant associations without material differences between high and low intakes. In the A allele, the pooled effects were below 1 (OR 0.79–0.97, P = 0.13–0.82). G allele effects suggested increased risk (OR 1.11–1.20, P = 0.40–0.65), and the AG genotype effects ranged from null (high intake: OR 1.00, P = 0.99) to increased risk (low intake: OR 1.14, P = 0.40).
Table 4.
Summary results of the folate intake analysis.
| GENETIC COMPARISON | HIGH
|
LOW
|
||||
|---|---|---|---|---|---|---|
| OR | 95% CI | P VALUE | OR | 95% CI | P VALUE | |
| A allele | 0.97 | 0.71–1.31 | 0.82 | 0.79 | 0.58–1.08 | 0.13 |
| G allele | 1.11 | 0.70–1.76 | 0.65 | 1.20 | 0.79–1.81 | 0.40 |
| A/G genotype | 1.00 | 0.59–1.68 | 0.99 | 1.14 | 0.84–1.53 | 0.40 |
Abbreviations: OR, odds ratio; CI, confidence interval.
Sensitivity treatment deemed that the low folate summary effects were robust but not the high folate pooled ORs on account of three studies.24,25,36
Discussion
With a sample size of 20,945, the main message of this meta-analysis is lack of evidence of an overall association between MTRR A66G and CRC. The appeal of meta-analysis is statistically detecting profiles of the component studies in regard to their contribution to the overall effect. Thus, in the A allele analysis, omission of the three studies deemed outliers33,34,36 resulted in a significant 9% protective effect with concomitant abolition of heterogeneity. Omitting both the outliers and the HWE-deviating studies23,29,33,34,36 increased the protective effect to 10% with significance and zero heterogeneity retained. In addition, sensitivity treatment did not alter the pooled post-outlier and post-outlier-HWE ORs, thus conferring reliability to the findings. Thus, in addition to the previous data and based on statistical significance, high methodological quality, and combinability of the studies, the modest A allele effects indicate protection from CRC. Also, omission of the HWE-deviating studies minimizes the chance of false-positive results,50 which further strengthens our A allele finding.
Study-specific ORs in the A allele analysis indicating reduced risk were observed in 13 studies and significant in one of them (OR 0.66, 95% CI 0.44–0.97). The remaining seven study-specific ORs indicated increased risk, significant (ORs 1.36–1.60, 95% CI 1.03–2.51) in two studies, which happen to be outliers.34,36 This spectrum of individual study effects suggests usefulness of the meta-analytical approach in examining broad trends of MTRR associations with CRC. This then, may avoid possible misleading conclusions based on only single-population studies.
Our post-outlier overall significant protective finding in the A allele and similar results from the high NOS subgroup as well as the post-outlier/HWE results agree with a study that found a significant 34% protective role of the MTRR A/A genotype in a European population.37 A functional explanation for the possible role of the MTRR 66A/A genotype in preventing colon carcinogenesis may be through regulation of MTR activity. This might consequently influence levels of s-adenosylmethionine (SAM) and DNA methylation reactions.37 Both MTR and MTRR regulate the reaction that produces methionine through the irreversible transfer of a methyl group from 5-methyltetrahydrofolate. MTR is maintained in its active form by MTRR, an enzyme that regenerates a functional MTR via reductive methylation.27 The MTR 2756A > G and MTRR 66A > G polymorphisms are putatively functional,7 but the variant enzyme of MTRR has a lower affinity for MTR.51,52 Although functional effects of the MTRR A66G variant have not been fully established, in vitro experiments suggest that the variant MTRR enzyme restores MTR activity less efficiently than wild type.51,53 The variant alleles of MTR 2756G and MTRR 66G are thought to affect enzymatic activity, with consequently reduced production of methyl groups and risk for CRC.7
In the Asian subgroup, the MTRR 1.1-fold homozygote (A/A) susceptibility contrasting with the heterozygous A/G significant 12% protective effects suggests molecular heterosis. This genetic phenomenon occurs when individuals have the heterozygote advantage over homozygotes.54 Thus, based on heterotic effects, Asian heterozygotes are protected from CRC. Although heterosis seems counterintuitive to the standard gene dosage effects, it is increasingly recognized in humans, up to 50% of all gene associations.54 Molecular heterosis has been demonstrated in other cancers.55,56
The folate analysis has the following features: (i) non-robustness of the high folate analysis as conferred to by sensitivity treatment; (ii) in contrast, this treatment conferred robustness to the low folate findings; (iii) in the G allele analysis, both low and high intakes showed 1.1–1.2-fold increased risk, indicating absence of material differences between the two subgroups. In the A allele analysis, the low folate subgroup showed 21% reduced risk. This finding agrees with a cohort study that found reduced CRC risk in subjects with low folate levels and another that found a decrease in number and size of induced CRC tumors in folate-deficient rats.57 Biochemical explanation for the protective effect of low folate may be that proliferating cancer cells have greater need for folate.58 Cancer cells tend to upregulate their membrane receptors that mediate their folate uptake for DNA synthesis.59 Low folate intake thus impedes cancer cell proliferation.
Because many genes are involved in folate metabolism, effect of multiple functional polymorphisms in genes encoding for enzymes in the pathway are expected to be stronger than the effect of any one individual polymorphism. Of the 17 publications, only two23,35 examined MTRR by itself; the remaining 15 articles investigated MTRR in concert with polymorphisms of other genes. Of the other genes, the most common was methylenetetrahydrofolate reductase (MTHFR) in 13 publications20–22,24–26,29,31,32,36–39 followed by MTR in 11 publications.21,22,25,26,29,31–34,36,37
In a Japanese population, Morita et al33 found a suggestive interaction for MTRR A66G and MTHFR A1298C (P = 0.07) and an adjusted 1.4-fold increased risk for MTHFR 1298C allele and MTRR 66A/A genotype compared with those having the MTHFR 1298A/A and MTRR 66A/A genotype. In an Eastern European population study, not only found a significant 1.3-fold increased risk (P = 0.04) from a combination of MTHFR 1298A and MTRR 66G haplotypes but also found a significant 22% protective effect (P = 0.04) of MTHFR 1298A with MTRR 66A haplotype.37 Jokic et al37 showed that polymorphisms MTRR A66G and MTHFR A1298C combined influence colon cancer risk. A functional explanation for the combined influence of these two SNPs seems to be that MTHFR Glu429Ala, which results from A1298C, is located near the binding site of SAM, the allosteric inhibitor of MTHFR.60 As MTRR influences homocysteine conversion to methionine, which in turn converts into SAM, A66G may influence SAM production, which could change MTHFR feedback inhibition.
We compare our meta-analysis findings with two recent ones (2012) that addressed associations of MTRR A66G with CRC. The study by Han et al27 examined CRC in subgroup that composed of seven studies. The other study by Zhou et al28 examined polymorphisms in three genes (MTHFR, MTRR, and MTR) with risk of CRC that composed of 12 studies. Given the 20 studies in our meta-analysis, we have higher sample sizes compared to both. Another difference is that both meta-analyses used the standard genetic models (eg, homozygous, recessive) while we used three genetic comparisons for the reason that the minor allele frequencies were non-uniform across the studies. Han et al27 did not find significant associations but Zhou et al28 did, at least for the G allele increasing Caucasian risk but not in Asians. In contrast, our G allele findings found no associations among Caucasians but found significant A allele and A/G genotype associations among Asians. Our Caucasian and Asian findings are based on double (and almost double) the number of studies (N = 12 and 5, respectively) compared to Zhou et al28 (N = 6 and 3, respectively).
This meta-analysis has a number of important strengths: (i) Large sample sizes in the overall analysis translate to high statistical power. Even without the outliers plus HWE-deviating studies where the resulting summary effects were significant, statistical power remained high. (ii) Thirteen (65%) of the studies had high NOS (7–9). (iii) Evidence of lack of publication bias in the A allele and A/G genotype analyses. (iv) Controls in 15 (75%) publications were matched with cases. (v) Twelve (60%) the studies were population-based, indicating that the findings could be extrapolated to the general population. These features render selection bias unlikely. Furthermore, (vi) outlier treatment rendered significance to the overall findings in the A allele analysis and erased its heterogeneity. This was confirmed with high NOS results and removal of the outlier-HWE studies, which generated remarkably similar pooled ORs in terms of significance and non-heterogeneity, suggesting consistency of the A allele summary effects. (vii) All significant findings were non-heterogeneous. (viii) Sensitivity treatment conferred robustness of all findings in the homozygote comparisons.
These strengths, however, are countered by the following limitations of our study: (i) effects of gene–gene and gene–environment interactions were not addressed; (ii) eight of the 17 articles (47%) mention healthy controls; (iii) evidence of publication bias in the G allele comparison warrants caution in interpretation of its findings; and (iv) folate analysis did not reveal material differences between low and high intakes. More studies may be needed to confirm or modify our findings.
Considered individually, this polymorphism may have little or no influence and would probably require haplotype analysis to discern combined effects. In addition, integrated pathway analysis61 may elucidate how genetic variations in several genes cooperate in the etiology of CRC.20 Such analyses may shed light on the complexities of the many pathways involved in one-carbon metabolism and CRC, providing hypotheses for future functional studies.
Supplementary Materials
Supplementary table 1. Quantitative features of the included studies that examined the A66G (rs1801394) polymorphism in the MTRR gene and its association with colorectal cancer.
Supplementary table 2. PRISMA checklist.
Footnotes
ACADEMIC EDITOR: Barbara Guinn, Editor in Chief
PEER REVIEW: Two peer reviewers contributed to the peer review report. Reviewers’ reports totaled 216 words, excluding any confidential comments to the academic editor.
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Study conceived and planned: NP. Performed the literature search, data collection, and analyses: NP, ES, and LT. Wrote the first draft of the manuscript: NP. Main preparation of the manuscript: NP, ES, LT, HJ, and NS. Agreed with manuscript results and conclusions: HJ and NS. Jointly developed the structure and arguments for the paper: NP, HJ, and NS. Critically revised and reviewed the subsequent drafts: HJ and NS. All authors reviewed and approved the final manuscript.
REFERENCES
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Bogaert J, Prenen H. Molecular genetics of colorectal cancer. Ann Gastroenterol. 2014;27(1):9–14. [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi H, Jin C, Rajabi H, et al. MUC1-C activates the TAK1 inflammatory pathway in colon cancer. Oncogene. 2015;34(40):5187–5197. doi: 10.1038/onc.2014.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLean MH, Murray GI, Stewart KN, et al. The inflammatory microenvironment in colorectal neoplasia. PLoS One. 2011;6(1):e15366. doi: 10.1371/journal.pone.0015366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dyson JK, Rutter MD. Colorectal cancer in inflammatory bowel disease: what is the real magnitude of the risk? World J Gastroenterol. 2012;18(29):3839–3848. doi: 10.3748/wjg.v18.i29.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmed FE. Gene-gene, gene-environment & multiple interactions in colorectal cancer. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2006;24(1):1–101. doi: 10.1080/10590500600614295. [DOI] [PubMed] [Google Scholar]
- 7.Sharp L, Little J. Polymorphisms in genes involved in folate metabolism and colorectal neoplasia: a HuGE review. Am J Epidemiol. 2004;159(5):423–443. doi: 10.1093/aje/kwh066. [DOI] [PubMed] [Google Scholar]
- 8.Kim YI. Folate and carcinogenesis: evidence, mechanisms, and implications. J Nutr Biochem. 1999;10(2):66–88. doi: 10.1016/s0955-2863(98)00074-6. [DOI] [PubMed] [Google Scholar]
- 9.Friso S, Choi SW. Gene-nutrient interactions in one-carbon metabolism. Curr Drug Metab. 2005;6(1):37–46. doi: 10.2174/1389200052997339. [DOI] [PubMed] [Google Scholar]
- 10.Leclerc D, Wilson A, Dumas R, et al. Cloning and mapping of a cDNA for methionine synthase reductase, a flavoprotein defective in patients with homocystinuria. Proc Natl Acad Sci U S A. 1998;95(6):3059–3064. doi: 10.1073/pnas.95.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson A, Platt R, Wu Q, et al. A common variant in methionine synthase reductase combined with low cobalamin (vitamin B12) increases risk for spina bifida. Mol Genet Metab. 1999;67(4):317–323. doi: 10.1006/mgme.1999.2879. [DOI] [PubMed] [Google Scholar]
- 12.Storch KJ, Wagner DA, Burke JF, Young VR. [1-13C; methyl-2H3]methionine kinetics in humans: methionine conservation and cystine sparing. Am J Physiol. 1990;258(5 pt 1):E790–E798. doi: 10.1152/ajpendo.1990.258.5.E790. [DOI] [PubMed] [Google Scholar]
- 13.Sanjoaquin MA, Allen N, Couto E, Roddam AW, Key TJ. Folate intake and colorectal cancer risk: a meta-analytical approach. Int J Cancer. 2005;113(5):825–828. doi: 10.1002/ijc.20648. [DOI] [PubMed] [Google Scholar]
- 14.Guerreiro CS, Carmona B, Gonçalves S, et al. Risk of colorectal cancer associated with the C677T polymorphism in 5,10-methylenetetrahydrofolate reductase in Portuguese patients depends on the intake of methyl-donor nutrients. Am J Clin Nutr. 2008;88(5):1413–1418. doi: 10.3945/ajcn.2008.25877. [DOI] [PubMed] [Google Scholar]
- 15.Su LJ, Arab L. Nutritional status of folate and colon cancer risk: evidence from NHANES I epidemiologic follow-up study. Ann Epidemiol. 2001;11(1):65–72. doi: 10.1016/s1047-2797(00)00188-5. [DOI] [PubMed] [Google Scholar]
- 16.Luebeck EG, Moolgavkar SH, Liu AY, Boynton A, Ulrich CM. Does folic acid supplementation prevent or promote colorectal cancer? Results from model-based predictions. Cancer Epidemiol Biomarkers Prev. 2008;17(6):1360–1367. doi: 10.1158/1055-9965.EPI-07-2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mason JB, Dickstein A, Jacques PF, et al. A temporal association between folic acid fortification and an increase in colorectal cancer rates may be illuminating important biological principles: a hypothesis. Cancer Epidemiol Biomarkers Prev. 2007;16(7):1325–1329. doi: 10.1158/1055-9965.EPI-07-0329. [DOI] [PubMed] [Google Scholar]
- 18.Giovannucci E. Epidemiologic studies of folate and colorectal neoplasia: a review. J Nutr. 2002;132(8 suppl):2350S–2355S. doi: 10.1093/jn/132.8.2350S. [DOI] [PubMed] [Google Scholar]
- 19.Van Guelpen B, Hultdin J, Johansson I, et al. Low folate levels may protect against colorectal cancer. Gut. 2006;55(10):1461–1466. doi: 10.1136/gut.2005.085480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hazra A, Wu K, Kraft P, Fuchs CS, Giovannucci EL, Hunter DJ. Twenty-four non-synonymous polymorphisms in the one-carbon metabolic pathway and risk of colorectal adenoma in the Nurses’ Health Study. Carcinogenesis. 2007;28(7):1510–1519. doi: 10.1093/carcin/bgm062. [DOI] [PubMed] [Google Scholar]
- 21.Koushik A, Kraft P, Fuchs CS, et al. Nonsynonymous polymorphisms in genes in the one-carbon metabolism pathway and associations with colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2006;15(12):2408–2417. doi: 10.1158/1055-9965.EPI-06-0624. [DOI] [PubMed] [Google Scholar]
- 22.Le Marchand L, Donlon T, Hankin JH, Kolonel LN, Wilkens LR, Seifried A. B-vitamin intake, metabolic genes, and colorectal cancer risk (United States) Cancer Causes Control. 2002;13(3):239–248. doi: 10.1023/a:1015057614870. [DOI] [PubMed] [Google Scholar]
- 23.Matsuo K, Hamajima N, Hirai T, et al. Methionine synthase reductase gene A66G polymorphism is associated with risk of colorectal cancer. Asian Pac J Cancer Prev. 2002;3(4):353–359. [PubMed] [Google Scholar]
- 24.Otani T, Iwasaki M, Hanaoka T, et al. Folate, vitamin B6, vitamin B12, and vitamin B2 intake, genetic polymorphisms of related enzymes, and risk of colorectal cancer in a hospital-based case-control study in Japan. Nutr Cancer. 2005;53(1):42–50. doi: 10.1207/s15327914nc5301_5. [DOI] [PubMed] [Google Scholar]
- 25.Steck SE, Keku T, Butler LM, et al. Polymorphisms in methionine synthase, methionine synthase reductase and serine hydroxymethyltransferase, folate and alcohol intake, and colon cancer risk. J Nutrigenet Nutrigenomics. 2008;1(4):196–204. doi: 10.1159/000136651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Theodoratou E, Farrington SM, Tenesa A, et al. Dietary vitamin B6 intake and the risk of colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2008;17(1):171–182. doi: 10.1158/1055-9965.EPI-07-0621. [DOI] [PubMed] [Google Scholar]
- 27.Han D, Shen C, Meng X, et al. Methionine synthase reductase A66G polymorphism contributes to tumor susceptibility: evidence from 35 case-control studies. Mol Biol Rep. 2012;39(2):805–816. doi: 10.1007/s11033-011-0802-6. [DOI] [PubMed] [Google Scholar]
- 28.Zhou D, Mei Q, Luo H, Tang B, Yu P. The polymorphisms in methylenetetra-hydrofolate reductase, methionine synthase, methionine synthase reductase, and the risk of colorectal cancer. Int J Biol Sci. 2012;8(6):819–830. doi: 10.7150/ijbs.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Vogel S, Wouters KA, Gottschalk RW, et al. Genetic variants of methyl metabolizing enzymes and epigenetic regulators: associations with promoter CpG island hypermethylation in colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2009;18(11):3086–3096. doi: 10.1158/1055-9965.EPI-09-0289. [DOI] [PubMed] [Google Scholar]
- 30.Wells GA, Shea B, O’Connell D, et al. The Newcastle-Ottawa Scale (NOS) for Assessing the Quality of Non-Randomised Studies in Meta-Analyses. Ottawa Hospital Research Institute; [Accessed May, 2015]. http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp. [Google Scholar]
- 31.Curtin K, Ulrich CM, Samowitz WS, et al. Candidate pathway polymorphisms in one-carbon metabolism and risk of rectal tumor mutations. Int J Mol Epidemiol Genet. 2011;2(1):1–8. [PMC free article] [PubMed] [Google Scholar]
- 32.Guimaraes JL, Ayrizono Mde L, Coy CS, Lima CS. Gene polymorphisms involved in folate and methionine metabolism and increased risk of sporadic colorectal adenocarcinoma. Tumour Biol. 2011;32(5):853–861. doi: 10.1007/s13277-011-0185-2. [DOI] [PubMed] [Google Scholar]
- 33.Morita M, Yin G, Yoshimitsu S, et al. Folate-related nutrients, genetic polymorphisms, and colorectal cancer risk: the fukuoka colorectal cancer study. Asian Pac J Cancer Prev. 2013;14(11):6249–6256. doi: 10.7314/apjcp.2013.14.11.6249. [DOI] [PubMed] [Google Scholar]
- 34.Yoshimitsu S, Morita M, Hamachi T, et al. Methionine synthase and thymidylate synthase gene polymorphisms and colorectal adenoma risk: the self defense forces study. Mol Carcinog. 2012;51(suppl 1):E151–E157. doi: 10.1002/mc.21895. [DOI] [PubMed] [Google Scholar]
- 35.Burcoş T, Toma M, Stavarachi M, et al. MTRR polymorphism and the risk for colorectal and breast cancer in Romanian patients—a preliminary study. Chirurgia. 2010;105(3):379–382. [PubMed] [Google Scholar]
- 36.Hubner RA, Muir KR, Liu JF, et al. Folate metabolism polymorphisms influence risk of colorectal adenoma recurrence. Cancer Epidemiol Biomarkers Prev. 2006;15(9):1607–1613. doi: 10.1158/1055-9965.EPI-06-0274. [DOI] [PubMed] [Google Scholar]
- 37.Jokić M, Brčić-Kostić K, Stefulj J, et al. Association of MTHFR MTR MTRR, RFC1, and DHFR gene polymorphisms with susceptibility to sporadic colon cancer. DNA Cell Biol. 2011;30(10):771–776. doi: 10.1089/dna.2010.1189. [DOI] [PubMed] [Google Scholar]
- 38.Liu AY, Scherer D, Poole E, et al. Gene-diet-interactions in folate-mediated one-carbon metabolism modify colon cancer risk. Mol Nutr Food Res. 2013;57(4):721–734. doi: 10.1002/mnfr.201200180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pardini B, Kumar R, Naccarati A, et al. MTHFR and MTRR genotype and haplotype analysis and colorectal cancer susceptibility in a case-control study from the Czech Republic. Mutat Res. 2011;721(1):74–80. doi: 10.1016/j.mrgentox.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 40.Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst. 1959;22(4):719–748. [PubMed] [Google Scholar]
- 41.DerSimonian R Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7(3):177–188. doi: 10.1016/0197-2456(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 42.Lau J, Ioannidis JP, Schmid CH. Quantitative synthesis in systematic reviews. Ann Intern Med. 1997;127(9):820–826. doi: 10.7326/0003-4819-127-9-199711010-00008. [DOI] [PubMed] [Google Scholar]
- 43.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21(11):1539–1558. doi: 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- 44.Galbraith RF. A note on graphical presentation of estimated odds ratios from several clinical trials. Stat Med. 1988;7(8):889–894. doi: 10.1002/sim.4780070807. [DOI] [PubMed] [Google Scholar]
- 45.Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327(7414):557–560. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zafarmand MH, van der Schouw YT, Grobbee DE, de Leeuw PW, Bots ML. The M235T polymorphism in the AGT gene and CHD risk: evidence of a Hardy-Weinberg equilibrium violation and publication bias in a meta-analysis. PLoS One. 2008;3(6):e2533. doi: 10.1371/journal.pone.0002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315(7109):629–634. doi: 10.1136/bmj.315.7109.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics. 1994;50(4):1088–1101. [PubMed] [Google Scholar]
- 49.Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097. doi: 10.1371/journal.pmed.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu J, Turner A, Little J, Bleecker ER, Meyers DA. Positive results in association studies are associated with departure from Hardy-Weinberg equilibrium: hint for genotyping error? Hum Genet. 2002;111(6):573–574. doi: 10.1007/s00439-002-0819-y. [DOI] [PubMed] [Google Scholar]
- 51.Olteanu H, Munson T, Banerjee R. Differences in the efficiency of reductive activation of methionine synthase and exogenous electron acceptors between the common polymorphic variants of human methionine synthase reductase. Biochemistry. 2002;41(45):13378–13385. doi: 10.1021/bi020536s. [DOI] [PubMed] [Google Scholar]
- 52.Brown CA, McKinney KQ, Kaufman JS, Gravel RA, Rozen R. A common polymorphism in methionine synthase reductase increases risk of premature coronary artery disease. J Cardiovasc Risk. 2000;7(3):197–200. doi: 10.1177/204748730000700306. [DOI] [PubMed] [Google Scholar]
- 53.Gaughan DJ, Kluijtmans LA, Barbaux S, et al. The methionine synthase reductase (MTRR) A66G polymorphism is a novel genetic determinant of plasma homocysteine concentrations. Atherosclerosis. 2001;157(2):451–456. doi: 10.1016/s0021-9150(00)00739-5. [DOI] [PubMed] [Google Scholar]
- 54.Comings DE, MacMurray JP. Molecular heterosis: a review. Mol Genet Metab. 2000;71(1–2):19–31. doi: 10.1006/mgme.2000.3015. [DOI] [PubMed] [Google Scholar]
- 55.Ashton KA, Meldrum CJ, McPhillips ML, et al. The association of the COMT V158M polymorphism with endometrial/ovarian cancer in HNPCC families adhering to the Amsterdam criteria. Hered Cancer Clin Pract. 2006;4(2):94–102. doi: 10.1186/1897-4287-4-2-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marian C, Tao M, Mason JB, et al. Single nucleotide polymorphisms in uracil-processing genes, intake of one-carbon nutrients and breast cancer risk. Eur J Clin Nutr. 2011;65(6):683–689. doi: 10.1038/ejcn.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Le Leu RK, Young GP, McIntosh GH. Folate deficiency reduces the development of colorectal cancer in rats. Carcinogenesis. 2000;21(12):2261–2265. doi: 10.1093/carcin/21.12.2261. [DOI] [PubMed] [Google Scholar]
- 58.Wagner C. Biochemical role of folate in cellular metabolism. In: Bailey L, editor. Folate in Health and Disease. New York, NY: Marcel Dekker; 1995. pp. 23–42. [Google Scholar]
- 59.Kelemen LE. The role of folate receptor alpha in cancer development, progression and treatment: cause, consequence or innocent bystander? Int J Cancer. 2006;119(2):243–250. doi: 10.1002/ijc.21712. [DOI] [PubMed] [Google Scholar]
- 60.Curtin K, Bigler J, Slattery ML, Caan B, Potter JD, Ulrich CM. MTHFR C677T and A1298C polymorphisms: diet, estrogen, and risk of colon cancer. Cancer Epidemiol Biomarkers Prev. 2004;13(2):285–292. doi: 10.1158/1055-9965.epi-03-0083. [DOI] [PubMed] [Google Scholar]
- 61.Ulrich CM. Nutrigenetics in cancer research—folate metabolism and colorectal cancer. J Nutr. 2005;135(11):2698–2702. doi: 10.1093/jn/135.11.2698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1. Quantitative features of the included studies that examined the A66G (rs1801394) polymorphism in the MTRR gene and its association with colorectal cancer.
Supplementary table 2. PRISMA checklist.