

Abstract
Treatment with erlotinib, an epidermal growth factor receptor tyrosine kinase inhibitor used for treating non-small-cell lung cancer (NSCLC) and other cancers, is frequently associated with adverse events (AE). We present a modeling and simulation framework for the most common erlotinib-induced AE, rash, and diarrhea, providing insights into erlotinib toxicity. We used the framework to investigate the safety of high-dose erlotinib pulses proposed to limit acquired resistance while treating NSCLC. Continuous-time Markov models were developed using rash and diarrhea AE data from 39 NSCLC patients treated with erlotinib (150 mg/day). Exposure and different covariates were investigated as predictors of variability. Rash was also tested as a survival predictor. Models developed were used in a simulation analysis to compare the toxicities of different regimens, including the previously mentioned pulsed strategy. Probabilities of experiencing rash or diarrhea were found to be highest early during treatment. Rash, but not diarrhea, was positively correlated with erlotinib exposure. In contrast with some common understandings, radiotherapy decreased transitioning to higher rash grades by 81% (p < 0.01), and experiencing rash was not correlated with positive survival outcomes. Model simulations predicted that the proposed pulsed regimen (1600 mg/week + 50 mg/day remaining week days) results in a maximum of 20% of the patients suffering from severe rash throughout the treatment course in comparison to 12% when treated with standard dosing (150 mg/day). In conclusion, the framework demonstrated that radiotherapy attenuates erlotinib-induced rash, providing an opportunity to use radiotherapy and erlotinib together, and demonstrated the tolerability of high-dose pulses intended to address acquired resistance to erlotinib.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-015-9815-8) contains supplementary material, which is available to authorized users.
KEY WORDS: adverse events, erlotinib, modeling, NSCLC
INTRODUCTION
Lung cancer, of which non-small-cell lung cancer (NSCLC) represents the majority of the cases, has been described as the most commonly diagnosed and fatal type of cancer (1). Better understanding of the tumor biology on a genetic basis, identifying genome alterations that can be targeted for improved outcomes, and the provision of diagnostic tools that can detect such alterations in the clinic have paved the way for genetically tailored targeted cancer therapy. Lung cancer is commonly considered as a prototype for this approach (2,3). Tyrosine kinase inhibitors (TKIs) such as erlotinib used for treating epidermal growth factor receptor (EGFR)-mutant lung adenocarcinomas (4), or crizotinib used for patients tested positive for the anaplastic lymphoma kinase (ALK) gene rearrangement (5) are examples of lung cancer therapeutics targeting signaling pathways relevant to these alterations.
However, patients treated with the previously mentioned drugs (and other related molecules) inevitably develop acquired resistance and relapse (6–8). Efforts to address this problem include the currently ongoing development of molecules that can selectively target both the sensitizing and resistant T790M mutant EGFR (known to be the most common resistance mechanism for EGFR inhibitors) (9,10). In another attempt to address acquired resistance, a mathematical modeling framework was developed proposing the optimization of the dosing regimens of the currently available EGFR-TKIs. Patients were suggested to be treated with high-dose pulses, which would slow down the emergence of resistance, together with a continuous low-dose inhibiting the drug-sensitive cells (11,12).
Based on data from advanced NSCLC patients treated with erlotinib (13), our primary objectives in this study were to develop a modeling and simulation framework describing the probabilities of transitioning of the patients between the different grades of the most commonly TKI-associated adverse events, namely rash and diarrhea, both impairing the quality of life which could eventually disrupt antineoplastic treatment. We also aimed to evaluate and quantify the impact of different patient covariates on the probabilities of adverse events incidence. Models developed were then used for simulations to investigate the toxicity of the proposed pulsed dosing regimens compared to the standard dosing regimen of erlotinib, 150 mg/day.
MATERIALS AND METHODS
Patients and Data
Data was available from a phase II trial that evaluated positron emission tomography (PET) for early predicting the survival of advanced NSCLC patients first treated with erlotinib; 150 mg was orally administered daily until disease progression or intolerable toxicity (NCT00568841) (13). The study was approved by the local ethics committee, the Federal Institute for Drugs and Medical Devices, the responsible federal state authorities of North Rhine-Westphalia, and the German Authority for Radiation Safety. All patients provided written informed consents. Forty stage- IV NSCLC patients were enrolled, yet one was excluded from this analysis since he died before taking the drug.
One thousand one hundred twenty-four adverse event incidents were documented. Dates of incidence and resolution of adverse events were recorded, and they were graded according to the National Cancer Institute-Common Toxicity Criteria (NCI-CTC) version 3.0 (grade 1 = mild, grade 2 = moderate, grade 3 = severe) (14). None of the patients experienced life-threatening (grade 4) toxicities, and none of the deaths were treatment-related (grade 5). According to the study protocol, and at the discretion of the treating clinician, dosage was reduced for grade 3 rash and re-escalated when grade was ≤2, while treatment was to be stopped for grade 3 diarrhea and restarted at a reduced dose after resolution to grade≤1. All rash and diarrhea incidents considered to be certainly, likely or possibly related to erlotinib, and that took place between starting erlotinib and the final scheduled follow-up visit within the study (12 months after starting therapy) were included for analysis.
Overall survival and covariates data including the mutational status, Eastern Cooperative Oncology Group (ECOG) performance status, histological sub-classification, PET uptakes, radiotherapy and co-medications histories, laboratory findings, and demographics were available (summarized in Table I).
Table I.
Patient Characteristics
| Characteristic | Number (%) |
|---|---|
| Age (years) | |
| Median (range) | 62 (38–78) |
| Gender | |
| Males | 18 (46.2%) |
| Females | 21 (53.8%) |
| Baseline ECOG performance status | |
| 0 | 17 (43.6%) |
| 1 | 17 (43.6%) |
| 2 | 5 (12.8%) |
| Histological sub-classification | |
| Adenocarcinoma | 29 (74.4%) |
| Bronchoalveolar carcinoma | 4 (10.3%) |
| Squamous cell carcinoma | 5 (12.8%) |
| Large cell carcinoma | 1 (2.5%) |
| Mutational status | |
| No mutation | 20 (51.3%) |
| EGFR mutation | 7 (17.9%) |
| KRAS mutation | 3 (7.7%) |
| No information available | 9 (23.1%) |
| Smoking status | |
| Non-smoker (0.0 pack-years) | 14 (35.9%) |
| Previous light smoker (0.1–10 pack-years) | 4 (10.2%) |
| Previous heavy smoker (>10 pack-years) | 5 (12.8%) |
| Current smoker | 13 (33.4%) |
| No data available | 3 (7.7%) |
| Radiation therapy | |
| No radiation therapy | 19 (48.8%) |
| Before starting erlotinib [mean total dose ± standard deviation] | 8 (20.5%) [40.4 Gy ± 8.8 ] |
| During erlotinib treatment [mean total dose ± standard deviation] | 7 (17.9%) [58.6 Gy ± 32.9] |
| After erlotinib treatment [mean total dose ± standard deviation] | 5 (12.8%) [64.4 Gy ± 31.7] |
| Survival during the first scheduled 12 months | |
| Died | 24 (61.5%) |
| Overall survival/daysa, medianb (range) | 165 (10–429) |
ECOG Eastern Cooperative Oncology Group
aOverall survival was defined as the length of time the patients lived after starting erlotinib therapy
bBased on Kaplan-Meier estimate
Since no erlotinib concentrations were measured in the study, a published one-compartment pharmacokinetic model (absorption rate constant = 0.95 h−1, oral volume of distribution = 233 L, and oral clearance = 3.95 L/h) for erlotinib was used to generate the patients’ exposure levels based on their dosing and covariates (15).
Data Analysis and Software
Non-linear mixed-effects modeling was used for analysis using the Laplacian method to approximate the marginal likelihood in NONMEM 7.3 (16). The likelihood-ratio test was used for discriminating nested models, while the Akaike information criteria were used for non-nested models. Using a parsimonious approach, i.e., selecting a model giving a sufficient goodness-of-fit with the least number of parameters, an extra parameter was included in the model if proved statistically significant with p ≤ 0.01. Visual predictive checks (described later) also assisted model selection. Covariate model building was carried out using the forward inclusion (α = 0.05) and backward elimination (α = 0.01) method, and covariates were included if proved statistically significant and clinically relevant.
R (versions 3.0.2 and higher) (17) were used for data management and graphical outputting. Scripts within PsN (version 4.2.0) assisted model development, and Pirana (version 2.9.0) was used as a front interface (18).
Adverse Events Models
Each day was considered as an observation and patients were classified not to have an adverse event, or experience one of the adverse event grades (1, 2, or 3). If a patient suffered from rash at two or more sites simultaneously, but with different grades, the grade was set to the highest.
A continuous-time Markov model (19,20) was developed to describe the ordinal data. A compartmental structure was used (Fig. 1) with four compartments, each representing a severity level. Probabilities of experiencing one of the adverse event severities were modeled as compartment amounts and were defined by four differential equations (Eq. 1). For each day, the probability of the severity (compartment amount in this setting) corresponding to the observed grade was set to 1 while other states were set to 0; this introduces the Markov property, meaning that a future state (on the following day) depends on the current state. The likelihood was directly maximized to observe each set of states. With the incidence (or resolution) of adverse events, and the subsequent setting of different “amounts” to 1 and others to 0, rate constants for movement of these amounts, reflecting probability transitions between different states, were estimated.
| 1 |
Fig. 1.
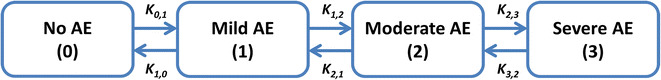
The compartmental structure for the continuous-time Markov model describing the temporal courses of probabilities of adverse event (AE) intensities (0 = no AE, 1 = mild AE, 2 = moderate AE, 3 = severe AE). Forward “worsening” rate constants are represented by K0,1, K1,2, and K2,3, while the backward “recovery” rate constants are represented by K1,0, K2,1, and K3,2
dPr(grade)/dt represent the rate of change of the probability of experiencing grades 0, 1, 2, or 3 with respect to time, Pr(grade) are the probabilities of experiencing each grade, Kgrade,grade+1 represent rate constants for worsening from grades 0, 1, or 2 to the higher grades 1, 2, or 3, respectively, while Kgrade,grade-1 are rate constants for recovery from grades 3, 2, or 1 to the lower grades 2, 1, or 0, respectively.
Constant, exponential, and Weibull models (supplemental 1) were tested to evaluate how the rate constants changed with time since treatment initiation. Furthermore, we tested whether erlotinib plasma concentrations can be linked to the incidence and deterioration of adverse events by exploring various relationships (linear, Emax, and Hill equations; supplemental 1). Finally, a covariate analysis was run to investigate the effect of different factors on adverse events. Continuous covariates were tested using linear, power, or exponential relationships, while fractional changes for each category of categorical covariates were evaluated (supplemental 1).
Survival Model
Since NSCLC is fatal (24 patients died during the analyzed period; median survival time = 165 days, range = 10–429 days), accounting for death in the framework is essential to properly evaluate it using simulation diagnostics. A parametric time-to-death model was included for this purpose (21). The time-to-death of the patients were assumed to follow a continuous distribution, where the distribution can be parameterized through an underlying hazard rate function, h(t) (Eq. 2), representing the instantaneous hazard of death at each time point conditioned that the patient lives to that point.
| 2 |
h0(t) specifies how the baseline hazard changes with time, and different underlying distributions in shape, namely exponential, Weibull, Gompertz, log-logistic, and log-normal distributions were tested. Different covariates, as well as experiencing rash, were evaluated as overall survival predictors where β1, β2, …, and βn are regression coefficients reflecting the magnitude of the impacts of x1, x2, …, xn predictors.
Model Evaluation
Simulation-based diagnostics, namely visual predictive checks, were mainly used to evaluate the ability of the models to capture the proportions of patients experiencing different intensities over time. Using the models developed, 1000 datasets were simulated, and the simulated proportions of the alive patients experiencing different intensities were compared to the observed proportions.
A bootstrap analysis was used to evaluate the robustness of the models, and the precision and bias of parameter estimates. One thousand replicate datasets were generated by sampling individuals from the original dataset with replacement, and the models were refit to the replicates to compute the median and the 95% confidence intervals.
Simulation Analysis
Using the models developed, a simulation analysis based on 1000 virtual patients, where the individual covariate sets were sampled from the original dataset with replacement, was carried out to compare the toxicity of different erlotinib dosing regimens; these are D1: the standard dosing regimen (150 mg/day), and D2: the pulsed dosing regimen proposed to address acquired resistance (1600 mg/week + 50 mg/day on the remaining days of the week) (12). Furthermore, four other regimens currently tested in an ongoing clinical trial, the purpose of which is to investigate the benefits and risks of the hypothesized pulsed erlotinib regimens, were included in this simulation analysis (22). These treatment arms have high doses administered on days 1 and 2 during the first week with no dosing for the rest of the week, and starting from the second week high doses are to be administered on days 1 and 2 with a continuous daily low dose of 50 mg for the rest of the week; high doses are D3: 600 mg, D4: 750 mg, D5: 900 mg, or D6: 1050 mg.
Since the main intention of the simulation analysis was to evaluate the toxicity (and not clinical efficacy), we made the assumption that patients lived for the entire period (12 months) to evaluate how would they move between different toxicity levels if they “perfectly” benefited from therapy. Grade 3 adverse event was of main interest as its incidence imposes the interruption of antineoplastic therapy by reducing or stopping dosing.
RESULTS
The final framework consisted of three components for rash, diarrhea, and survival described in the following three subsections (model code and data structure are provided in supplemental files 2 and 3, respectively):
Rash
Twenty-three patients experienced at least one rash incident, and in total 33 mild (mean duration = 74 days, range = 4–283 days), 34 moderate (mean duration= 39 days, range = 3–150 days), and 9 severe (mean duration = 28 days, range = 8–47 days) incidents were recorded. Rash imposed dose reduction to 100 mg/day in two incidents and treatment suspension/withdrawal in six incidents.
“Worsening” rate constants for movement from lower to higher rash grades were best described using a decreasing exponential relationship with time since starting treatment and were found to linearly increase with erlotinib exposure as an additive component. Furthermore, receiving radiotherapy prior or during erlotinib systemic treatment was found to decrease the worsening constants by 81% (Fig. 2; Fig. 2a shows the temporal courses of the probabilities of experiencing rash of any grade, and Fig. 2b shows the probability of experiencing severe rash which may result in treatment suspension). Equation 3 shows the final model for the worsening rate constants
| 3 |
Fig. 2.
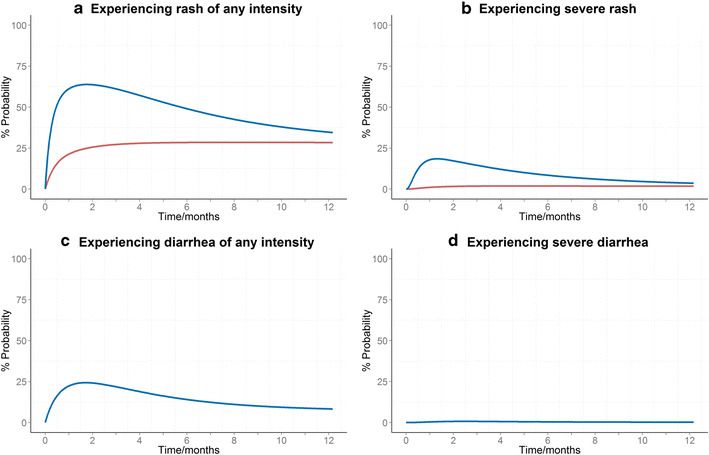
The temporal course of the probabilities of experiencing a any rash intensity, b severe rash, c any diarrhea intensity, and d severe diarrhea with the administration of erlotinib (150 mg/day). Figures a, b show the attenuating effect of receiving radiotherapy prior or during treatment with erlotinib (red) in comparison to other patients (blue)
Where BASEgrade,grade+1 represents the baseline worsening rate constant, KTF is the constant by which the worsening rate constants decrease with time since starting erlotinib, and RTX is 1 for those who received radiotherapy prior or during treatment with erlotinib, and equal to zero for those who did not. Cp is the model generated plasma erlotinib concentration which in turn is related by the SLOPE linearly to the worsening rate constants. Parameter estimates together with their relative standard errors and 95% bootstrap confidence intervals, both of which demonstrating adequate precision except for BASE1,2, are listed in Table II. BASE0,1 and BASE2,3 were found to be almost equal and estimating separate parameters was not statistically significant, and therefore were combined.
Table II.
Parameter Estimates
| Original dataset results | Bootstrap results | |||
|---|---|---|---|---|
| Parameter | Estimate | Relative standard error (%) | Median | 95% confidence interval |
| Rash model parameters | ||||
| BASE0,1 & BASE2,3 (day−1) | 0.0810 | 27.2 | 0.0818 | 0.0458 - 0.134 |
| BASE1,2 (day−1) | 0.798 | 67.8 | 0.879 | 0.244 - 6.11 |
| BASE1,0, BASE2,1 & BASE3,2 (day−1) | 0.0389 | 35.0 | 0.0404 | 0.0208 - 0.0901 |
| SLOPE (mg−1L) | 0.00235 | 29.7 | 0.00233 | 0.00106 - 0.00417 |
| BEXP (mg−1L) | 0.701 | 40.1 | 0.714 | 0.243 - 1.41 |
| KTF (day−1) | 0.0823 | 20.4 | 0.0862 | 0.0567 - 0.125 |
| Effect of RTX on K0,1, K1,2 & K2,3 | -0.806 | 12.7 | -0.822 | -0.985 - -0.543 |
| Diarrhea model parameters | ||||
| BASE0,1, BASE1,2 & BASE2,3 (day−1) | 0.0172 | 31.3 | 0.0177 | 0.00879 - 0.0337 |
| BASE1,0, BASE2,1 & BASE3,2 (day−1) | 0.0274 | 54.7 | 0.0280 | 0.00909 - 0.0911 |
| KTF (day−1) | 0.0244 | 29.4 | 0.0248 | 0.0149 - 0.0527 |
| KTB (day−1) | 0.00740 | 48.1 | 0.00729 | 0.00127 - 0.0200 |
| Survival model parameters | ||||
| BHAZ (day−1) | 0.00207 | 27.0 | 0.00207 | 0.00117 - 0.00341 |
| Hazard ratio for ECOG=2 | 5.75 | 22.4 | 5.88 | 2.66 – 14.1 |
Parameter estimates (and 95% confidence intervals based on a bootstrapping analysis with 1000 samples) for the rash, diarrhea and survival models. BASE0,1, BASE1,2, & BASE2,3 are baseline worsening transition probability constants for movement from NCI-CTC rash grades 0, 1 & 2 to NCI-CTC rash grades 1, 2, & 3, respectively. BASE1,0, BASE2,1, & BASE3,2 are baseline recovery transition probability constants for movement from NCI-CTC rash grades 1, 2 & 3 to NCI-CTC rash grades 0, 1, & 2, respectively. SLOPE is the constant linearly relating plasma exposure to worsening of rash, BEXP is the constant by which the recovery constants for rash decrease exponentially with the increase in exposure, KTF and KTB are constants by which worsening and recovery transition probability constants, respectively, decrease with time since starting erlotinib, and BHAZ is the hazard constant for the exponential distribution describing overall survival.
“Recovery” rate constants for moving from high to lower rash grades were found to decrease exponentially with erlotinib exposure (Eq. 4).
| 4 |
Where BASEgrade,grade-1 is the baseline recovery rate constant, and BEXP is the constant by which the recovery constants decrease exponentially with the increase in exposure.
Temporal courses of the simulated proportions of patients experiencing each state adequately captured the observed temporal courses as seen in the visual predictive checks (Fig. 3a), demonstrating the adequate predictive model performance.
Fig. 3.
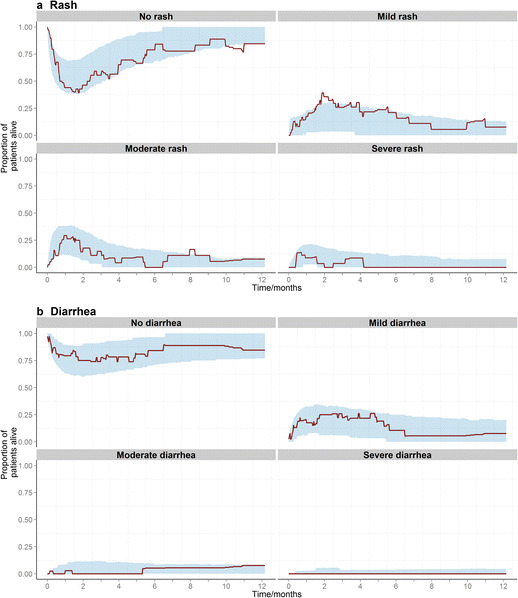
Visual predictive checks showing the proportions of alive patients experiencing each adverse event grade of a rash, and b diarrhea throughout the first year since starting erlotinib, in observed data (solid line), and in simulated data with the corresponding 95% prediction intervals (shaded regions) based on 1000 datasets simulated using the models developed. (n.b. even though none of the patients recruited experienced severe diarrhea, the plot is shown here since a worsening rate constant has been estimated for all possible transitions)
Diarrhea
Eighteen patients experienced at least one diarrhea incident, and in total 19 mild (mean duration = 63 days, range = 1–405 days), and 3 moderate (mean duration = 84 days, range = 7–232 days) incidents were recorded, with no patients experiencing severe diarrhea. One incident of diarrhea imposed treatment suspension.
Worsening and recovery rate constants (Eqs. 5 and 6, respectively) were found to decrease exponentially with time, yet changes in erlotinib exposure had no effect on them.
| 5 |
| 6 |
where KTB is the constant by which the recovery from diarrhea rate constants decrease with time. None of the covariates tested were found to affect any of the model parameters. Parameter estimates (listed in Table II) were estimated precisely, and visual predictive checks for the model (Fig. 3b) demonstrated adequate predictive capability of the model.
Survival
An exponential distribution (with a constant underlying baseline hazard rate, h0(t)) was selected after adequately describing the time-to-death distribution, and after none of the other distributions (all with an extra degree of freedom) gave a statistically significant better fit.
The hazard for death was found to dramatically increase when the ECOG score increased to 2; hazard ratio = 5.75, 95% CI = 2.66–14.0). Experiencing rash with any grade decreased the baseline hazard for death by 56%, yet the relationship was not statistically significant (p = 0.0997), and none of the other covariates were statistically significant. Parameter estimates are presented in Table II.
Simulation Analysis
Since exposure was not found to significantly impact diarrhea, the analysis was done only for rash. Based on the models developed and on the results of the simulation analysis (Fig. 4), it was clear that exposure played a negligible role in the first few weeks as the proportions of patients suffering from rash were similar for the different dosing regimens. Later on, patients got more tolerant, and exposure became the only determinant. Comparing the maximum proportion of patients suffering from severe rash at any time point for the different dosing regimens tested, D1, D2, D3, D4, D5, and D6 resulted in 12.1, 19.6, 14.4, 19.2, 25.7, and 31.7%, respectively.
Fig. 4.
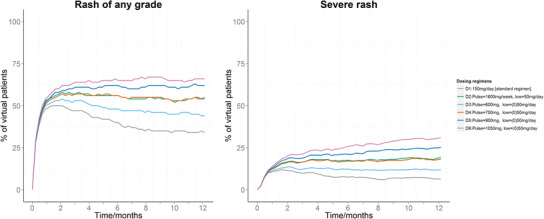
Simulated profiles of the proportions of patients suffering from any grade of rash (left) or severe rash (right) for the different dosing regimens explained in the legend; “pulse” refers to the weekly pulsed high erlotinib dose (D3–D6 have the pulses administered on two consecutive days every week), and “low” refers to the continuous low dose to be administered daily for the rest of the week (which was not administered in the first week for D3–D6, and therefore assigned with (0) in the legend)
DISCUSSION
A modeling framework providing insights into erlotinib-induced adverse events was developed. The framework showed that radiotherapy attenuates erlotinib-induced rash, and it was also used to simulate the toxicity profiles with different dosing regimens proposed to address acquired resistance.
The conventional means of studying the toxicity of a drug would be to link exposure to the highest severity of an adverse event while discarding information by ignoring its times of incidence and resolution, as was done already for erlotinib (15). One alternative to retain the time scale which is important for temporal predictions and simulations and to handle the discrete nature of the data would have been the proportional odds model; however, this model does not assume dependency between observations, and simulations based on it could be inconsistent with observed data. For this reason a continuous-time Markov model, which has demonstrated its usefulness in modeling efficacy and adverse events data of similar type (20,23,24), and which assumes dependency between an observation and the next one was used.
According to the final models developed, the probability to experience rash or diarrhea was highest early after starting treatment with erlotinib at the standard dosing of 150 mg/day, yet later on with further dosing patients got more tolerant. In contrast with others (25), we were not able to show a significant correlation between rash and clinical efficacy in terms of survival. This suggests to rely on rash merely as a pharmacokinetic marker demonstrating adequate bioavailability rather than a pharmacodynamic or clinical marker.
In agreement with previous results (15), erlotinib exposure was found to be positively linearly correlated with experiencing higher rash grades, yet no relationship was found between exposure and diarrhea. It needs to be mentioned however that a major limitation of our analysis was the lack of observed plasma erlotinib concentrations and the inability to investigate the link between true exposure and adverse events. This mandated the use of a published model (15), developed using data from 591 patients recruited in 5 trials, as an alternative to generate exposure using the actual set of individual covariate substituted into the model. Furthermore, we were not able to investigate whether lower oral bioavailability were associated with higher risks of gastrointestinal irritation and diarrhea. Another limitation is the limited tested exposure range, meaning the linearity assumption which proved the most adequate within the tested range, might be over simplistic when used for extrapolations for higher doses tested in the simulation analysis. Testing in patients will be the only way to investigate such limitations of the models.
Remarkably, receiving radiotherapy before or during systemic treatment with erlotinib was found to decrease the transitioning probability to higher rash grades by 81% (p < 0.01), which to the best of our knowledge, is the first time to demonstrate the effect of radiotherapy against erlotinib-induced rash based on data from a cohort of patients. This finding comes in counter-intuition with the notion that radiotherapy and erlotinib would aggravate the rash typically caused by each other when administered alone, yet comes in agreement with previously reported cases (26,27). A clear explanation for this observation is not available, yet it has been suggested that EGFR-harboring basal keratinocytes in the skin, which are negatively affected by EGFR inhibitors giving rise to cutaneous toxicity, are decreased or depleted following irradiation (28). Further exploration in the clinic is needed in a larger number of patients to confirm this effect.
It needs to be mentioned that even though the survival model was based on data from the same cohort of patients used in previous analyses (13,29), ECOG was the only significant overall survival predictor included and neither baseline tumor burden nor changes in PET uptakes showed statistical significance. The reason for this discrepancy is because of the way the analyses were carried out. The previous analyses intended to evaluate covariates as “early” predictors of survival. For that purpose, these analyses used landmark survival analyses with landmark time points set to baseline or one week after treatment initiation, meaning that only the ECOG score at that landmark was used and not the updated time varying ECOG scores which change over time as in this case (masking the effect of all other covariates).
Based on the simulation analysis, it was found that exposure had a limited effect on determining the probability of rash incidence in comparison to the intrinsic effect of administering erlotinib in the first few weeks, but later on exposure became the determinant. Furthermore, the framework provided insights by comparing different dosing regimens, including various pulsed ones. Considering severe rash which usually prompts a clinical decision to modify the dose or suspend the treatment, the proposed pulsed regimen (1600 mg/week + 50 mg/day remaining days of the week (12)) resulted in almost a maximum of 20% of the patients in the simulated cohort to suffer from severe rash at any time point in comparison to 12% for the arm treated with the standard 150 mg/day dosing. Such results, in addition to these of the other simulated arms, could be used in outcomes research and by clinicians to balance between the positive clinical effects of these regimens against cancer, and the negative physical and psycho-social effects possibly caused by them and eventually disrupting treatment.
Model-based approaches such as described in this work, or in (12,29–31), provide an opportunity to maximize the amount of information extracted out of data, and to use this information in rationalizing decision making in lung cancer therapy. Furthermore, in addition to the efforts invested in developing moieties targeting new targets or addressing resistance mechanisms, dosing and scheduling should be further explored for better use of treatments available. With comparatively limited investments in time and money, mathematical modeling provides an appealing alternative for such an exploration to eventually streamline drug development and clinical management.
CONCLUSIONS
In conclusion, we have been successful in developing mathematical models characterizing the temporal courses of experiencing common adverse events with erlotinib. Radiotherapy was shown to mitigate erlotinib-induced rash, providing an opportunity to use both radiotherapy and erlotinib systemic treatment simultaneously in treatment protocols. The modeling framework has also demonstrated the tolerability of high-dose pulses intended to address acquired resistance to erlotinib.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(DOCX 18 kb)
(TXT 4 kb)
(CSV 1 kb)
Acknowledgments
Conflicts of Interest
None of the authors declared conflicting interests which are relevant to this manuscript or which can influence the judgments made in it. However, a grant was received from Roche for the study, the data generated by which were used to develop the models presented in this analysis.
REFERENCES
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Buettner R, Wolf J, Thomas RK. Lessons learned from lung cancer genomics: the emerging concept of individualized diagnostics and treatment. J Clin Oncol. 2013;31:1858–65. doi: 10.1200/JCO.2012.45.9867. [DOI] [PubMed] [Google Scholar]
- 3.Clinical Lung Cancer Genome Project (CLCGP) Network Genomic Medicine (NGM) A genomics-based classification of human lung tumors. Sci Transl Med. 2013;5:209ra153. doi: 10.1126/scitranslmed.3006802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–46. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 5.Kwak EL, Bang Y-J, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 7.Engelman JA, Jänne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14:2895–9. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 8.Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70:10038–43. doi: 10.1158/0008-5472.CAN-10-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yun C-H, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong K-K, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–5. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med. 2011;3:90ra59. doi: 10.1126/scitranslmed.3002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foo J, Chmielecki J, Pao W, Michor F. Effects of pharmacokinetic processes and varied dosing schedules on the dynamics of acquired resistance to erlotinib in EGFR-mutant lung cancer. J Thorac Oncol. 2012;7:1583–93. doi: 10.1097/JTO.0b013e31826146ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zander T, Scheffler M, Nogova L, Kobe C, Engel-Riedel W, Hellmich M, et al. Early prediction of nonprogression in advanced non-small-cell lung cancer treated with erlotinib by using [(18)F]fluorodeoxyglucose and [(18)F]fluorothymidine positron emission tomography. J Clin Oncol. 2011;29:1701–8. doi: 10.1200/JCO.2010.32.4939. [DOI] [PubMed] [Google Scholar]
- 14.National Cancer Institute. Common terminology criteria for adverse events v3.0 (CTCAE). 2006 [cited 2014 Nov 26]. Available from: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf
- 15.Lu J-F, Eppler SM, Wolf J, Hamilton M, Rakhit A, Bruno R, et al. Clinical pharmacokinetics of erlotinib in patients with solid tumors and exposure-safety relationship in patients with non-small cell lung cancer. Clin Pharmacol Ther. 2006;80:136–45. doi: 10.1016/j.clpt.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 16.Beal S, Sheiner L, Boeckmann A, Bauer R. NONMEM User’s Guides. (1989-2009), Icon Development Solutions, Ellicott City, MD, USA, 2009.
- 17.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Available from http://www.R-project.org/.
- 18.Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on pirana, PsN, and xpose. CPT Pharmacometrics Syst Pharmacol. 2013;2:e50. doi: 10.1038/psp.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergstrand M, Söderlind E, Weitschies W, Karlsson MO. Mechanistic modeling of a magnetic marker monitoring study linking gastrointestinal tablet transit, in vivo drug release, and pharmacokinetics. Clin Pharmacol Ther. 2009;86:77–83. doi: 10.1038/clpt.2009.43. [DOI] [PubMed] [Google Scholar]
- 20.Pilla Reddy V, Petersson KJ, Suleiman AA, Vermeulen A, Proost JH, Friberg LE. Pharmacokinetic-pharmacodynamic modeling of severity levels of extrapyramidal side effects with markov elements. CPT Pharmacometrics Syst Pharmacol. 2012;1:e1. doi: 10.1038/psp.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holford N. A time to event tutorial for pharmacometricians. CPT Pharmacometrics Syst Pharmacol. 2013;2:e43. doi: 10.1038/psp.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.NCT01967095. Low Dose Daily Erlotinib in Combination With High Dose Twice Weekly Erlotinib in Patients With EGFR-Mutant Lung Cancer. 2015 [cited 2014 Dec 17]. Available from: https://clinicaltrials.gov/ct2/show/record/NCT01967095.
- 23.Hansson EK, Ma G, Amantea MA, French J, Milligan PA, Friberg LE, et al. PKPD modeling of predictors for adverse effects and overall survival in Sunitinib-treated patients with GIST. CPT Pharmacometrics Syst Pharmacol. 2013;2:e85. doi: 10.1038/psp.2013.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lacroix BD, Karlsson MO, Friberg LE. Simultaneous exposure-response modeling of ACR20, ACR50, and ACR70 improvement scores in rheumatoid arthritis patients treated with certolizumab pegol. CPT Pharmacometrics Syst Pharmacol. 2014;3:e143. doi: 10.1038/psp.2014.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee SM, Khan I, Upadhyay S, Lewanski C, Falk S, Skailes G, et al. First-line erlotinib in patients with advanced non-small-cell lung cancer unsuitable for chemotherapy (TOPICAL): a double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2012;13:1161–70. doi: 10.1016/S1470-2045(12)70412-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitra SS, Simcock R. Erlotinib induced skin rash spares skin in previous radiotherapy field. J Clin Oncol. 2006;24:e28–9. doi: 10.1200/JCO.2005.05.1631. [DOI] [PubMed] [Google Scholar]
- 27.Lips IM, Koster MEY, Houwing RH, Vonk EJA. Erlotinib-induced rash spares previously irradiated skin. Strahlenther Onkol. 2011;187:499–501. doi: 10.1007/s00066-011-2232-8. [DOI] [PubMed] [Google Scholar]
- 28.Lacouture ME. Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat Rev Cancer. 2006;6:803–12. doi: 10.1038/nrc1970. [DOI] [PubMed] [Google Scholar]
- 29.Suleiman AA, Frechen S, Scheffler M, Zander T, Kahraman D, Kobe C, et al. Modeling tumor dynamics and overall survival in advanced non-small-cell lung cancer treated with erlotinib. J Thorac Oncol. 2015;10:84–92. doi: 10.1097/JTO.0000000000000330. [DOI] [PubMed] [Google Scholar]
- 30.Suleiman AA, Nogova L, Fuhr U. Modeling NSCLC progression: recent advances and opportunities available. AAPS J. 2013;15:542–50. doi: 10.1208/s12248-013-9461-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Sung C, Dartois C, Ramchandani R, Booth BP, Rock E, et al. Elucidation of relationship between tumor size and survival in non-small-cell lung cancer patients can aid early decision making in clinical drug development. Clin Pharmacol Ther. 2009;86:167–74. doi: 10.1038/clpt.2009.64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 18 kb)
(TXT 4 kb)
(CSV 1 kb)