

Abstract
Various drug delivery approaches can be used to maximize therapeutic efficacy and minimize side effects, by impacting absorption, distribution, metabolism, and elimination (ADME) of a drug compound. For those drugs with poor water solubility or low permeability, techniques such as amorphous solid dispersion, liposomes, and complexations have been used to improve their oral bioavailability. Modified release (MR) formulations have been widely used to improve patient compliance, as well as to reduce side effects, especially for those drugs with short half-lives or narrow therapeutic windows. More than ten drugs using sterile long-acting release (LAR) formulations with clear clinical benefit have been successfully marketed. Furthermore, drug delivery systems have been used in delaying drug clearance processes. Additionally, modifying the in vivo drug distribution using targeted delivery systems has significantly improved oncology treatments. All the drug delivery approaches have their advantages and limitations. For both brand and generic drugs, the achievement of consistent quality and therapeutic performance using drug delivery systems can also pose serious challenges in developing a drug for the market, which requires close collaboration among industry, academia, and regulatory agencies. With the advent of personalized medicines, there will be great opportunities and challenges in utilizing drug delivery systems to provide better products and services for patients.
KEY WORDS: absorption, distribution, metabolism, and elimination (ADME); adverse effects; bioequivalence; clinical pharmacology; drug delivery; formulation design; local delivery; long-acting release; modified release; personalized medicine; pharmacokinetic profiles; prodrug; quality; regulatory; targeted delivery; therapeutic performance
INTRODUCTION
Drug delivery (1) refers to various approaches for delivering a pharmaceutical compound in the human body to achieve and/or optimize the desired therapeutic effect(s), while minimizing its adverse effect(s) if possible (2,3). Pharmaceutical compounds include, but are not limited to, chemicals, peptides, antibodies, and vaccines, as well as gene-based drugs. Drug delivery systems can be classified into different categories based on the route of administration. In addition to the traditional approaches like oral, injectable, transdermal, inhalation, implant, suppository, ophthalmic, and otic dosage forms, novel drug delivery systems like targeted delivery and drug-device combinations are now attracting more and more attention in drug development.
Since drug delivery approaches have been so widely explored to address pharmacology-related issues from some many different angles, it is hard to categorize them without a clear criterion. Except prodrug approach, drug delivery approaches generally do not involve chemical modification of the active ingredient. These approaches include solubilization, permeability enhancement, modified release (MR), and other special drug delivery mechanisms, such as targeted delivery, reduced local irritation, and drug-device combination. Prodrug delivery, which involves chemical modification of the pharmacologically active moiety, is discussed in a separate section.
Drug delivery approaches do not change the fundamental pharmacodynamic properties of a pharmaceutical compound, but they may change its pharmacokinetic properties to impact its pharmacodynamic performance. For both drug development and subsequent regulatory evaluation, a desired drug target product profile (DTPP) has to be well defined (4). Clinical pharmacology-related issues that may influence the desired therapeutic outcomes include, but are not limited to, the following:
Dose, dosing frequency, and dose adjustments
Pharmacokinetic profiles
Local drug irritation
Patient compliance
Inter-patient variation
Among different pharmacokinetic properties, area under the curve of concentration-time profile (AUC), maximum serum concentration (Cmax), time to reach Cmax (Tmax), and elimination half-life (T1/2) are the four parameters that have been most commonly evaluated and optimized in drug development and subsequent regulatory evaluation. Aimed at maximizing therapeutic efficacy and minimizing side effects, drug delivery approaches may be used to achieve the preferred AUC, Cmax, Tmax, and T1/2 by adjusting absorption, distribution, metabolism, and elimination (ADME) of a pharmaceutical compound (5,6).
In addition to poor aqueous solubility and low permeability, it is important to mention that presystemic clearance is another common cause for poor oral bioavailability. Orally administered drug may be prematurely cleared off the system due to hydrolysis in the stomach, enzymatic digestion in the gastric and small intestinal fluids, metabolism in the brush border of the gut wall, metabolism by microorganisms in the colon, and metabolism in the liver prior to entering the systemic circulation (first pass effect), etc., gastrointestinal (GI) tract site-specific delivery approaches, such as enteric coating, colonic delivery, and gastric-retention device, or altered routes of administrations, such as transdermal, intravenous (IV), buccal, inhalation, colon, suppository, can be used to address this issue of presystemic clearance to achieve the desired bioavailability.
For certain drugs, localized drug exposure rather than systemic exposure is preferred in order to deliver a high dose to the site of action to minimize or even avoid systemic adverse effect(s). Local drug exposure may be realized by applying the drug product directly to the site of action through conventional local drug delivery systems, such as topical, transdermal, ophthalmic, pulmonary, and intrathecal drug delivery systems. It is worth noting that some drug delivery systems applied locally may also provide systemic exposure which may or may not be desirable. Similar to but different from local delivery, targeted delivery is another way to achieve the goal of maximizing the local drug exposure and minimizing systemic side effect(s). As can be seen in the literature and drug product approval history, targeted therapy has been attracting increasing attention in oncology treatments, as it provides a way to overcome the notorious systemic side effects of traditional chemotherapy. In addition to designing chemicals with high selectivity and immunotherapy, drug delivery systems such as nanomedicines and antibody-drug conjugates (ADC) have been successfully applied for a few marketed drugs (3).
Patient compliance is frequently part of the success story of any good drug product in the marketplace. Minimal side effects and ease of use are two major ways to promote patient compliance. As mentioned above, drug delivery approaches that help to minimize side effects, both systemically and locally, certainly will improve patient compliance. Other drug delivery approaches to further ease the use of a drug product are modified release approaches for decreased dosing frequencies and patient-friendly delivery devices to facilitate self-administration (5,6). Considering such approaches is essential as drug delivery systems are only effective if (the) patients take them.
SOLUBILITY ENHANCEMENT
Considering the screen process for lead compounds in drug discovery stage, more and more drug candidates moving into development stage are water insoluble, especially for oncology compounds. Based on drug aqueous solubility and permeability, drugs have been classified into four biopharmaceutical classes (7–9). For a drug product intended for injection, especially for IV administration, achieving sufficient solubility at a physiological compatible environment is required. For a drug product that is intended for oral administration, the aqueous solubility typically refers to drug solubility under the human GI tract pH which ranges from 1.0 to 7.4. To achieve sufficient oral bioavailability for a water-insoluble pharmaceutical compound, improving dissolution of the compound in the human GI tract is generally the focus during/of pharmaceutical development (10). Commonly used drug delivery approaches for solubilizing drugs with poor aqueous solubility are listed as follows:
pH adjustment and salt formation
Co-solvent
Micelles (surfactants)
Microemulsions and emulsions
Particle size reduction
Nanosuspensions
Co-crystal
Solid dispersion (amorphous)
Liposomes
Complexation
Each drug delivery approach has its advantages and disadvantages, but selecting the most suitable drug delivery systems is still achieved through a combination of science and art. Consistent quality and therapeutic performance during scale-up and storage in selecting a suitable solubilization approach have to be taken into consideration for drug development and subsequent regulatory evaluation.
pH Adjustment and Salt Formation
It is well known that the aqueous solubility of acidic, basic, and amphoteric drugs can be impacted by dissolution media pH values (11,12). As shown in Fig. 1 (13), when the pH value of the medium is close to or higher than the pKa of an acidic drug, the drug’s solubility will increase significantly as the pH value increases; when the pH value of the medium is close to or higher than the pKb of a basic drug, the drug’s solubility will decrease significantly as the pH value increases. For amphoteric drugs that can react as an acid as well as a base, such as sulfadimethoxine and cefadroxil, the aqueous solubility will be higher at either acidic or basic media.
Fig. 1.
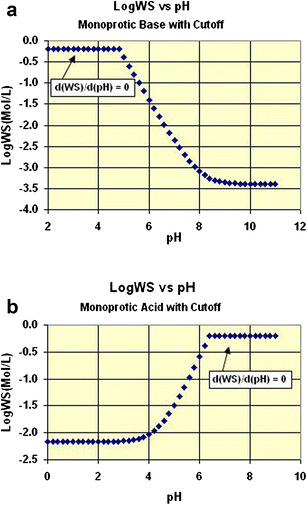
The relationship between drug aqueous solubility and pH values: a monoprotic acid, b monoprotic base
In addition to liquid formulation in which the pH value can be directly adjusted to achieve desired solubility, it is also a common practice to enhance drug dissolution rate by adjusting microenvironment pH in solid dosage forms especially in MR formulations. For example, in an osmotic pump-based MR formulation, the pH value can be fixed by choosing suitable excipients, because no other ions except water can enter the osmotic pump after oral administration. Therefore, adjusting the pH inside the osmotic pump-based formulation can be very useful for maintaining drug solubility and/or stability. However, one of the main disadvantages of using pH adjustment is that the solubilized drug may precipitate out with environmental pH changes in the human GI tract, or upon injection. For example, sodium salts of warfarin and phenytoin may convert back to insoluble acids during dissolution in acidic stomach environment (14,15). Therefore, the pH stability profile of a drug should be evaluated before using the pH adjustment approach.
For ionizable compounds with poor water solubility, forming salts is common practice in the pharmaceutical industry and Morris et al. (16) have proposed an integrated approach to the selection of the optimal salt form for a new drug candidate. The Handbook of Pharmaceutical Salts (17) has summarized all commonly used counterions in forming salts with free acids or free base, as well as their related properties. For free acids, the counterions used in salt formation include, but are not limited to, sodium, potassium, calcium, and zinc. For free bases, the counterions used in salt formation include, but are not limited to, phosphate, hydrochloride, malonate, mesylate, succinate, and acetate. Even though salt formation can improve the dissolution rate of drugs with poor water solubility, it is important to note that the salt does not necessary change their pH solubility profiles. The impact of counterions on pH solubility profiles can be complex, and the negative impact of the counterion in dissolution media cannot be ignored (18).
Co-Solvents
Use of co-solvent is one of the simplest and most common approaches in solubilizing drugs with poor water solubility. The most commonly used co-solvents in oral and parenteral formulation designs are propylene glycol (PPG), polyethylene glycol (PEG), ethanol, and glycerin (19). Yalkowsky et al. have proposed one of the simplest and most useful equations to predict drug solubility in co-solvent systems:
where Sm is the solubility of the drug in the binary mixture, f is the volume fraction of the co-solvent, Sc is the solubility of the drug in the pure co-solvent, and Sw is the solubility of the drug in water.
Trivedi (20) has summarized commercially available oral and injectable drug products using co-solvents in his comprehensive review of drug solubilization using the co-solvent approach. Commercially available solubilized oral products include, but are not limited to, phenobarbital solubilized by ethanol andetoposide by PEG-400 and glycerin, as well as rapamycin and ester derivatives by ethanol. One major disadvantage is precipitation on dilution. When a formulation is diluted with blood or other aqueous media, the concentrations of both the drug and the co-solvent will be reduced to the same extent. If the drug concentration in the diluted media is higher than the drug solubility in the new medium, the drug may become supersaturated in the diluted medium and even precipitate out. Such precipitation may result in therapeutic failure, and if the precipitation occurs in IV infusion, may result in tissue damage, or even organ failure. Other limitations include toxicity and the allowable maximum amount that can be used safely. For example, PEG can have toxic effects on the kidney (21). Note that the requirements by different regulatory agencies may be different.
Micelles
Drug solubilization using surfactants above the critical micelle concentration (CMC) has been widely used for enhancing bioavailability of drugs with poor water solubility (22–24). Traditionally used pharmaceutical surfactants can be classified into four groups: nonionic, anionic, cationic, and zwitterionic surfactants. In addition to traditional surfactants, such as sodium lauryl sulfate, quaternary ammonium, and polysorbates, diblock polymers and triblock polymers have also been designed and used for polymeric micelle-based drug delivery. Micelles, an aggregate of surfactant molecules dispersed in a liquid colloid, form only when the surfactant concentration is higher than the CMC, and the medium temperature is also greater than the critical micelle temperature. As shown in Fig. 2, in a micelle, the hydrophilic head of a surfactant molecule is exposed to the aqueous solution, while its hydrophobic tail stays within the micelle. The hydrophobic portion of micelles can interact with the hydrophobic portion of poorly soluble drugs, and solubilize them.
Fig. 2.
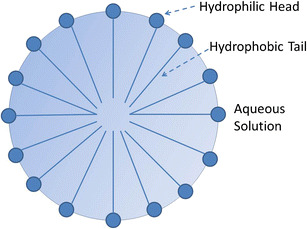
Scheme of a micelle formed by phospholipids in an aqueous solution
The water-insoluble drug will exist in both the aqueous phase and micellar phase and reach equilibrium between the two phases. There are many factors contributing to micellization and micellar solubilization, such as temperature, pH value, electrolytes and ionic strength, nature and concentration of surfactant and solute, as well as other ingredients. While enhancing solubilization capability, surfactants can also cause toxicity and even disrupt the membrane structure. As the toxicity of a surfactant is directly related to its concentration, the surfactant toxicity has to be taken into consideration during drug development. In self-micellizing solid dispersion (SMSD) systems, surfactants or amphiphilic block copolymers have been used to form micelles in aqueous media to solubilize drugs with poor water solubility and to prevent drug precipitation during dissolution process (25,26). Many drugs with poor water solubility have been successfully developed and commercialized using micellar solubilization, such as paclitaxel injection and cyclosporine injection, as well as griseofulvin-PEG-dispersion.
In addition to solubilization for water-insoluble drugs, micelles have also been explored to achieve passive targeting, stimuli sensitivity, ligand-mediated targeting, intracellular delivery of micelles, and intracellular trafficking (27). Like many advanced drug delivery systems, targeted and stimuli-sensitive micelles have not been widely used in actual drug development due to various reasons. Among them, the communication gap between clinical demands and drug delivery research has clearly limited the application of advanced drug delivery in resolving practical challenges faced by pharmaceutical industry.
Emulsions and Microemulsions
As lipid-based drug delivery systems, emulsions and microemulsions have been widely studied and applied for drug solubilization (28,29). An emulsion is a mixture of two immiscible liquids (typically, water and oil) stabilized by an emulsifier. Microemulsions (oil/water, water/oil) are clear, stable, isotropic liquid mixtures of oil/lipids, water, emulsifiers (surfactants), and/or co-solvents (e.g., ethanol, PPG, PEG). The surfactant molecules may form a monolayer at the interface between the oil and water, with the hydrophobic tails of the surfactant molecules dissolved in the oil phase and the hydrophilic head groups in the aqueous phase, and stabilize the emulsion by reducing the interfacial tension between the oil and aqueous phase. Among the multiple advantages of emulsions and microemulsions, two of them should be noted in particular: (1) the potential to avoid precipitation upon dilution, and (2) the combined approaches with other formulation strategies. In addition to traditional emulsions and microemulsions, Gao et al. (30–32) have developed supersaturated self-emulsifying drug delivery system (S-SEDDS) and have significantly improved oral absorption as compared to the conventional SEDDS formulation. S-SEDDS represents a new stable formulation approach which contains reduced amount of a surfactant and effective polymeric crystallization inhibitor (like hydroxypropyl methycellulose [HPMC] and polyvinylpyrrolidone [PVP]) to generate and maintain an in vivo supersaturated drug solution for drugs with poor water solubility. They have successfully applied S-SEDDS for multiple water-insoluble drugs and have achieved significant improvement of in vivo bioavailability in both animals and humans.
There are not many commercial drug products using emulsions or microemulsions due to physical and chemical stability challenges. However, in the pharmaceutical industry, emulsions and microemulsions are commonly used during the early development stage for bioavailability enhancement, especially for animal toxicity studies, since emulsion and microemulsions can help deliver the maximum amount of drug to achieve the highest bioavailability.
Particle Size Reduction and Nanosuspensions
Particle size reduction is commonly used to achieve faster dissolution and higher bioavailability. Based on Fick’s first law of diffusion, the drug release rate is determined by the drug release surface area, the thickness of transport barrier, and the concentration difference between drug donor and receptor, that is, the difference between the drug dosage surface and the dissolution bulk media. In the pharmaceutical industry, to enhance drug dissolution rates, water-insoluble drugs are typically micronized to ∼10 μm or less for oral dosage forms. It should be noted that reducing particle size will not only increase surface area but also affect drug substance solubility through changing surface free energy. For particles larger than a few microns, the solubility (also referred to as macroscopic solubility) will not change as the interfacial energy change is negligible. When particle size is smaller than 100 nm (i.e., nanoparticles), the high particle surface free energy will contribute to a higher solubility (also called microscopic solubility). Overall, particle size reduction to less than 1 μm will enhance drug dissolution rates by larger surface areas and/or higher solubility.
The methods for particle size reduction include, but are not limited to, fluid energy milling (dry milling), ball milling, media milling (stirred ball milling), and microfluidization (high pressure). The challenges of nanosuspension development stem from not only reproducible scale-up and manufacturing process but also potential stability issues like surface area increase and particle aggregation (33). Table I below lists the nanotechnologies and some of the successfully marketed drugs using those technologies. Although nanotechnology has been used successfully to improve dissolution and oral bioavailability, considering the cost involved in overcoming the challenges of processing and stability compared to other methods for drug solubilization (e.g., co-solvents), nanotechnology is not the most widely used for drug solubilization.
Table I.
A Brief List of Nanotechnology and Marketed Drugs
| Name | Technology | Marketed drugs |
|---|---|---|
| NanoCrystal technology of Alkermes (developed by Elan) | Wet media milling | Aprepitant, fenofibrate, sirolimus, megestrol acetate, paliperidone palmitate |
| IDD® (insoluble drug delivery) of Skyepharma | Microparticles and phospholipids | Fenofibrate |
| Biorise® of Actavis (developed by Eurand) | Dry milling and stabilizing agent | Not available |
| SoluMatrix™ of iCeutica | Dry milling | Not available |
In addition to solubilization, nanotechnology has attracted more and more attention in targeted drug delivery and one of the most successful drugs is Abraxane™ (protein-bound nanoparticle formulation of paclitaxel) (33). Even though the complexity of nanoparticles have posed challenges in setting regulatory standards, FDA has published the draft BE guidance for Abraxane™ (34), which has recommended both in vivo bioequivalence study with pharmacokinetic (PK) endpoints and in vitro particle size distribution.
Co-Crystals and Amorphous Solid Dispersion
Modifying the solid state by co-crystal or amorphous solid dispersion can also enhance drug dissolution. In April 2013, FDA published the Guidance for Industry, “Regulatory Classification of Pharmaceutical Co-Crystals” (35), in which co-crystals are clearly defined as “solids that are crystalline materials composed of two or more molecules in the same crystal lattice.” Among multiple benefits, co-crystals can achieve higher oral bioavailability through improved dissolution rate and allow formulations to be manufactured by traditional granulation processes which are more environmentally friendly than solid dispersion. However, the risk of converting to free drug especially at slurry cannot be underestimated. Since the last decade, there have been many drug development studies based on co-crystal approach (36–39). For example, Novartis’ LCZ696, an investigational combination drug consisting of two antihypertensives (blood pressure-lowering drugs), valsartan and sacubitril (40), granted priority review designation by the FDA (41), has been approved by FDA to reduce risk of cardiovascular death and heart failure hospitalization (42).
In amorphous solid dispersion(s), drug molecules change from crystalline form to amorphous form, and the change causes their free energy level to increase significantly (43,44). The higher free energy level as well as increased surface area contributes to the enhanced dissolution rate of amorphous solid dispersion drug products. Typical polymers used in amorphous solid dispersion preparation are PEG, HPMC, PVP, hypromellose acetate succinate (HPMCAS), and sometimes two or more polymers can be used together in the amorphous solid dispersion. The marketed solid dispersion drugs include, but are not limited to, griseofulvin, nabilone, itraconazole, tacrolimus, and lopinavir/ritonavir. Roche’s vemurafenib, a practically insoluble drug, has been successfully developed using an amorphous polymer-stabilized solid dispersion prepared by a solvent-controlled co-precipitation process (45).
During storage of amorphous solid dispersions, drug may not only recrystallize from the amorphous state but also face more/further chemical stability challenges due to the higher free energy. In addition to the physical and chemical stability challenges during storage, drug may precipitate out from solution during dissolution because dissolved drug typically exists as supersaturated solution. In spite of the great potential for enhancing bioavailability using solid dispersion, the/its commercial application is limited and only a few products have been marketed to date. To ensure consistent quality and clinical performance, it is critical to deepen the understanding of amorphous solid dispersions, especially the impact of quality change on therapeutic performance.
Liposomes
Liposomal delivery, an exciting drug delivery system especially for drugs with poor water solubility, such as anticancer and anti-HIV compounds, has been widely studied. Liposomes are bilayer phospholipid systems that carry drugs by entrapping them in tiny vesicles and can provide controlled release (CR) delivery as well as targeted delivery. Typical routes of administration for liposome products are IV, intramuscular (IM), and subcutaneous (SC). However, compared to other drug delivery systems, fewer drugs have been successfully developed and marketed using liposomal drug delivery systems (2).
Some of the challenges in developing liposomal delivery systems are the difficulty of retaining drugs inside the liposomes, the rapid clearance of liposomes by the body’s mononuclear phagocyte system, the delivery to intracellular sites, and scale-up issues (46). Despite these challenges, there are a number of successfully marketed drug products using liposomes. For example, incorporating cholesterol or sphingomyelin in the phospholipid bilayer helped to retain the drug inside the liposomes (47,48). Vincristine, a widely used anticancer drug, has high neurotoxicity and has rapid clearance using the traditional delivery system. The sphingomyelin/cholesterol liposomes of Marqibo® (vincristine sulfate liposome injection) provided slower clearance to enhance exposure to the tumor cells and received accelerated approval by the FDA in 2012 (49). In the case of amphotericin b, a highly renal toxic drug, incorporation of cholesterol in the liposomal delivery system greatly improved tolerability by stabilizing the drug in the liposomes (50). Attaching PEG to the liposomes (PEGylation) increases the circulation half-life of the liposomes (51). Antibody-targeted liposomes bode well for targeted therapy, although the delivery system involves tedious manipulations (52,53). Liposomal delivery systems can be designed to be heat-sensitive, pH-sensitive, photosensitive, enzyme-triggered, or even magnetically responsive to deliver the drug to a target organ or tumor (46). Table II provides a list of some of the marketed products with a brief description of the liposomal technologies that are used. There is a lot of opportunity for developing successful drug products with positive clinical outcomes, especially for anticancer drugs.
Table II.
Examples of FDA-Approved Drugs that Use Liposome Technologies
| Marketed drugs | Description |
|---|---|
| Doxil® (doxorubicin HCl liposome injection) | Uses STEALTH® which are PEG-liposomes;drug is entrapped within the bilayer |
| AmBisome® (amphotericin b injection, powder, lyophilized, for injection) | Drug is intercalated within the membrane of phospholipids/cholesterol liposomes; liposomes are less than 100 nm in diameter. |
| DaunoXome® (daunorubicin citrate liposome injection) | Drug is encapsulated in the aqueous core of the liposomes; liposomes are 35–65 nm in diameter. |
| Marqibo® (vincristine sulfate liposome injection) | Drug is encapsulated in sphingomyelin/cholesterol liposomes; mean liposome diameter is 100 nm. |
For both nanoparticles and liposomes, their particle size distribution may impact in vivo drug performance through not only drug release but also drug deposition on targeted tissue which has not been fully understood. To further understand the impact of particle size distribution on in vivo drug performance, multiple questions have to be addressed, such as how to measure particle size especially those small particles, measurement reproducibility, batch reproducibility, the relationship between particle size distribution and drug release, as well as the relationship between particle size distribution and drug deposition.
Complexations
Cyclodextrins (CDs) are commonly used excipients for solubility enhancing through forming complexation with drugs with poor aqueous solubility (54). α-, β-, and γ-CDs, which respectively consist of six, seven, and eight D-glucose units, have been used in pharmaceutical applications. The cavity of CDs is relatively apolar, where drugs with poor water solubility can form complexation with CDs. The internal cavity diameters are approximately 5.7, 7.8, and 9.5 Å for α-, β-, and γ–CDs, respectively. In addition to being a solubilization drug delivery system, complexation has also been used to increase drug stability, reduce irritation to the human GI tract (55), and mask unpleasant taste [US 20030215503 A1].
There are several advantages in using CD complexation for solubility enhancement (54). Firstly, the dissociation between complexing agent and drug is very rapid. Secondly, 2-hydroxypropyl-β-cyclodextrin (HP-β-CD) and sulfobutyl ether-β-cyclodextrin (SBE-β-CD) are less toxic when compared to other solubilizing agents such as surfactants and co-solvents. Thirdly, because most of the complexation are 1:1 complexes, dilution of the complex will not result in a supersaturated solution for water-insoluble drugs; thus, precipitation upon dilution is not a concern as commonly observed in other solubilization approaches such as amorphous solid dispersion, co-solvent, and pH adjustment. Lastly, complexation can be combined with other approaches such as solid dispersion and pH adjustment to improve the bioavailability of water-insoluble drugs (56,57).
As expected, there are also disadvantages using complexation (54). To form complexes with complexing agents, drugs with poor water solubility sometimes have to first be dissolved in the medium. For compounds with very limited solubility, the solubility enhancement will be very limited by taking the binding constants of complexes into consideration. The second limitation is that for the complexes of Ap type which have formed higher order complexes (58), dilution of a system may still result in precipitation. Other limitations are the potential toxicity, regulatory issues arising from the toxicity, and quality control issues related to the presence of the ligand, which may add complication and cost to the development process. Lastly, compared with other approaches, the complexation efficiency is often rather low, thus either relatively large amount of CDs are typically required to achieve desirable solubilization effect, or a combination with other drug delivery systems is preferred.
As reported by CycloLab, a CD-manufacturing company, in the February 2013 issue of the cyclodextrin news, there was a total of 49 marketed CD pharmaceutical products across the world, mostly in India, Europe, and Japan, with a few in the USA (59). Among the 49 products, 45 used β-CD and its derivatives. The drugs approved in the USA and around the world utilizing SBE-β-CD (Captisol®) include, but are not limited to, voriconazole and ziprasidone mesylate by Pfizer, aripiprazole by Bristol-Myers Squibb, and amiodarone hydrochloride by Baxter. The drugs approved in the USA and around the world utilizing HP-β-CD include, but are not limited to, itraconazole by Janssen, indomethacin by Chauvin, and mitomycin by Novartis.
PERMEATION ENHANCEMENT
For drugs with low permeability, such as metformin and aliskiren, higher doses have been used in their approved drugs to compensate for their relatively low bioavailability. For example, the strengths of metformin can go as high as 1 g in drug products. Improving bioavailability for drugs with low permeability, i.e., Biopharmaceutical Classification System (BCS) class III and IV drugs, is more complex than drugs with poor water solubility. Common approaches for enhancing bioavailability for drugs with low permeability are listed below:
Drugs with a “window for absorption” refer to those drugs which are absorbed only from the human upper GI tract. In addition to adjusting the drug release rate at different parts of the human GI tract, retaining drugs above the upper GI tract may also be a choice for achieving desired bioavailability. The two main approaches for retaining those drugs are bioadhesive microspheres that have slow intestinal transit and gastroretentive dosage systems (51,70,71). For the drug delivery systems used to enhance bioavailability of drugs with poor permeability, their disadvantages cannot be underestimated. For example, some intestinal membrane permeation enhancers like saponin cannot be considered as a generally recognized as safe (GRAS) excipient due to their toxicities (62). Similarly, not all PGP inhibitors like piperine can be considered as a GRAS excipient (63).
The absorption of those drugs with poor water solubility but high permeability (i.e., BCS class II) is mainly controlled by their dissolution rate, rather than their local concentration in the human GI tract or retention time in different parts of the human GI tract. However, for drugs with high water solubility but poor permeability (i.e., BCS class III), in addition to their dissolution rate, their absorption will also be impacted by their local concentration in the human GI tract as well as their retention time in different parts of the human GI tract. All the factors impacting absorption have contributed to the high pharmacokinetic variability for BCS class III drugs; thus, BCS class III drugs typically require a greater number of subjects in bioequivalence studies than typical BCS class II drugs. For the BCS class III drugs, due to those factors contributing to absorption, formulation changes may induce different “subject-by-formulation” interaction. In May 2015, FDA has published a draft guidance about “Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System,” in which biowaiver has been extended to BCS class III drug products (72).
MODIFIED RELEASE FORMULATIONS
Modified release (MR) delivery systems include CR, delayed release (DR), sustained release (SR), extended release (ER), and pulsatile delivery systems. MR formulations include both oral MR formulations and sterile long-acting release (LAR) formulations, and the number of marketed oral MR drugs far out-weighs the number of LAR drugs. Typically, a drug can produce both desired and adverse effects. The ratio of the minimum plasma drug concentration that produces toxic effects (minimum toxic concentration (MTC)) and the minimum plasma concentration that produces the desired effect (minimum effective concentration (MEC)) is used to determine the therapeutic index. The term “therapeutic window” refers to the drug plasma concentration range between the MEC and MTC. A narrower therapeutic window for a drug leads to a greater possibility of the drug causing unwanted toxicity in patients, such as narrow therapeutic index (NTI) drugs. MR formulations may help to achieve the desired plasma drug concentration and maintain it for a certain period, typically for a longer time than immediate release (IR) formulations.
Oral MR Formulations
The benefits of oral MR formulations have been well studied and utilized (73). By modifying drug pharmacokinetic profiles, oral MR formulations have been widely used to improve patient compliance, as well as reduce side effects, especially for those drugs with short half-lives or narrow therapeutic windows. Drugs with short half-lives typically require multiple doses, and the MR formulation can improve patient compliance by reducing the dosing frequency. By reducing Cmax of a drug with a narrow therapeutic window, the adverse effects of the drug can be minimized while maintaining its therapeutic effects. MR formulations can reduce peak-to-trough variations, which also help improve the patient’s drug tolerance. However, the higher doses in MR formulations make them more attractive as the targets for abuse. On November 20, 2014, Purdue Pharma L.P. announced that the FDA had approved Hysingla™ ER (hydrocodone bitartrate) tablets CII, a once-daily opioid analgesic formulated with abuse-deterrent properties (74). In April 2015, FDA published the Guidance for Industry: Abuse-Deterrent Opioids—Evaluation and Labeling (75).
The drug release mechanisms of oral MR formulations include dissolution, diffusion, osmosis, and ion-exchange CR. Common oral MR formulations include, but are not limited to, soluble matrix, insoluble matrix, coated beads (reservoir, dissolution-controlled), coated tablets (diffusion-controlled), DR tablets or beads, osmotic pump tablets, combinations of different MR formulations designs, as well as combinations of MR formulation design with IR formulation design. Typically, as MR formulations contain larger amount of drugs than IR formulations, they may result in serious adverse effects if dose dumping occurs. In addition to achieving the desired pharmacokinetic profiles, MR formulation designs have to take this potential clinical risk into consideration. For example, although coated tablets (with reservoir system) have been used in some approved drugs, any damage on the coated film can induce dose dumping which makes this type of formulation design more risky from safety perspective.
Out of concern regarding dose dumping from MR drug products when taken with alcohol, additional dissolution testing using various concentrations of ethanol, typically up to 40% (v/v) of ethanol in the dissolution medium are recommended in their specific bioequivalence guidances. The FDA has recommended an alcohol dose dumping test for MR formulations in multiple guidances for industry:
Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs—General Considerations (76)
Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA (77)
For MR drugs, their critical material attributes (CMAs) and critical process parameters (CPPs) on drug product quality have to be evaluated and optimized thoroughly to ensure consistent critical quality attributes (CQAs). For example, there are many more marketed drugs based on IR formulations than MR formulations. However, from January 2005 to June 2014, MR formulation drugs occupied approximately 52% of the dissolution-related Field Alert Reports (FARs) with 45% from ER formulations and 7% from DR formulations, which is also higher than the percentage of reports for IR formulation drugs (44%) (78). The results of the investigation suggest that the complex formulation designs and related manufacturing processes contribute to higher likelihood of quality failures.
Complex formulation designs are often under patent protection and/or are expensive to manufacture. Therefore, generic products of such innovator products sometimes use different delivery mechanisms to achieve similar AUC and Cmax to those of the innovator products. However, for drugs with very complex formulation designs, whether AUC and Cmax are enough to establish bioequivalence needs to be carefully evaluated. For example, the BE guidance for Ambien CR (zolpidem oral ER tablet) recommended AUC0–1.5, AUC1.5-t, AUC0-∞, and Cmax in fasting PK study (79).
Sterile LAR Formulations
As listed in Table III, more than ten drugs including small molecules, peptides, and proteins using sterile LAR formulations have been successfully developed and marketed with clear clinical benefits. Poly(lactic-co-glycolic acid) (PLGA) is the biodegradable polymer most commonly used in the LAR formulations. By adjusting the ratio between lactic acid and glycolic acid in PLGA, drug dissolution rate from PLGA can be adjusted. In general, a higher content of glycolide units in a PLGA polymer leads to faster dissolution of the PLGA polymer. Many other factors like end-cap group, molecular weight, and polydisperse index (PI) may affect drug release and clinical performance, as well as their quality and clinical performance consistency. During drug development of a LAR peptide (unpublished work), a significant change in pharmacokinetic profile was observed from an approximately two times scale-up while keeping the other factors consistent. For LAR formulations based on PLGA, more research is needed to fully understand the impact of manufacturing processes and excipient properties on drug release and related pharmacokinetic profiles.
Table III.
Examples of Marketed Long-Acting Release (LAR) Drugs
| Drug product | Release period | API | Comment |
|---|---|---|---|
| Zoladex | 1 and 3 months | Goserelin acetate (peptide) | Depot |
| Lupron | 1, 3, 4 and 6 months | Leuprolide acetate (peptide) | Depot |
| Sandostatin LAR | 1 month | Octreotide acetate (peptide) | Depot |
| Nutropin | 1 month | Somatropin (rDNA origin protein) | Depot; withdrawn |
| Trelstar/Decapeptyl | 1, 2 and 6 months | Triptorelin pamoate (peptide) | Injectable suspension |
| Suprefact | 2 and 3 months | Buserelin acetate (peptide) | Depot |
| Somatuline Depot | 1 month | Lanreotide (peptide) | Depot |
| Arestin | 2 weeks | Minocycline HCl | Microsphere |
| Eligard | 1, 3, 4, and 6 months | Leuprolide acetate (peptide) | Injectable suspension |
| Risperdal Consta | 2 weeks | Risperidone | Long-acting injection |
| Vivitrol | 1 month | Naltrexone | ER-injectable injection |
| Ozurdex | 3 months | Dexamethasone | Intravitreal implant |
| Bydureon | 1 week | Exenatide (peptide) | ER-injectable suspension |
These LAR products tend to replace daily oral dosing with injections with long intervals, further improving patient compliance. In spite of the benefits of LAR formulations, the risks of LAR formulations especially during scale-up and manufacturing process cannot be ignored. For example, Zyprexa Relprevv (olanzapine pamoate), a long-acting antipsychotic, is administered by a health care professional every 2 to 4 weeks by deep IM gluteal injection (80). In 2013, the FDA initiated an investigation on Zyprexa Relprevv due to two deaths following injection (81). Both patients died 3–4 days after receiving an appropriate dose of the drug, well after the 3-h post-injection monitoring period required under the Zyprexa Relprevv Risk Evaluation and Mitigation Strategy (REMS). Both patients were found to have very high olanzapine blood levels after death. In December 2014, amended “Warnings and Precautions” were added to the drug’s prescribing information (80). MedWatch provided the following safety information regarding Zyprexa Relprevv (82): “FDA has concluded a review of a study undertaken to determine the cause of elevated levels of the injectable schizophrenia drug Zyprexa Relprevv (olanzapine pamoate) in two patients who died. The study results were inconclusive. FDA is unable to exclude the possibility that the deaths were caused by rapid, but delayed, entry of the drug into the bloodstream following intramuscular injection.” Therefore, when designing LAR formulations, one must consider the therapeutic benefit as well as the risk of overdose.
PRODRUGS
A prodrug refers to a chemically modified pharmaceutical compound that must undergo enzymatic and/or chemical transformation in vivo to release the active parent molecule in order for the desired pharmacology action to be exhibited (65,83–85). A pharmaceutical compound with undesirable pharmacokinetic properties can be modified with additional chemical group(s) to form the prodrug which has enhanced ADME profiles to facilitate the delivery of desired therapeutic effect. As can be seen in the abundant publications and marketed drug products, prodrug approaches can be and have been successfully applied to enhance solubility or dissolution rate, enhance permeability, target delivery, reduce side effect, protect from presystemic metabolism, and delay clearance of a pharmaceutical compound (86–89). Potential modifications are as follows:
Hydrophilic groups such as phosphate and sulfate esters
Amino acids like lysine, γ-glutamyl derivatives
Hydrophobic groups
Polymers and macromolecules
Stella and Nti-Addae have published a comprehensive review of prodrug strategies to overcome poor water solubility, using successfully marketed prodrugs like fosamprenavir, fosphenytoin, and bortezomib for illustrative purposes (88). It was well studied that the water solubility of a parent molecule may be improved by adding ionizable group(s) or non-ionizable polar group(s). Phosphate, sulfate, amino acids, sugar moieties, etc. have been attached to low solubility compounds to make successful drug products (88,89). Furthermore, prodrugs can enhance oral availability by decreasing crystal packing, through the solubilization effects of bile salts and lecithin in the human GI tract. One of the significant challenges for prodrugs is chemical stability especially for parenteral formulations; thus, many parenteral prodrugs are lyophilized for reconstitution.
For orally administered drug products, the active ingredient has to have sufficient permeability to be absorbed to achieve the desired therapeutic efficacy, including overcoming the “blood–brain barrier” (BBB) for central nervous system therapies (65,83,90–92). Many drugs of a highly polar nature or charged at physiological pH typically have very low permeabilities, such as carboxylic acids, strongly basic compounds such as amidines and guanidines, nucleosides, and nucleotides. Hans Maag systematically reviewed prodrug approaches of enhancing permeability of different types of parent drugs (90). For example, esterification is recommended to be tried first for carboxylic acids. As the most common route of drug absorption is passive transportation, suitable lipophilicity of a drug is very important for oral drug absorption. An oral novel potent reversible nonpeptide inhibitor of thrombin, dabigatran, a very polar and permanently charged molecule, has no bioavailability after oral administration (93,94). However, by masking the amidinium moiety as a carbamate ester and by turning the carboxylate into an ester group, the prodrug, dabigatran etexilate, has achieved an absolute bioavailability of about 7%.
Prodrugs can help improve tumor targetability and efficiency by incorporating tumor-targeting ligands, transporter-associated ligands and polymers (83,84,87,95). In addition to overcoming physiochemical or biopharmaceutical problems, prodrugs can help achieve targeted therapy by taking advantage of tumor-specific antigens on tumor cell surfaces, transporter-associated ligands, low extracellular pH inside the tumor core, etc. For example, reductase enzyme present in the hypoxic cell to convert prodrugs into its cytotoxic form can lead to a selective antitumor effect. For prodrugs with site-selective properties, their toxicity level can also be reduced by lowering dosing levels. Although the first FDA-approved antibody-drug conjugate (Mylotarg) was withdrawn from the market in 2010 due to safety concerns and lacking efficacy (96), numerous targeted prodrugs are under clinical trials (www.clinicaltrials.gov).
With all the benefits of prodrugs, multiple risks and limitations such as chemical stability and specificity do exists. Fundamentally, a prodrug is a new chemical entity, although it can achieve many desired properties during drug delivery processes compared to the pharmacologically active moiety. As a new chemical entity, the prodrug will produce much higher development cost and require more time than a typical 505(b)(2) drug (97). The FDA Guidance for Industry (76) “Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs—General Considerations” published in 2014 clarified: “The active ingredient that is released from the dosage form or its active moiety and, when appropriate, its active metabolites should be measured in biological fluids collected in BA studies…Measurement of the active ingredient or the active moiety, rather than metabolites, is generally recommended for BE studies because the concentration-time profile of the active ingredient or the active moiety is more sensitive to changes in formulation performance than that of the metabolite, which is more reflective of metabolite formation, distribution, and elimination.” The guidance further clarified the two instances when an active metabolite(s) should be measured. Finally, when a scientist plans to develop a prodrug, two more questions still need to be taken into consideration even if they cannot be answered immediately:
Does a prodrug molecule itself have biological activities in human?
Will a prodrug be degraded into the core active in human based on design?
Overall, the in vivo performance of prodrugs may not readily turn out as predicted and will require comprehensive studies as a new chemical compounds.
PEGylation for Delaying Drug Clearance Processes
In addition to prodrug and peptide terminal modification, the most common approach to delay drug clearance process is PEGylation for peptide and protein drugs (98–100). Conjugation of PEG to therapeutics has proven to be an effective approach to delay the drug clearance process and increase drug serum half-life. However, the increased use of PEGylated therapeutics has resulted in unexpected immune-mediated side effects (101–103). There are claims that these adverse effects are due to anti-PEG antibodies inducing rapid clearance. These claims are hampered due to the lack of standardized and well-validated antibody assays. However, it is still recommended that the immunogenicity of PEGylated substances should be tested in the development stage, and that the titer of anti-PEG antibodies in patients should be pre-screened and monitored prior to and throughout a course of treatment with a PEGylated substance.
In 2013, for Omontys (peginesatide), a PEGylated peptide drug used to treat anemia caused by chronic kidney disease, there were reports of multiple serious hypersensitivity reactions including anaphylaxis, a life-threatening allergic reaction (104). An investigation in conjunction with Affymax’s marketing partner Takeda found no quality or manufacturing issues with Omontys (105). However, it also failed to identify any specific root cause for the allergic reactions, so Affymax and Takeda were left with no option but to withdraw the product. No clear evidence has been identified to link the serious hypersensitivity reactions with PEG.
OTHER SPECIAL DRUG DELIVERY APPROACHES
In addition to the drug delivery systems discussed above, many novel drug delivery systems have been developed to address different clinical pharmacology related issues. Some of them are listed as follows:
Targeted drug delivery
Local drug delivery
Drug delivery systems for delaying drug clearance processes
Drug delivery systems for reducing local irritation
Drug device combination
In contrast to targeted drug delivery systems, local drug delivery systems are typically administered to the sites of action directly. Most local drug delivery systems are for local therapeutic effects with only a few exceptions, such as transdermal nitroglycerin whose indication is the prevention of angina pectoris due to coronary artery disease. Local drug delivery systems include, but are not limited to, transdermal, ophthalmic, otic, intrathecal, and pulmonary drug delivery systems. Locally GI acting drugs such as Asacol HD are also considered to fall under the local drug delivery category even though not administered to the sites of action directly. Because typical local drug delivery systems are designed for local therapy rather than systemic exposure, plasma drug concentrations, i.e., pharmacokinetic profiles, may not be available for establishing bioequivalence like those drugs with systemic exposure.
Targeted Drug Delivery
To achieve desired targeted therapy, in addition to designing pharmaceutical compounds with higher receptor selectivity, drug delivery systems have been explored extensively (3,106,107). Using targeted drug delivery to modify the in vivo drug distribution, oncology treatment has been significantly improved recently. Dwidczyk et al. (3) reviewed the design strategies of the two FDA-approved antibody-drug conjugates (Brentuximab vedotin and Trastuzumab emtansine) and the four FDA-approved nanoparticle-based drug delivery platforms (Doxil, DaunoXome, Marqibo, and Abraxane). In addition to targeting efficiency, the authors have discussed physiological imposed design constraints, such as circulation (distribution in the vascular system and peripheral tissues), the mononuclear phagocyte system, enhanced permeability and retention effect, drug loading, and drug release, as well as toxicity. A comprehensive list of design rules for targeted nanoparticle drug delivery systems have been proposed, which have taken circulation, distribution, elimination, tumor accumulation, tumor cell uptake, and drug release into consideration, together with their design requirements and possible strategies. Even though facing numerous challenges, targeted drug delivery is still in the early stages and has great potential in the near future.
Drug Delivery Systems for Reducing Local Irritation
In the human GI tract, many drugs such as aspirin and nonsteroidal anti-inflammatory drugs (NSAIDs) can induce local irritation. In addition to the human GI tract, local irritation can also occur for transdermal and ocular drug delivery systems. Depending on the cause of the local irritation, different approaches such as DR, complexation with CD, microsphere, and drug-device combination have been applied to reduce local irritation (108–110). However, there are still many reports of drug-related local irritation. Many factors, such as the knowledge gap and insufficient communication between formulation scientists and clinicians, have contributed to the failure of developing drugs with reduced local irritation. Below is a good example of using the drug delivery approach to address GI local irritation.
Undesired local irritation caused by some drugs, such as the GI irritation caused by (NSAIDs can be mitigated by pre-associating drugs with lipids) (111–113). In the human GI tract, NSAIDs disrupt the mucosal gel layer and let in acid which causes local irritation including erosions, ulcers, bleeding, and perforation. The NSAID-lipid pre-association effectively sequesters the NSAID in a lipid matrix, allowing the NSAID to reach the blood stream with less disruption of the GI lining. This technology allows the same effective dose of the NSAID to reach its site of action in the body, while significantly reducing the serious and common GI adverse effects caused by NSAIDs. NSAID products utilizing this technology are bioequivalent to their currently marketed counterpart. In addition, the lipids used in the formulations are a nonpharmaceutical natural product derived from soybeans.
Drug-Device Combinations
Since the last decade, drug-device combinations have attracted increasing attention from the pharmaceutical and healthcare industries, as drug-device combinations can provide clear clinical benefits. Dry powder inhaler (DPI) drugs (114–117), drug-eluting stents (118,119), and insulin pumps (120,121) are examples of widely used drug-device combinations, with many reports on their benefits and limitations. Although most drugs used in drug-device combinations are small molecules, significant progress has been made for delivering peptides and proteins, while maintaining their stability and activity. For example, Intarcia developed a proprietary SC delivery system, which is comprised of a small, matchstick-sized osmotic pump that is placed subdermally to deliver a slow and consistent flow of medication (122). Each device contains an appropriate volume of drug product to treat a patient for a predetermined, extended duration of time, and the device can be placed quickly during a reimbursable in-office procedure. Once placed under the skin, water from the extracellular fluid enters the device at one end, by diffusing through a semi-permeable membrane directly into a salt osmotic engine that expands to drive a piston at a controlled rate, which forces the drug to be released in a consistent fashion at the other end of the device. Delivering drugs this way avoids unwanted peaks and troughs of drug levels often associated with adverse events and potentially suboptimal therapeutic effects. Proteins and peptides can be maintained stable at human body temperature by suitable formulation in the device and achieve continuous delivery of therapy up to 12 months. Finally, the metallic implant can be quickly withdrawn if unscheduled removal of the device is required.
REGULATORY PERSPECTIVES
For drug products to be marketed, they must be approved by the regulatory body. Regulatory agencies will not only consider clinical benefits and risks but will also focus on ensuring consistent quality and therapeutic performance of drug products. How to achieve consistent performance poses serious challenges in drug development especially for complex drug delivery systems, and requires close collaboration between industry, academia, and regulatory agencies. The regulatory considerations on quality and therapeutic performance apply to both brand and generic drugs, although there are some obvious differences. In June 2013, FDA published the Guidance for Industry “ANDAs: Stability Testing of Drug Substances and Products” (123), which follows the stability recommendations provided in the International Conference on Harmonisation (ICH) stability guidelines. The guidance helps to ensure consistent quality requirements between generic drugs and brand drugs. Currently, there are ongoing efforts in harmonizing regulatory guidances among FDA, EMA, and other regulatory agencies, while inconsistencies still exist.
Although drugs are developed and approved based on the best knowledge and scientific understanding, occasional reports on individual products’ quality failures and adverse events can still appear in the post-marketing period. Systematic monitoring on drug quality and adverse events will not only help protect public health but are also valuable in developing new drugs with improved quality features. Below are a few useful resources from which drug quality and therapeutic performance information can be found:
Drug Quality Reporting System (DQRS) (124)
FARs, Biological Product Defect Reports (BPDRs)
- Medwatch, the FDA safety information and adverse event reporting system (125)
- In 2012, Adverse Event Reporting System (AERS) was upgraded to FDA Adverse Event Reporting System (FAERS)
Recalls, market withdrawals, and safety alerts (126)
Publications of core reports
Media reports
For drug delivery systems using complex formulation designs and manufacturing processes, the task of ensuring consistent quality becomes more challenging than traditional simple dosage forms. To ensure consistent quality of a drug product, in addition to suitable formulation design and manufacturing process selection, their CMAs and CPPs have to be identified, and their impacts on the CQAs have to be thoroughly evaluated (127). Consistent quality during commercial manufacturing, storage, and transportation has to be taken into consideration while using drug delivery systems to address clinical pharmacology related issues.
To support the Generic Drug User Fee Amendments (GDUFA) commitments, the FDA Office of Generic Drugs (OGD) developed the following fiscal year (FY) 2015 regulatory science priorities for generic drugs (128):
Post-market evaluation of generic drugs
Equivalence of complex products
Equivalence of locally-acting products
Therapeutic equivalence evaluation and standards
Computational and analytical tools
These regulatory science priorities clearly show that confirming pharmaceutical equivalence and therapeutic equivalence based on the current scientific knowledge is not always straightforward. Although therapeutic equivalence of most orally administered generic drug products is established based on in vivo bioequivalence studies, for many complex drugs, additional tests such as studies with clinical endpoints and even in vitro tests may be needed to confirm therapeutic equivalence.
FUTURE OF DRUG DELIVERY
With advancing diagnostic technologies, more and more drugs targeting patients with specifically identified genetic defects have been developed and approved. For example, the FDA recently approved a test made by 23andMe™, a personal genetics company, for a gene that can cause a rare disorder called Bloom syndrome, which causes short stature and a heightened risk of cancer (129). With the advent of personalized medicines, there will be great opportunities and challenges in utilizing drug delivery systems to provide better products and services for patients. Due to intrinsic patient factors and disease characteristics, drugs may exhibit different therapeutic performance in different patients, although all approved drugs have statistically met the desired balance of benefits and risks. A one-size-fits-all approach will not only fail to provide suitable treatments for some patients but also waste a lot of resources. By integrating progresses in different fields such as those in information technology and diagnostics, drug delivery systems which can take individual pharmacokinetic properties into consideration will help to significantly improve therapeutic performance for individual patients.
To achieve personalized medicines, compared with traditional drug delivery designs, dynamic drug delivery systems seem more attractive and realistic. The widely used insulin pumps are a successful example of personalized drug delivery systems. Last year, Novartis and Google started working together on a smart contact lens that monitors blood-sugar levels and corrects vision in a new way, the latest in a series of technology products designed to monitor body functions (130).
In recent years, information technology has been combined increasingly closely with pharmaceutical and health care fields, and the combination has started to establish a foundation for personalized medicines: collecting information, processing information, and providing feedback. Proteus Digital Health™ developed an ingestible sensor which is powered by human body, and this is the first such device approved by the FDA (131). The sensor can gather information about the time at which medication(s) are taken and relate information such as pH and temperature in the human GI tract. By capturing real-time physiological and behavioral data emitted by the sensor, caregivers and clinicians can track patient compliance during clinical studies and ensure that the clinical results and clinical pharmacology data are reliable. Although the sensor is simple in concept, it shows a direction of drug delivery systems to achieve personalized medicine.
SUMMARY AND OUTLOOK
There are many ways to ensure drug delivery to achieve desired clinical outcome. Some of the more traditional approaches include solubility enhancements for poorly soluble drugs, permeation enhancements for poorly permeable drugs, and simple MR formulations. Some of the more modern approaches include complex MR formulations, prodrug delivery, and targeted drug delivery. There are regulatory and product developmental challenges associated especially with complex dosage forms. Understanding the relationship among the drug delivery design, pharmacokinetics, and the clinical effect would require cross-discipline collaboration, as well as collaboration among the industry, academia, regulatory agencies, patient advocacy organizations, and other stakeholders. However, the future of drug delivery is more exciting than ever. Continuing advances in understanding of traditional and modern delivery systems and emerging technology in personalized medicine are expected to provide greater clinical improvement in all therapeutic areas.
Acknowledgments
The authors would like acknowledge scientific discussions and support of Larissa Lapteva, Wenlei Jiang, and Robert Lionberger.
Disclaimer
This article reflects the views of the authors and should not be construed to represent the FDA’s views or policies.
References
- 1.Drug delivery. Available from: http://en.wikipedia.org/wiki/Drug_delivery.
- 2.Anselmo AC, Mitragotri S. An overview of clinical and commercial impact of drug delivery systems. J Control Release. 2014;190:15–28. doi: 10.1016/j.jconrel.2014.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dawidczyk CM, et al. State-of-the-art in design rules for drug delivery platforms: Lessons learned from FDA-approved nanomedicines. J Control Release. 2014;187:133–44. doi: 10.1016/j.jconrel.2014.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guidance for industry and review staff: target product profile—a strategic development process tool. 2007; Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm080593.pdf.
- 5.Advanced delivery devices - wearable bolus injectors - a new class of patient-friendly drug delivery systems 2014; Available from: http://www.drug-dev.com/Main/Back-Issues/ADVANCED-DELIVERY-DEVICES-Wearable-Bolus-Injectors-688.aspx.
- 6.Next-generation self-administered drug-device combinations new technologies and business opportunities. Available from: http://www.technology-catalysts.com/reports_services/ddc.asp.
- 7.Amidon GL, et al. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 8.Yu LX, et al. Biopharmaceutics classification system: the scientific basis for biowaiver extensions. Pharm Res. 2002;19(7):921–5. doi: 10.1023/A:1016473601633. [DOI] [PubMed] [Google Scholar]
- 9.Yu LX, et al. Transport approaches to the biopharmaceutical design of oral drug delivery systems: prediction of intestinal absorption. Adv Drug Deliv Rev. 1996;19(3):359–76. doi: 10.1016/0169-409X(96)00009-9. [DOI] [PubMed] [Google Scholar]
- 10.Shi Y, et al. Recent advances in intravenous delivery of poorly water-soluble compounds. Expert Opin Drug Deliv. 2009;6(12):1261–82. doi: 10.1517/17425240903307423. [DOI] [PubMed] [Google Scholar]
- 11.Shoghi E, et al. Solubility–pH profiles of some acidic, basic and amphoteric drugs. Eur J Pharm Sci. 2013;48:291–300. doi: 10.1016/j.ejps.2012.10.028. [DOI] [PubMed] [Google Scholar]
- 12.Voelgyi G, et al. Study of pH-dependent solubility of organic bases. Revisit of Henderson-Hasselbalch relationship. Anal Chim Acta. 2010;673(1):40–6. doi: 10.1016/j.aca.2010.05.022. [DOI] [PubMed] [Google Scholar]
- 13.The relationship between drug aqueous solubility and pH values. Available from: http://www.chemsilico.com/CS_prWS/WSintro.html (Chemsilico LLC closed).
- 14.Walker JE, et al. In vivo and in vitro availability of commercial warfarin tablets. J Pharm Sci. 1971;60(5):666–77. doi: 10.1002/jps.2600600503. [DOI] [PubMed] [Google Scholar]
- 15.Serajuddin ATM, Jarowski CI. Influence of pH on release of phenytoin sodium from slow-release dosage forms. J Pharm Sci. 1993;82(3):306–10. doi: 10.1002/jps.2600820318. [DOI] [PubMed] [Google Scholar]
- 16.Morris KR, et al. An integrated approach to the selection of optimal salt form for a new drug candidate. Int J Pharm. 1994;105(3):209–17. doi: 10.1016/0378-5173(94)90104-X. [DOI] [Google Scholar]
- 17.Pharmaceutical salts: properties, selection, and use. 2011
- 18.Li S, et al. Effect of chloride ion on dissolution of different salt forms of haloperidol, a model basic drug. J Pharm Sci. 2005;94(10):2224–31. doi: 10.1002/jps.20440. [DOI] [PubMed] [Google Scholar]
- 19.Yalkowsky SH, Roseman TJ. Solubilization of drugs by cosolvents. Drugs Pharm Sci. 1981;12(Tech. Solubilization Drugs):91–134. [Google Scholar]
- 20.Trivedi JS. Solubilization using cosolvent approach. In: Water-insoluble drug formulation (2nd Ed.), 2008. p. 161–194.
- 21.Ethylene glycol: human health aspects by WHO. 2002.
- 22.Florence AT. Drug solubilization in surfactant systems. Drugs Pharm Sci. 1981;12(Tech. Solubilization Drugs):15–89. [Google Scholar]
- 23.Liu R, Dannenfelser R-M, Li S. Micellization and drug solubility enhancement. In: Water-insoluble drug formulation (2nd Ed.), 2008. p. 255–306.
- 24.Liu R, Forrest ML, Kwon GS. Micellization and drug solubility enhancement part II: polymeric micelles. In: Water-insoluble drug formulation (2nd Ed.), 2008. p. 307–374.
- 25.Frank KJ, et al. What is the mechanism behind increased permeation rate of a poorly soluble drug from aqueous dispersions of an amorphous solid dispersion? J Pharm Sci. 2014;103(6):1779–86. doi: 10.1002/jps.23979. [DOI] [PubMed] [Google Scholar]
- 26.Onoue S, et al. Self-micellizing solid dispersion of cyclosporine A with improved dissolution and oral bioavailability. Eur J Pharm Sci. 2014;62:16–22. doi: 10.1016/j.ejps.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Torchilin VP. Micellar nanocarriers: pharmaceutical perspectives. Pharm Res. 2007;24(1):1–16. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 28.Cannon JB, Long MA. Emulsions, microemulsions, and lipid-based drug delivery systems for drug solubilization and delivery-part II: oral applications. In: Water-insoluble drug formulation (2nd Ed.), 2008: p. 227–254.
- 29.Cannon JB, Shi Y, Gupta P. Emulsions, microemulsions, and lipid-based drug delivery systems for drug solubilization and delivery-part I: parenteral applications. In: Water-insoluble drug formulation (2nd Ed.), 2008. p. 195–226.
- 30.Gao P. Fundamental concept, design and development of supersaturatable S-SEDDS: a case study. Bull Tech Gattefosse. 2011;104:51–60. [Google Scholar]
- 31.Gao P, et al. Characterization and optimization of AMG 517 supersaturatable self-emulsifying drug delivery system (S-SEDDS) for improved oral absorption. J Pharm Sci. 2009;98(2):516–28. doi: 10.1002/jps.21451. [DOI] [PubMed] [Google Scholar]
- 32.Shi Y, et al. Application of a biphasic test for characterization of in vitro drug release of immediate release formulations of celecoxib and its relevance to in vivo absorption. Mol Pharma. 2010;7(5):1458–65. doi: 10.1021/mp100114a. [DOI] [PubMed] [Google Scholar]
- 33.Desai N. Challenges in development of nanoparticle-based therapeutics. AAPS J. 2012;14(2):282–95. doi: 10.1208/s12248-012-9339-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Draft guidance on paclitaxel. 2012; Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM320015.pdf.
- 35.Guidance for industry: regulatory classification of pharmaceutical co-crystals. 2013; Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM281764.pdf.
- 36.Blagden N, et al. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv Drug Deliv Rev. 2007;59(7):617–30. doi: 10.1016/j.addr.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 37.Chaudhari PK, Dudhat M, Thosar M. Co-crystallization: a novel approach for enhancing drug properties. Inventig Impact Pharm Tech. 2012;2:58–61. [Google Scholar]
- 38.Hoffman M, Lindeman JA. Co-crystals: commercial opportunities and patent considerations. RSC Drug Discov Ser. 2012;16(Pharmaceutical Salts and Co-crystals):318–29. [Google Scholar]
- 39.Sekhon BS. Drug-drug co-crystals. Daru J Pharm Sci. 2012;20:45. doi: 10.1186/2008-2231-20-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gradman AH. LCZ696: the next step in improving RAS inhibition? Curr Hypertens Rep. 2015;17(5):1–9. doi: 10.1007/s11906-015-0548-y. [DOI] [PubMed] [Google Scholar]
- 41.Novartis’ heart failure medicine LCZ696 granted FDA priority review. 2015; Available from: http://www.novartis.com/newsroom/media-releases/en/2015/1894530.shtml.
- 42.Novartis’ new heart failure medicine LCZ696, now called Entresto(TM), approved by FDA to reduce risk of cardiovascular death and heart failure hospitalization. 2015; Available from: https://www.novartis.com/news/media-releases/novartis-new-heart-failure-medicine-lcz696-now-called-entrestotm-approved-fda.
- 43.Serajuddin ATM. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88(10):1058–66. doi: 10.1021/js980403l. [DOI] [PubMed] [Google Scholar]
- 44.Vasanthavada M, Tong WQ, Serajuddin ATM. Development of solid dispersion for poorly water-soluble drugs. In: Water-insoluble drug formulation (2nd Ed.), 2008. p. 499–529.
- 45.Shah N, et al. Improved human bioavailability of vemurafenib, a practically insoluble drug, using an amorphous polymer-stabilized solid dispersion prepared by a solvent-controlled coprecipitation process. J Pharm Sci. 2013;102(3):967–81. doi: 10.1002/jps.23425. [DOI] [PubMed] [Google Scholar]
- 46.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 47.Kirby C, Clarke J, Gregoriadis G. Effect of the cholesterol content of small unilamellar liposomes on their stability in vivo and in vitro. Biochem J. 1980;186(2):591–8. doi: 10.1042/bj1860591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Allen TM, Hansen C, Rutledge J. Liposomes with prolonged circulation times: factors affecting uptake by reticuloendothelial and other tissues. Biochim Biophys Acta Biomembr. 1989;981(1):27–35. doi: 10.1016/0005-2736(89)90078-3. [DOI] [PubMed] [Google Scholar]
- 49.Silverman JA, Deitcher SR. Marqibo (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother Pharmacol. 2013;71(3):555–64. doi: 10.1007/s00280-012-2042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adler-Moore JP, Proffitt RT. Development, characterization, efficacy and mode of action of Am Bisome, a unilamellar liposomal formulation of amphotericin B. J Liposome Res. 1993;3(3):429–50. doi: 10.3109/08982109309150729. [DOI] [Google Scholar]
- 51.Kharia AA, et al. Formulation strategy for low absorption window antihypertensive agent. Res J Pharm Technol. 2010;3(1):113–7. [Google Scholar]
- 52.Rotman M, et al. Enhanced glutathione PEGylated liposomal brain delivery of an anti-amyloid single domain antibody fragment in a mouse model for Alzheimer’s disease. J Control Release. 2015;203:40–50. doi: 10.1016/j.jconrel.2015.02.012. [DOI] [PubMed] [Google Scholar]
- 53.Park JW, et al. Development of anti-p185HER2 immunoliposomes for cancer therapy. Proc Natl Acad Sci U S A. 1995;92(5):1327–31. doi: 10.1073/pnas.92.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tong WQ, Wen H. Applications of complexation in the formulation of insoluble compounds. In: Water-insoluble drug formulation (2nd Ed.), 2008. p. 133–159.
- 55.Szekely-Szentmiklosi B, Tokes B. Characterization and molecular modelling of cyclodextrin/fluoroquinolone inclusion complexes. Acta Med Marisiensis. 2011;57(2):116–20. [Google Scholar]
- 56.Zoeller T, Dressman JB, Klein S. Application of a ternary HP-β-CD-complex approach to improve the dissolution performance of a poorly soluble weak acid under biorelevant conditions. Int J Pharm (Amsterdam, Neth) 2012;430(1–2):176–83. doi: 10.1016/j.ijpharm.2012.04.029. [DOI] [PubMed] [Google Scholar]
- 57.Sistla A, Kertelj A, Shenoy N. Development of an intravenous formulation of SU010382 (prodrug of SU5416, an anti-angiogenesis agent) PDA J Pharm Sci Technol. 2008;62(3):200–10. [PubMed] [Google Scholar]
- 58.Badr-Eldin SM, Elkheshen SA, Ghorab MM. Inclusion complexes of tadalafil with natural and chemically modified β-cyclodextrins. I: Preparation and in-vitro evaluation. Eur J Pharm Biopharm. 2008;70(3):819–27. doi: 10.1016/j.ejpb.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 59.Approved pharmaceutical products containing cyclodextrins. 2013; Available from: http://cyclolab.hu/images/CDNews/cdn_2013_feb.pdf.
- 60.Sun J. Intestinal absorption of low permeability drugs: A transporter- and enzyme-targeted approach. 2010. p. 95.
- 61.Aungst BJ. Absorption enhancers: applications and advances. AAPS J. 2012;14(1):10–8. doi: 10.1208/s12248-011-9307-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kumar A, et al. Saponin as absorption enhancer. World J Pharm Res. 2014;3(6):2099–110. [Google Scholar]
- 63.Singh DV, Godbole MM, Misra K. A plausible explanation for enhanced bioavailability of P-gp substrates in presence of piperine: simulation for next generation of P-gp inhibitors. J Mol Model. 2013;19(1):227–38. doi: 10.1007/s00894-012-1535-8. [DOI] [PubMed] [Google Scholar]
- 64.Woo JS, et al. Enhanced oral bioavailability of paclitaxel by coadministration of the P-glycoprotein inhibitor KR30031. Pharm Res. 2003;20(1):24–30. doi: 10.1023/A:1022286422439. [DOI] [PubMed] [Google Scholar]
- 65.Beaumont K, et al. Design of ester prodrugs to enhance oral absorption of poorly permeable compounds: challenges to the discovery scientist. Curr Drug Metab. 2003;4(6):461–85. doi: 10.2174/1389200033489253. [DOI] [PubMed] [Google Scholar]
- 66.Sapra B, et al. A critical appraisal of microemulsions for drug delivery: part I. Ther Deliv. 2013;4(12):1547–64. doi: 10.4155/tde.13.116. [DOI] [PubMed] [Google Scholar]
- 67.Sapra B, et al. A critical appraisal of microemulsions for drug delivery: part II. Ther Deliv. 2014;5(1):83–94. doi: 10.4155/tde.13.125. [DOI] [PubMed] [Google Scholar]
- 68.Wang K, et al. Enhancement of oral bioavailability of cyclosporine A: comparison of various nanoscale drug-delivery systems. Int J Nanomed. 2014;9:4991–9. doi: 10.2147/IJN.S72560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miller JM. The impact of molecular complexation on intestinal membrane permeation. 2009. p. 114.
- 70.Heinig R, et al. Pharmacokinetics of the controlled-release nisoldipine coat-core tablet formulation. Int J Clin Pharmacol Ther. 1997;35(8):341–51. [PubMed] [Google Scholar]
- 71.Davis SS. Formulation strategies for absorption windows. Drug Discov Today. 2005;10(4):249–57. doi: 10.1016/S1359-6446(04)03351-3. [DOI] [PubMed] [Google Scholar]
- 72.Draft guidance for industry: waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. 2015; Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070246.pdf.
- 73.Tungaraza Tongeji E, Talapan-Manikoth P, Jenkins R. Curse of the ghost pills: the role of oral controlled-release formulations in the passage of empty intact shells in faeces. Two case reports and a literature review relevant to psychiatry. Ther Adv Drug Saf. 2013;4(2):63–71. doi: 10.1177/2042098612474681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Purdue Pharma L.P. receives FDA approval for HysinglaTM ER (hydrocodone bitartrate) extended-release tablets CII, a once-daily opioid analgesic formulated with abuse-deterrent properties. 2014.
- 75.Guidance for industry: abuse-deterrent opioids — evaluation and labeling. 2015; Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm334743.pdf.
- 76.Guidance for industry: bioavailability and bioequivalence studies submitted in NDAs or INDs — general considerations. 2014; Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm389370.pdf.
- 77.Guidance for industry: bioequivalence studies with pharmacokinetic endpoints for drugs submitted under an ANDA. 2013; Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm377465.pdf.
- 78.Wen H, et al. Survey of failed dissolution related field alerts of oral drug products. In: American Association of Pharmaceutical Scientists. 2014. San Diego, California.
- 79.Guidance on Zolpidem. 2011; Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM175029.pdf.
- 80.Label: ZYPREXA RELPREVV (olanzapine) for extended release injectable suspension. 2014; Available from: http://pi.lilly.com/us/zyprexa_relprevv.pdf.
- 81.Zyprexa Relprevv (Olanzapine Pamoate): drug safety communication - FDA investigating two deaths following injection. 2013; Available from: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm357601.htm.
- 82.Zyprexa Relprevv (olanzapine pamoate): drug safety communication - FDA review of study sheds light on two deaths associated with the injectable schizophrenia drug. 2015; Available from: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm439472.htm.
- 83.Dahan A, Zimmermann EM, Ben-Shabat S. Modern prodrug design for targeted oral drug delivery. Molecules. 2014;19(10):16489–505. doi: 10.3390/molecules191016489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mahato R, Tai W, Cheng K. Prodrugs for improving tumor targetability and efficiency. Adv Drug Deliv Rev. 2011;63(8):659–70. doi: 10.1016/j.addr.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zawilska JB, Wojcieszak J, Olejniczak AB. Prodrugs: a challenge for the drug development. Pharmacol Rep. 2013;65(1):1–14. doi: 10.1016/S1734-1140(13)70959-9. [DOI] [PubMed] [Google Scholar]
- 86.Rautio J, and Editor, Prodrugs and targeted delivery: towards better ADME properties. [In: Methods Princ. Med. Chem., 2011; 47]. 2011. 496 pp.
- 87.Huttunen KM, Raunio H, Rautio J. Prodrugs - from serendipity to rational design. Pharmacol Rev. 2011;63(3):750–71. doi: 10.1124/pr.110.003459. [DOI] [PubMed] [Google Scholar]
- 88.Stella VJ, Nti-Addae KW. Prodrug strategies to overcome poor water solubility. Adv Drug Deliv Rev. 2007;59(7):677–94. doi: 10.1016/j.addr.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 89.Velazquez S, et al. Dipeptidyl-peptidase IV (DPP IV/CD26)-activated prodrugs: a successful strategy for improving water solubility and oral bioavailability. Curr Med Chem. 2015;22(8):1041–54. doi: 10.2174/0929867322666150114163449. [DOI] [PubMed] [Google Scholar]
- 90.Maag H. Overcoming poor permeability - the role of prodrugs for oral drug delivery. Drug Discov Today Technol. 2012;9(2):e121–30. doi: 10.1016/j.ddtec.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 91.Chin CM, et al. Peptide prodrugs for the treatment of CNS disorders: a perspective for new drugs. Curr Med Chem. 2014;21(23):2599–609. doi: 10.2174/0929867321666140217125526. [DOI] [PubMed] [Google Scholar]
- 92.Stockwell J, et al. Novel central nervous system drug delivery systems. Chem Biol Drug Des. 2014;83(5):507–20. doi: 10.1111/cbdd.12268. [DOI] [PubMed] [Google Scholar]
- 93.Eisert WG, et al. Dabigatran: an oral novel potent reversible nonpeptide inhibitor of thrombin. Arterioscler Thromb Vasc Biol. 2010;30(10):1885–9. doi: 10.1161/ATVBAHA.110.203604. [DOI] [PubMed] [Google Scholar]
- 94.Blech S, et al. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug Metab Dispos. 2008;36(2):386–99. doi: 10.1124/dmd.107.019083. [DOI] [PubMed] [Google Scholar]
- 95.Singh Y, Palombo M, Sinko PJ. Recent trends in targeted anticancer prodrug and conjugate design. Curr Med Chem. 2008;15(18):1802–26. doi: 10.2174/092986708785132997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.FDA: Pfizer voluntarily withdraws cancer treatment Mylotarg from U.S. market. 2010.
- 97.Guidance for industry: applications covered by section 505(b)(2). 1999; Available from: http://www.fda.gov/downloads/Drugs/Guidances/ucm079345.pdf.
- 98.Culhane JC, Dawson PE. Peptide pharmaceuticals: modifications to the peptide backbone that enhance stability and targeting. Chim Oggi. 2014;32(4):17–8. [Google Scholar]
- 99.Milla P, Dosio F, Cattel L. PEGylation of proteins and liposomes: a powerful and flexible strategy to improve the drug delivery. Curr Drug Metab. 2012;13(1):105–19. doi: 10.2174/138920012798356934. [DOI] [PubMed] [Google Scholar]
- 100.Pasut G, Morpurgo M, Veronese FM. Basic strategies for PEGylation of peptide and protein drugs. Deliv Prot Pept Drugs Cancer. 2006; 53–84.
- 101.Ishida T, Kiwada H. Anti-polyethyleneglycol antibody response to PEGylated substances. Biol Pharm Bull. 2013;36(6):889–91. doi: 10.1248/bpb.b13-00107. [DOI] [PubMed] [Google Scholar]
- 102.Schellekens H, Hennink WE, Brinks V. The Immunogenicity of polyethylene glycol: facts and fiction. Pharm Res. 2013;30(7):1729–34. doi: 10.1007/s11095-013-1067-7. [DOI] [PubMed] [Google Scholar]
- 103.Verhoef JJF, et al. Potential induction of anti-PEG antibodies and complement activation toward PEGylated therapeutics. Drug Discov Today. 2014;19(12):1945–52. doi: 10.1016/j.drudis.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 104.Gever J. Fatal reactions prompt omontys recall. 2013; Available from: http://www.medpagetoday.com/HematologyOncology/Anemia/37509.
- 105.Broadwith P. Affymax folds after drug withdrawal. 2014; Available from: http://www.rsc.org/chemistryworld/2014/06/affymax-folds-after-drug-withdrawal.
- 106.Dawidczyk Charlene M, Russell Luisa M, Searson Peter C. Nanomedicines for cancer therapy: state-of-the-art and limitations to pre-clinical studies that hinder future developments. Front Chem. 2014;2:69. doi: 10.3389/fchem.2014.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Onoue S, Yamada S, Chan H-K. Nanodrugs: pharmacokinetics and safety. Int J Nanomed. 2014;9:1025–37. doi: 10.2147/IJN.S38378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tiwari G, Tiwari R, Rai AK. Cyclodextrins in delivery systems: applications. J Pharm BioAllied Sci. 2010;2(2):72–9. doi: 10.4103/0975-7406.67003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Andersson SB, Jonn S, Landh T. Nicotine compositions and methods of formulation thereof. 1999, (Pharmacia & Upjohn AB, Swed.). WO. p. 33.
- 110.Stefansson E, Loftsson T. Cyclodextrins in eye drop formulations. J Incl Phenom Macrocycl Chem. 2003;44(1–4):23–7. [Google Scholar]
- 111.Ahuja M, et al. Topical ocular delivery of NSAIDs. AAPS J. 2008;10(2):229–41. doi: 10.1208/s12248-008-9024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Abdel-Tawab M, Zettl H, Schubert-Zsilavecz M. Nonsteroidal anti-inflammatory drugs: a critical review on current concepts applied to reduce gastrointestinal toxicity. Curr Med Chem. 2009;16(16):2042–63. doi: 10.2174/092986709788682209. [DOI] [PubMed] [Google Scholar]
- 113.; Available from: http://www.plxpharma.com/.
- 114.Chan JGY, et al. Advances in Device and Formulation Technologies for Pulmonary Drug Delivery. AAPS PharmSciTech. 2014;15(4):882–97. doi: 10.1208/s12249-014-0114-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cheng YS. Mechanisms of pharmaceutical aerosol deposition in the respiratory tract. AAPS PharmSciTech. 2014;15(3):630–40. doi: 10.1208/s12249-014-0092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cordts E, Steckel H. Formulation considerations for dry powder inhalers. Ther Deliv. 2014;5(6):675–89. doi: 10.4155/tde.14.35. [DOI] [PubMed] [Google Scholar]
- 117.Cui Y, et al. Towards the optimization and adaptation of dry powder inhalers. Int J Pharm (Amsterdam, Neth) 2014;470(1–2):120–32. doi: 10.1016/j.ijpharm.2014.04.065. [DOI] [PubMed] [Google Scholar]
- 118.Escarcega RO, et al. Current application and bioavailability of drug-eluting stents. Expert Opin Drug Deliv. 2014;11(5):689–709. doi: 10.1517/17425247.2014.888054. [DOI] [PubMed] [Google Scholar]
- 119.Wiebe J, Nef HM, Hamm CW. Current status of bioresorbable scaffolds in the treatment of coronary artery disease. J Am Coll Cardiol. 2014;64(23):2541–51. doi: 10.1016/j.jacc.2014.09.041. [DOI] [PubMed] [Google Scholar]
- 120.Atkinson Mark A, Eisenbarth George S, Michels Aaron W. Type 1 diabetes. Lancet. 2014;383(9911):69–82. doi: 10.1016/S0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tauschmann M, Hovorka R. Insulin pump therapy in youth with type 1 diabetes: toward closed-loop systems. Expert Opin Drug Deliv. 2014;11(6):943–55. doi: 10.1517/17425247.2014.910192. [DOI] [PubMed] [Google Scholar]
- 122.Intarcia therapeutics home page. Available from: http://www.intarcia.com.
- 123.Guidance for industry: ANDAs: stability testing of drug substances and products. 2013; Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM320590.pdf.
- 124.Drug Quality Reporting System (DQRS). Available from: http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ucm082071.htm.
- 125.MedWatch: the FDA safety information and adverse event reporting program. Available from: http://www.fda.gov/Safety/MedWatch/default.htm.
- 126.Recalls, market withdrawals, & safety alerts. Available from: http://www.fda.gov/Safety/Recalls/.
- 127.Yu LX, et al. Understanding pharmaceutical quality by design. AAPS J. 2014;16(4):771–83. doi: 10.1208/s12248-014-9598-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.GDUFA regulatory science priorities for fiscal year 2015. Available from: http://www.fda.gov/downloads/ForIndustry/UserFees/GenericDrugUserFees/UCM417234.pdf.
- 129.Herper M. What 23andMe’s FDA approval means for the future of genomics. 2015.
- 130.Morse A. Novartis and Google to work on smart contact lenses. 2014.
- 131.Proteus home page. Available from: http://www.proteus.com/.