

Abstract
Rationale: Bronchopulmonary dysplasia remains a significant cause of neonatal morbidity; however, the identification of novel targets to predict or prevent the development of bronchopulmonary dysplasia remains elusive. Proper microRNA (miR)-17∼92 cluster is necessary for normal lung development, and alterations in expression are reported in other pulmonary diseases. The overall hypothesis for our work is that altered miR-17∼92 cluster expression contributes to the molecular pathogenesis of bronchopulmonary dysplasia.
Objectives: The current studies tested the hypothesis that alterations in miR-17∼92 cluster and DNA methyltransferase expression are present in bronchopulmonary dysplasia.
Methods: miR-17∼92 cluster expression, promoter methylation, and DNA methyltransferase expression were determined in autopsy lung samples obtained from premature infants who died with bronchopulmonary dysplasia, or from term/near-term infants who died from nonrespiratory causes. Expression of miR-17∼92 cluster members miR-17 and -19b was measured in plasma samples collected in the first week of life from a separate cohort of preterm infants at a second institution in whom bronchopulmonary dysplasia was diagnosed subsequently.
Measurements and Main Results: Autopsy tissue data indicated that miR-17∼92 expression is significantly lower in bronchopulmonary dysplasia lungs and is inversely correlated with promoter methylation and DNA methyltransferase expression when compared with that of control subjects without bronchopulmonary dysplasia. Plasma sample analyses indicated that miR-17 and -19b expression was decreased in infants who subsequently developed bronchopulmonary dysplasia.
Conclusions: Our data are the first to demonstrate altered expression of the miR-17∼92 cluster in bronchopulmonary dysplasia. The consistency between our autopsy and plasma findings further support our working hypothesis that the miR-17∼92 cluster contributes to the molecular pathogenesis of bronchopulmonary dysplasia.
Keywords: bronchopulmonary dysplasia, microRNA, miR-17∼92 cluster
Bronchopulmonary dysplasia (BPD), a chronic lung disease associated with prematurity, is characterized by altered lung growth, cellular proliferation and differentiation, and persistent respiratory morbidities (1–6). The identification of novel targets to predict or prevent the development of BPD remains elusive. MicroRNAs (miRs) are small noncoding RNAs that can alter gene expression by either blocking translation or degrading target mRNA. miRs can be encoded in intronic or exonic DNA regions and in controlled DNA promoter elements (7). Transgenic murine models have demonstrated the importance of the miR-17∼92 cluster in normal lung growth and development (8). Processing of the polycistronic miR-17∼92 cluster results in six individual miRs (miR-17, -18a, -19a, -19b, -20a, and -92a) that are predicted to suppress the expression of several genes implicated in BPD pathogenesis, including those responsible for collagen (COL) production and matrix rearrangement (7, 9). In idiopathic pulmonary fibrosis (IPF), attenuated miR-17∼92 cluster expression is directly correlated with deficits in pulmonary function; however, the expression of the miR-17∼92 cluster in BPD is unknown (7). Genetic disruption of the miR-17∼92 cluster in mice causes death from respiratory insufficiency in the neonatal period (10–12).
DNA methylation, the covalent addition of a methyl group to the carbon-5 position of cytosine, predominantly in the cytosine-phosphate-guanine (CpG) dinucleotide, is the major form of epigenetic regulation of gene expression in mammalian cells (13). More than 80% of the miR- 17∼92 cluster promoter contains a large CpG island (7). In IPF, the miR∼17–92 cluster appears to be regulated epigenetically via methylation of its promoter CpG islands by DNA methyltransferase (DNMT) enzymes (7). Three DNMTs are expressed in humans; DNMT-1 is responsible for the epigenetic repair of injured tissues, and DNMT-3a and -3b are responsible for the maintenance of methylation (14).
Given the similarities in the pathways involved in the pathogenesis of BPD and IPF, the current studies tested the hypothesis that alterations in miR-17∼92 cluster and DNMT expression are present in BPD. Members of the miR-17∼92 cluster and DNMTs were measured using lung autopsy tissues from preterm infants who died with BPD, or from term/near-term infants who died from nonrespiratory causes. Our data indicate that miR-17∼92 cluster expression was decreased and was inversely correlated with DNMT expression and promoter methylation. Routine acquisition of lung biopsy samples, which is standard of care in many lung diseases, is not feasible in extremely preterm neonates. Thus, there exists a need to identify suitable alternatives to biopsy that can be used as novel targets to predict or prevent the development of BPD. Using novel lipoplex nanoparticle technology, additional studies were performed with plasma samples obtained in the first week of life from a separate cohort of extremely preterm infants cared for at a separate, nonrelated institution. These data revealed that plasma miR-17 and -19b levels were decreased in infants who subsequently developed BPD compared with those who did not. Some of the results of these studies have been reported previously in abstract form (15, 16).
Methods
Human Tissues
Samples were obtained under protocols approved by the institutional review board and privacy board of the University of Rochester. Consent for autopsy, including a release of tissue for research, was obtained before tissue collection. The study was Health Insurance Portability and Accountability Act compliant. All tissues and data were deidentified before release to investigators. Data from this collection, including eligibility guidelines, exclusion criteria, and methodology of sample collection/storage, have been published previously (17–22). Fifteen samples (eight BPD) were analyzed for miR-17∼92 cluster expression and promoter methylation analyses. DNMT analyses were performed on only 10 samples (4 BPD) because of limitations in tissue availability (Table 1).
Table 1.
Subject demographics including age and pathological diagnosis
| Sample ID | Phenotype | Diagnosis | GA at Birth (wk) | GA at Death (wk) |
|---|---|---|---|---|
| Group 1: control subjects | ||||
| 4 | Control | Metabolic disease, NLD | 41 | 41.4 |
| 8 | Control | Neuromuscular abnormality, NLD | 40 | 40.6 |
| 21 | Control | HIE, NLD to very mild RDS | 36 | 36.1 |
| 30* | Control | HIE, NLD | 38.5 | 38.6 |
| 50* | Control | HIE, NLD | 41 | 41.4 |
| 51* | Control | NLD to mild RDS, central line event; chorioamnionitis | 32.3 | 32.7 |
| 56* | Control | NLD to mild RDS, central line event | 26.6 | 28.5 |
| Group 2: BPD | ||||
| 10* | BPD | BPD | 26 | 27.7 |
| 11* | BPD | BPD, NEC | 26 | 34.1 |
| 17* | BPD | BPD; cytomegalovirus + | 25 | 34.1 |
| 18 | BPD | BPD | 27 | 40.7 |
| 33* | BPD | BPD; cytomegalovirus + | 29.2 | 43.1 |
| 44* | BPD | BPD | 28 | 45 |
| 47 | BPD | BPD; cor pulmonale | 29.1 | 46.5 |
| 49* | BPD | BPD; rhinovirus + | 24.7 | 31.8 |
Definition of abbreviations: BPD = bronchopulmonary dysplasia; GA = gestational age; HIE = hypoxic ischemic encephalopathy; NEC = necrotizing enterocolitis; NLD = no lung disease; RDS = respiratory distress syndrome.
Used for DNA methyltransferase analyses.
Human Plasma
Samples were obtained under protocols approved by the institutional review board of Nationwide Children’s Hospital. Between May 2005 and December 2010, patients from neonatal intensive care units at Nationwide Children’s Hospital and Ohio State University Hospital were enrolled by written informed consent of the parents. The study was Health Insurance Portability and Accountability Act compliant. All tissues and data were deidentified before release to investigators. Data from this collection, including eligibility guidelines, exclusion criteria, and methodology of sample collection/storage, have been published previously (23, 24). The BPD group was defined as any infant requiring any respiratory support (supplemental oxygen, mechanical ventilation, nasal continuous positive airway pressure or cannula) at 36 weeks’ corrected gestational age.
Quantitative Real-time Polymerase Chain Reaction Analyses
RNA and miR were isolated from human and murine tissue samples (QIAGEN RNA Easy kit; QIAGEN, Valencia, CA) and analyses were performed (7). For miR analyses, small nucleolar RNA 202, RNU38B, and RNU6 samples were evaluated and the control with the most consistent threshold cycle (Ct) value and smallest variation was used for normalization. For mRNA analyses, control adenylyl cyclase-associated protein-1 was used for normalization.
In Situ Hybridization
Studies were performed as described (25). In brief, locked nucleic acid (LNA)-modified and 5′ digoxigenin–tagged probes specific for miR-let-7c, -19b, and -20a were used. The probe/target complex was visualized after the alkaline phosphatase-linked conjugate reacted with the chromogen, nitroblue tetrazolium and bromochloroindolyl phosphate with a nuclear fast red counterstain. miR-let-7c was used as a positive control because it is expressed consistently in human lung (7). Negative controls included omission of the probe and the use of a scrambled probe.
DNA Methylation Studies
DNA was isolated from tissue samples using the QIAamp DNA Mini Kit (QIAGEN), and methylation was analyzed using methyl-profiler assays according to manufacturer instructions.
Preparation of Cationic Lipoplex Nanoparticles Containing Molecular Beacons
Tethered cationic lipoplex nanoparticles (tCLN) containing molecular beacons (MBs) for miR-17 or -19b were prepared by injecting an MBs/lipids mixture into phosphate-buffered saline, using mixed thiol self-assembled monolayers as an anchoring membrane (26, 27). As reported previously, tCLN and quantitative real-time polymerase chain reaction (qRT-PCR) provide comparable results for miR detection. Data are also highly reproducible; previous studies have shown little well-to-well difference on the same chip, with chip-to-chip variability of less than 10% for the same sample (27). Glass slides were incubated with an avidin derivative (NeutrAvidin; Thermo Scientific, Waltham, MA) at room temperature for 5 min. MBs were tethered onto glass slides by biotin−avidin linkage. Samples were added on a tCLN biochip and incubated at 37°C for 2 h. Total internal reflection fluorescence microscopy was used to detect the fluorescence signals, and images were analyzed (27).
Statistics
Primary analyses compared all BPD subjects with all control subjects, using unpaired t tests or Mann-Whitney U tests (GraphPad PRISM 6; GraphPad Software, La Jolla, CA), and results are expressed as mean ± SD. Significance is noted at P < 0.05. For plasma analytes, receiver operating characteristic (ROC) curves were estimated to determine the extent to which miR concentrations predicted BPD at 36 weeks’ postmenstrual age.
Results
Human Autopsy Subject Demographics
miR-17∼92 cluster expression and promoter methylation were assessed in autopsy lung tissues from infants cared for at the University of Rochester Medical Center (n = 15) (Table 1). Gestational age at birth in the BPD group was significantly lower than in the control subjects (26.9 ± 0.6 vs. 36.5 ± 1.9 wk, P < 0.05); however, there were no differences in gestational age at death (37.9 ± 2.4 vs. 37.0 ± 1.9 wk, P = 0.794). In autopsy tissues used for DNMT analyses (n = 10) (Table 1), gestational age at birth in the BPD group was significantly lower than in the control subjects (26.5 ± 0.7 vs. 34.6 ± 3.2 wk, P < 0.05). Gestational age at death was not different (36.0 ± 2.7 vs. 35.3 ± 2.9 wk, P = 0.876).
miR-17∼92 Cluster Expression Is Decreased and Localization Is Altered in BPD Lung Tissues
Lung miR-17∼92 cluster expression was globally attenuated in infants who died with BPD (Figure 1). Compared with that of control subjects, expression of miR-17, -18a, -19a, -19b, and -92a was five times lower. Expression of miR-20a was two times lower in lung samples from BPD infants. Nonquantitative in situ hybridization studies were performed to characterize cluster localization in control and BPD tissues. A probe recognizing miR- let-7c, which is known to be present in adult lung, was used as a positive control. Analyses of non-BPD lungs revealed that mir-19b and -20a were expressed primarily in bronchial and alveolar epithelia (Figure 2). Conversely, in BPD lungs, miR-19b and -20a expression was localized predominantly to stromal cells.
Figure 1.

Decreased miR-17∼92 cluster expression in bronchopulmonary dysplasia (BPD). Expression was determined by quantitative real-time polymerase chain reaction from control (n = 7) and BPD (n = 8) autopsy lung samples. Data, normalized to RNU38B, are expressed as fold expression ± SD vs. control tissue (*P < 0.05).
Figure 2.

Altered localization of miR-19b and -20a in bronchopulmonary dysplasia (BPD). In situ hybridization was performed on autopsy tissues from four BPD and three term/near-term control patients using locked nucleic acid–modified DNA probes for miR-let-7c (positive control), -19b, and -20a. Scrambled probes were used as a negative control. Representative images are shown and each contains a terminal respiratory bronchiole (1,000×). Positively stained cells for miR-19b or -20a (blue, indicated by arrows) in control lungs are mostly epithelial, whereas positively stained cells in BPD lungs are predominantly stromal and not epithelial.
miR-17∼92 Cluster Promoter Methylation and DNMT Expression Is Increased in BPD Lung Tissues
In autopsy tissues, promoter methylation was approximately 50% in control subjects, compared with >90% in BPD lungs (Figure 3). Compared with that of control subjects, DNMT-1 expression was more than four times greater in BPD tissues (Figure 4A), whereas DNMT-3a expression was approximately three times greater (Figure 4B), and DNMT-3b was nearly six times greater (Figure 4C).
Figure 3.

Increased miR-17∼92 promoter methylation in bronchopulmonary dysplasia (BPD). Data are presented as average percent of unmethylated and methylated DNA in control (n = 7) and BPD (n = 8) tissues.
Figure 4.
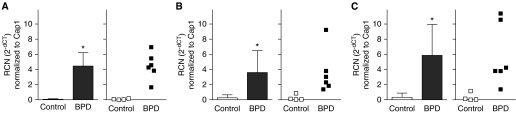
Increased DNA methyltransferase (DNMT) expression in bronchopulmonary dysplasia (BPD). Because of sample limitations, expression of (A) DNMT-1, (B) -3a, and (C) -3b was determined by quantitative real-time polymerase chain reaction in a subset of control (n = 4) and BPD (n = 6) tissues. Using adenylate cyclase-associated protein (Cap1) as an endogenous control, the average relative copy number (RCN) ± SD was calculated (*P < 0.01).
Plasma miR-17 and -19b Expression in the First Week of Life Individually Correlate With Subsequent BPD Development
Using miR-17 and -19b as surrogate markers of miR-17∼92 cluster expression, we analyzed 21 plasma samples obtained at day of life 5–7 from a cohort of patients cared for at Nationwide Children’s Hospital and The Ohio State University Medical Center in whom a diagnosis of BPD (n = 11) was later established (23). Birth weight (Figure 5A), gestational age at birth (Figure 5B), and Fio2 at the time of sample acquisition (Figure 5C) were not different between the control and BPD groups.
Figure 5.
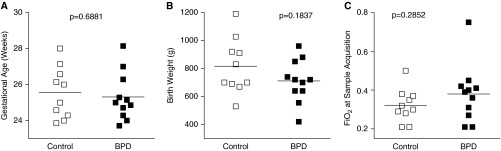
For plasma analyses, no differences were detected between control and bronchopulmonary dysplasia (BPD) groups in (A) birth weight, (B) gestational age at birth, and (C) Fio2 at the time of sample acquisition.
Plasma miR-17 expression was 3.4 times lower (Figure 6A) in infants who subsequently developed BPD than in infants who did not. This finding was confirmed using miR-19b, which was three times lower in infants who developed BPD (Figure 6B). Additional analyses indicated no effects of gestational age on plasma miR-17 or -19a expression (data not shown). ROC curves were estimated to determine the extent to which miR concentrations predicted BPD at 36 weeks’ postmenstrual age. The areas under the curve for miR-17 (Figure 6C) and miR-19b (Figure 6D) were 88% and 85%, respectively.
Figure 6.

Decreased plasma miR-17 and -19b expression in the first week of life in infants who subsequently developed bronchopulmonary dysplasia (BPD). (A and B) Expression (mean ± SD) of miR-17 and - 19b in control (n = 10) and BPD (n = 11) plasma samples was determined using lipoplex nanoparticles containing molecular beacons. (C and D) Receiver operating characteristic curves were estimated to determine the extent to which miR concentrations predicted BPD at 36 weeks’ postmenstrual age.
Discussion
Proper miR-17∼92 cluster expression is necessary for normal lung growth and development, whereas abnormal expression is associated with pulmonary fibrosis and cancers (8, 28). The cluster is highly expressed in the lungs during embryonic development, before declining in adulthood (11). Whole-genome miR-17∼92 cluster deletion is perinatally lethal, causing lung hypoplasia resulting from the absence of lung epithelial cells, which form the alveolar units (10). Epithelial cell–specific miR-17∼92 overexpression is similarly lethal because of epithelial cell hyperplasia and inhibition of normal epithelial cell differentiation (12).
In IPF, miR-17∼92 cluster expression is directly correlated with pulmonary function and inversely correlated with expression of transforming growth factor (TGF)-β, a predicted target of miR- 17, -19a, -19b, and -20a. In premature infants, tracheal aspirate TGF-β levels correlate directly with BPD severity and the need for home oxygen therapy (29–31). Individual members of the miR-17∼92 cluster regulate the pathways implicated in BPD development beyond TGF-β and its receptors (32), including hypoxia-inducible factor (33), matrix metalloproteinases, COL, and p53 (34).
Although not quantitative, our in situ analyses revealed marked differences in miR-19b and -20a localization in BPD lungs when compared with control lungs from term/near-term infants who died of nonrespiratory causes (Figure 2). Stromal localization in tissues from infants who died with BPD differed from the primarily epithelial localization in term/near-term control infants. The contribution of lung mesenchymal stromal cells to aberrant lung development in BPD and other lung diseases is incompletely understood. A recent comprehensive review by Collins and Thébaud concluded that this very heterogeneous population of cells is involved in BPD pathogenesis either by a change in numbers or by displaying an abnormal myofibroblast phenotype (35). Among the profibrotic factors produced by lung mesenchymal stromal cells, TGF-β, matrix metalloproteinases, and COL3A1 are predicted targets of members of the miR-17∼92 cluster (7). Collectively, our qRT-PCR demonstrating decreased miR-17∼92 cluster expression and in situ data demonstrating altered localization support a scenario in which attenuated miR-17∼92 cluster expression could conceivably contribute to enhanced production of profibrotic genes in BPD. A similar relationship has been demonstrated recently for miR-206, in which attenuated expression inversely correlated with enhanced expression of its predicted target fibronectin-1 in blood samples from infants with BPD. Mechanistically, a reduction in miR-206 enhanced fibronectin-1 function in vitro (36). Although not the focus of the current manuscript, our findings support the need for future studies in vitro to investigate the effects of attenuated miR-17∼92 cluster expression on downstream profibrotic genes.
Expression levels of all six individual miRs are lower in human autopsy lung tissues from infants who died with BPD when compared with term/near-term infant lung tissues (Figure 1). A recent metaanalysis revealed significant increases in miR-21, -34a, -431, and -let-7f expression in BPD lung tissues (37). Although our hypothesis focused on alterations in miR-17∼92 cluster expression, the findings in the metaanalysis suggest that decreased miR-17∼92 cluster expression is unlikely to represent global suppression of miR expression in BPD. Our findings led us to hypothesize that suppression of the miR-17∼92 cluster is associated with methylation of the promoter region of the parent transcript.
Three DNMTs are expressed in humans: DNMT-1, associated with repair of injured tissues, and DNMT-3a and -3b, which are critical for the maintenance of DNA methylation (14). Furthermore, DNMT-1 is regulated directly by the miR-17∼92 cluster, specifically miR-17, -20a, and -92a. Increases in DNMT activity result in increases in DNA methylation and transcriptional silencing of the affected genes. An alternative and/or complementary interpretation of the reciprocal expression of DNMT-1 and its regulatory miRs is that enhanced DNMT-1 expression represents validation of a change in the expression of a downstream target of the miR-17∼92 cluster. These relationships highlight the complexity between miRs and their targets, namely, which alteration is initially responsible for the alteration of the other.
Our human autopsy data confirmed hypermethylation of the promoter region of the miR-17∼92 gene (Figure 3) and dramatic increases in expression of the methyltransferases DNMT-1, -3a, and -3b in BPD lung tissues when compared with control tissues (Figure 4). The present data are consistent with a working model in which an epigenetic modification (methylation) in the promoter region of the miR-17∼92 cluster suppresses the transcription and expression of all members of the miR-17∼92 cluster. Cuna and coworkers recently reported the presence of differential methylation of 23 genes with reciprocal changes in gene expression in human lungs from patients with BPD (38). Genes identified included those responsible for lung development, extracellular matrix, and immune and antioxidant defenses. In this context, our findings in lung samples from infants who died with BPD suggest that enhanced DNMT expression in BPD likely mediates the methylation of many genes beyond miR-17∼92 alone.
The choice of the control group for our studies of autopsy tissues was driven by both practical and experimental reasons. The ideal control group would use tissues from prematurely born infants without BPD; however, lung specimens from these infants are virtually impossible to obtain. We also wanted to compare infants with BPD with infants without lung disease at a similar gestational age. The gestational age at death was not different between the groups (Table 1). Nonetheless, our findings are limited by the inability to determine the impact on cluster expression, DNA methylation, or DNMT expression of premature delivery, subsequent exposures and treatments, and/or differences in degradation in RNA in postmortem tissues. It is possible that these and other unidentified variables contributed to the observed differences reported in the present manuscript.
Our study is, to our knowledge, the first to demonstrate an association between altered plasma miR-17∼92 cluster expression and subsequent BPD development in predominantly extremely low–birth-weight infants (<1,000 g), those with the greatest risk of developing BPD. Recently, Wu and coworkers reported an association between BPD development and aberrant peripheral blood expression of miR-152, -30a-3p, -133, and -let-7f in the first 2 weeks of life in very low–birth-weight infants (<1,500 g) (39). The identification of relevant and specific analytes to predict the development of BPD in at-risk infants and/or to evaluate the effectiveness of novel therapeutic strategies has been hindered by a lack of access to relevant biologic compartments and/or sample volume limitations (40).
Although limited in volume, blood samples are obtained routinely from extremely preterm infants during the course of clinical management. The use of typically discarded leftover samples offers a potential source for sampling. The use of tCLN technology enabled us to overcome the sample volume limitations often encountered in neonatal research by permitting miR assessments using less than 50 μl of unprocessed plasma (27). As reported previously, tCLN and qRT-PCR provide comparable results for miR detection. tCLN data are highly reproducible, with little well-to-well difference on the same chip and chip-to-chip and variability of less than 10% for the same sample. An advantage of tCLN detection is the ability to detect a mixture of intact large molecules and smaller mRNA fragments that are present in extracellular vesicles, permitting simultaneous analyses of both miR and mRNA using the same unprocessed sample. MBs used in tCLN detection hybridize about 20 to 30 bases of the target exon, which may provide higher sensitivity than qRT-PCR with a much smaller sample volume. We used this technique to measure miR-17∼92 expression in plasma obtained at 4–6 days of life from 21 preterm infants (mean gestational age, 25 wk). Our data revealed significant decreases in plasma miR-17 and -19b expression in the first week of life in infants in whom BPD at 36 weeks’ postmenstrual age was diagnosed subsequently (Figures 6A and 6B). We chose to include all plasma values in our analyses, even though some were identified as statistical outliers because these data points came from different samples in each analysis. Nonetheless, the significance of the differences between groups remained even when excluding statistical outliers. Although our studies are preliminary, the significance of our ROC analyses (Figures 6C and 6D) supports the conducting of additional appropriately powered studies investigating early alterations in plasma miR-17∼92 expression and the relationship to the risk of subsequent BPD development.
Conclusions
In conclusion, our data indicate that decreased miR-17∼92 cluster expression in the developing lung is associated with the development of BPD. It is likely that differences in maternal and neonatal clinical management influence miR expression. Although we were unable to consider these variables in the present studies because of a lack of comprehensive clinical data, the influence of these variables will need to be examined in future studies. Our plasma data provide compelling evidence that the miR-17∼92 cluster is a measurable predictor for BPD development and may offer an avenue to test the therapeutic efficacy of treatments designed to preserve normal lung growth and prevent BPD. We believe our studies are the first to demonstrate the presence of altered expression of the miR-17∼92 cluster in BPD and suggest that these alterations are detectable early in life in low-volume samples. The consistency of our findings in different compartments (lung and plasma) at separate time points in separate cohorts of patients from different institutions further strengthens the association between altered miR-17∼92 cluster expression and BPD. Collectively, our data support miR-17∼92 as a potential target for predicting and/or preventing BPD development in at-risk infants.
Acknowledgments
Acknowledgment
Words cannot adequately convey the essential contributions of Melissa G. Piper, Ph.D., toward the design, execution, and interpretation of many of the studies in this manuscript. Dr. Piper’s tragic death preceded study and manuscript completion. The authors also acknowledge the excellent technical assistance provided by Cynthia Hill and Yijie Wang.
Footnotes
Supported by awards UL1TR001070 from National Center for Advancing Translational Sciences (T.E.T.) and K08HL093365 from the NHLBI (T.E.T.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NCATS, the NHLBI, or the National Institutes of Health.
Author Contributions: L.K.R., C.B.M., and T.E.T. conceived the ideas and designed and wrote the paper; L.K.R, M.R., D.D., and T.E.T. performed, oversaw, and analyzed studies; M.M. and G.N. assisted in in situ hybridization; Z.Y. and L.J.L. performed nanoparticle-based microRNA expression studies; G.S.P. performed pathologic analysis of lung autopsy tissues and assisted in data interpretation; G.M. assisted with statistical analyses.
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.McLeod A, Ross P, Mitchell S, Tay D, Hunter L, Hall A, Paton J, Mutch L. Respiratory health in a total very low birthweight cohort and their classroom controls. Arch Dis Child. 1996;74:188–194. doi: 10.1136/adc.74.3.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thébaud B, Abman SH. Bronchopulmonary dysplasia: where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med. 2007;175:978–985. doi: 10.1164/rccm.200611-1660PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thébaud B, Ladha F, Michelakis ED, Sawicka M, Thurston G, Eaton F, Hashimoto K, Harry G, Haromy A, Korbutt G, et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation. 2005;112:2477–2486. doi: 10.1161/CIRCULATIONAHA.105.541524. [DOI] [PubMed] [Google Scholar]
- 4.Van Marter LJ. Epidemiology of bronchopulmonary dysplasia. Semin Fetal Neonatal Med. 2009;14:358–366. doi: 10.1016/j.siny.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Wu S, Platteau A, Chen S, McNamara G, Whitsett J, Bancalari E. Conditional overexpression of connective tissue growth factor disrupts postnatal lung development. Am J Respir Cell Mol Biol. 2010;42:552–563. doi: 10.1165/rcmb.2009-0068OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambalavanan N, Carlo WA. Bronchopulmonary dysplasia: new insights. Clin Perinatol. 2004;31:613–628. doi: 10.1016/j.clp.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Dakhlallah D, Batte K, Wang Y, Cantemir-Stone CZ, Yan P, Nuovo G, Mikhail A, Hitchcock CL, Wright VP, Nana-Sinkam SP, et al. Epigenetic regulation of miR-17∼92 contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;187:397–405. doi: 10.1164/rccm.201205-0888OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonauer A, Dimmeler S. The microRNA-17-92 cluster: still a miRacle? Cell Cycle. 2009;8:3866–3873. doi: 10.4161/cc.8.23.9994. [DOI] [PubMed] [Google Scholar]
- 9.Morty RE, Königshoff M, Eickelberg O. Transforming growth factor-beta signaling across ages: from distorted lung development to chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:607–613. doi: 10.1513/pats.200908-087RM. [DOI] [PubMed] [Google Scholar]
- 10.Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nana-Sinkam SP, Karsies T, Riscili B, Ezzie M, Piper M. Lung microRNA: from development to disease. Expert Rev Respir Med. 2009;3:373–385. doi: 10.1586/ers.09.30. [DOI] [PubMed] [Google Scholar]
- 12.Lu Y, Thomson JM, Wong HY, Hammond SM, Hogan BL. Transgenic over-expression of the microRNA miR-17-92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev Biol. 2007;310:442–453. doi: 10.1016/j.ydbio.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daniel FI, Cherubini K, Yurgel LS, de Figueiredo MA, Salum FG. The role of epigenetic transcription repression and DNA methyltransferases in cancer. Cancer. 2011;117:677–687. doi: 10.1002/cncr.25482. [DOI] [PubMed] [Google Scholar]
- 14.Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, Jones PA. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 1999;27:2291–2298. doi: 10.1093/nar/27.11.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tipple TE, Robbins M, Dakhlallah D, Rogers LK, Piper MG. MiR-17∼92 cluster expression is decreased in bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2013;187:A5358. [Google Scholar]
- 16.Tipple TE, Robbins M, Rogers LK, Dakhlallah D, Piper MG.MiR-17∼92 cluster expression is decreased in lungs from infants with bronchopulmonary dysplasia Pediatric Academic Societies 2013. EPAS: 2825.2822
- 17.Reynolds SD, Reynolds PR, Pryhuber GS, Finder JD, Stripp BR. Secretoglobins SCGB3A1 and SCGB3A2 define secretory cell subsets in mouse and human airways. Am J Respir Crit Care Med. 2002;166:1498–1509. doi: 10.1164/rccm.200204-285OC. [DOI] [PubMed] [Google Scholar]
- 18.Maniscalco WM, Watkins RH, O’Reilly MA, Shea CP. Increased epithelial cell proliferation in very premature baboons with chronic lung disease. Am J Physiol Lung Cell Mol Physiol. 2002;283:L991–L1001. doi: 10.1152/ajplung.00050.2002. [DOI] [PubMed] [Google Scholar]
- 19.Lee MK, Pryhuber GS, Schwarz MA, Smith SM, Pavlova Z, Sunday ME. Developmental regulation of p66Shc is altered by bronchopulmonary dysplasia in baboons and humans. Am J Respir Crit Care Med. 2005;171:1384–1394. doi: 10.1164/rccm.200406-776OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhatt AJ, Pryhuber GS, Huyck H, Watkins RH, Metlay LA, Maniscalco WM. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2001;164:1971–1980. doi: 10.1164/ajrccm.164.10.2101140. [DOI] [PubMed] [Google Scholar]
- 21.Pryhuber GS, Huyck HL, Staversky RJ, Finkelstein JN, O’Reilly MA. Tumor necrosis factor-alpha-induced lung cell expression of antiapoptotic genes TRAF1 and cIAP2. Am J Respir Cell Mol Biol. 2000;22:150–156. doi: 10.1165/ajrcmb.22.2.3783. [DOI] [PubMed] [Google Scholar]
- 22.Bhattacharya S, Go D, Krenitsky DL, Huyck HL, Solleti SK, Lunger VA, Metlay L, Srisuma S, Wert SE, Mariani TJ, et al. Genome-wide transcriptional profiling reveals connective tissue mast cell accumulation in bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2012;186:349–358. doi: 10.1164/rccm.201203-0406OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogers LK, Young CM, Pennell ML, Tipple TE, Leonhart KL, Welty SE. Plasma lipid metabolites are associated with gestational age but not bronchopulmonary dysplasia. Acta Paediatr. 2012;101:e321–e326. doi: 10.1111/j.1651-2227.2012.02694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rogers LK, Graf AE, Bhatia A, Leonhart KL, Oza-Frank R. Associations between maternal and infant morbidities and sRAGE within the first week of life in extremely preterm infants. PLoS One. 2013;8:e82537. doi: 10.1371/journal.pone.0082537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nuovo GJ, Garofalo M, Valeri N, Roulstone V, Volinia S, Cohn DE, Phelps M, Harrington KJ, Vile R, Melcher A, et al. Reovirus-associated reduction of microRNA-let-7d is related to the increased apoptotic death of cancer cells in clinical samples. Mod Pathol. 2012;25:1333–1344. doi: 10.1038/modpathol.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwak KJ, Valincius G, Liao WC, Hu X, Wen X, Lee A, Yu B, Vanderah DJ, Lu W, Lee LJ. Formation and finite element analysis of tethered bilayer lipid structures. Langmuir. 2010;26:18199–18208. doi: 10.1021/la1021802. [DOI] [PubMed] [Google Scholar]
- 27.Wu Y, Kwak KJ, Agarwal K, Marras A, Wang C, Mao Y, Huang X, Ma J, Yu B, Lee R, et al. Detection of extracellular RNAs in cancer and viral infection via tethered cationic lipoplex nanoparticles containing molecular beacons. Anal Chem. 2013;85:11265–11274. doi: 10.1021/ac401983w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu Y, Okubo T, Rawlins E, Hogan BL. Epithelial progenitor cells of the embryonic lung and the role of microRNAs in their proliferation. Proc Am Thorac Soc. 2008;5:300–304. doi: 10.1513/pats.200710-162DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kotecha S, Wangoo A, Silverman M, Shaw RJ. Increase in the concentration of transforming growth factor beta-1 in bronchoalveolar lavage fluid before development of chronic lung disease of prematurity. J Pediatr. 1996;128:464–469. doi: 10.1016/s0022-3476(96)70355-4. [DOI] [PubMed] [Google Scholar]
- 30.Lecart C, Cayabyab R, Buckley S, Morrison J, Kwong KY, Warburton D, Ramanathan R, Jones CA, Minoo P. Bioactive transforming growth factor-beta in the lungs of extremely low birthweight neonates predicts the need for home oxygen supplementation. Biol Neonate. 2000;77:217–223. doi: 10.1159/000014219. [DOI] [PubMed] [Google Scholar]
- 31.Saito M, Ichiba H, Yokoi T, Hirai C, Yamano T, Kusuda S. Mitogenic activity of tracheal effluents from premature infants with chronic lung disease. Pediatr Res. 2004;55:960–965. doi: 10.1203/01.PDR.0000125257.55596.97. [DOI] [PubMed] [Google Scholar]
- 32.Li L, Shi JY, Zhu GQ, Shi B. MiR-17-92 cluster regulates cell proliferation and collagen synthesis by targeting TGFB pathway in mouse palatal mesenchymal cells. J Cell Biochem. 2012;113:1235–1244. doi: 10.1002/jcb.23457. [DOI] [PubMed] [Google Scholar]
- 33.Poitz DM, Augstein A, Gradehand C, Ende G, Schmeisser A, Strasser RH. Regulation of the Hif-system by micro-RNA 17 and 20a - role during monocyte-to-macrophage differentiation. Mol Immunol. 2013;56:442–451. doi: 10.1016/j.molimm.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 34.Oeztuerk-Winder F, Guinot A, Ochalek A, Ventura JJ. Regulation of human lung alveolar multipotent cells by a novel p38α MAPK/miR-17-92 axis. EMBO J. 2012;31:3431–3441. doi: 10.1038/emboj.2012.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins JJ, Thébaud B. Lung mesenchymal stromal cells in development and disease: to serve and protect? Antioxid Redox Signal. 2014;21:1849–1862. doi: 10.1089/ars.2013.5781. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Xu J, Wang J, Gortner L, Zhang S, Wei X, Song J, Zhang Y, Li Q, Feng Z. Reduction of microRNA-206 contributes to the development of bronchopulmonary dysplasia through up-regulation of fibronectin 1. PLoS One. 2013;8:e74750. doi: 10.1371/journal.pone.0074750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, Qiu J, Kan Q, Zhou XG, Zhou XY. MicroRNA expression profiling studies on bronchopulmonary dysplasia: a systematic review and meta-analysis. Genet Mol Res. 2013;12:5195–5206. doi: 10.4238/2013.October.30.4. [DOI] [PubMed] [Google Scholar]
- 38.Cuna A, Halloran B, Faye-Petersen O, Kelly D, Crossman DK, Cui X, Pandit K, Kaminski N, Bhattacharya S, Ahmad A, et al. Alterations in gene expression and DNA Methylation during murine and human lung alveolar septation. Am J Respir Cell Mol Biol. 2015;53:60–73. doi: 10.1165/rcmb.2014-0160OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu YT, Chen WJ, Hsieh WS, Tsao PN, Yu SL, Lai CY, Lee WC, Jeng SF. MicroRNA expression aberration associated with bronchopulmonary dysplasia in preterm infants: a preliminary study. Respir Care. 2013;58:1527–1535. doi: 10.4187/respcare.02166. [DOI] [PubMed] [Google Scholar]
- 40.Bhandari A, Bhandari V. Pitfalls, problems, and progress in bronchopulmonary dysplasia. Pediatrics. 2009;123:1562–1573. doi: 10.1542/peds.2008-1962. [DOI] [PubMed] [Google Scholar]