

Abstract
Purpose
The mutation S163R in complement C1q tumor necrosis factor–related protein-5 (C1QTNF5) causes an autosomal dominant disorder known as late-onset retinal degeneration (L-ORD). In this study, our goal is to evaluate the consequences of mutant S163R C1QTNF5 expression in mouse RPE following its delivery using an adeno-associated viral (AAV) vector.
Methods
We generated AAV vectors containing either human wild-type C1QTNF5 or mutant S163R C1QTNF5 driven by an RPE-specific BEST1 promoter, and delivered them subretinally into one eye of adult C57BL/6 mice. Transgene expression was detected by immunohistochemistry. Retinal function was assessed by full-field ERG. Pathological changes were further examined by digital fundus imaging and spectral-domain optical coherence tomography (SD-OCT).
Results
We show that the AAV-expressed mutant S163R leads to pathological effects similar to some of those found in patients with advanced L-ORD, including RPE thinning, RPE cell loss, and retinal degeneration. In addition, we provide in vivo evidence that mutant S163R C1QTNF5 can form large, transparent, spherical intracellular aggregates throughout the RPE, which are detectable by light microscopy. In contrast to AAV-expressed wild-type C1QTNF5, which is secreted apically from the RPE toward the photoreceptor cells and the outer limiting membrane, the S163R mutant is primarily routed toward the basal side of RPE, where it forms thick, extracellular deposits over time.
Conclusions
Adeno-associated viral–targeted expression of mutant S163R in the RPE represents a useful approach for quickly generating animal models that mimic pathological features of L-ORD and offers the potential to understand disease mechanisms and develop therapeutic strategies.
Keywords: retina, adeno-associated virus, age-related macular degeneration, retinal pigment epithelium, late-onset retinal degeneration
The retinal pigment epithelium is a polarized monolayer of postmitotic epithelial cells located between the neural retina and the vascular choroid. On its apical side, it contains numerous microvilli extending into the interphotoreceptor matrix, and surrounding the outer segments of rods and cones. On the basal side is Bruch's membrane, a thin, multilayered, extracellular matrix1 in which pathological changes are integral to AMD.2,3 The retinal pigment epithelium performs functions essential for both the structural integrity and survival of photoreceptor cells, including transport of nutrients, retinoids and waste products, phagocytosis of outer segments, synthesis of the visual chromophore11-cis retinal, and production of growth factors.4,5
The mutation S163R in the globular gC1q domain of complement C1q tumor necrosis factor–related protein-5 (C1QTNF5/CTRP5) causes a rare autosomal dominant disorder known as late-onset retinal degeneration (L-ORD).6 The earliest symptoms appear in midlife, manifested as difficulties in dark adaptation and loss of night vision.7–9 Over time, the disorder progresses to severe loss of both central and peripheral vision, with some patients also developing choroidal neovascularization in late stages of the disease10 that mimic neovascular AMD. In addition, patients also present with abnormally long anterior zonular insertions on the lens capsule which serve as an early disease phenotype.11,12 The hallmark of L-ORD is the presence of abnormal extracellular deposits between the RPE and Bruch's membrane.7,13 These thick deposits, which are histopathologically similar to those found in AMD patients, have a widespread distribution, extending outside the macular region into the peripheral retina.14 Although there is evidence that supplementation with vitamin A may be beneficial early in the disease course when patients exhibit dark-adaptation abnormalities,11 there is no current treatment that prevents the progressive retinal degeneration leading to vision loss.
C1QTNF5 belongs to the C1q and tumor necrosis factor-related superfamily (C1q/TNF), characterized by an N-terminal signal peptide, a short variable region, a collagen repeat domain, and a C-terminal globular domain (gC1q) that is homologous to the immune complement component 1q. All members of the C1q/TNF family are secreted multimeric proteins, with specific tissue expression profiles, and many are found circulating in plasma, with levels influenced by genetic background, sex, and metabolic status. Members of the C1q/TNF family are of broad clinical interest since they play numerous roles in immunity and inflammation, glucose and lipid metabolism, and vascular maintenance.15,16 C1QTNF5 is a membrane-associated and secreted protein of unknown function, whose expression is the highest in the RPE and ciliary epithelium.11,17,18 It colocalizes with ZO1, an RPE tight-junction protein, at the hexagonal RPE membrane, and is also secreted into the interphotoreceptor matrix.17,19 C1QTNF5 can also be found in other ocular tissues, including the lens.17 It is secreted by the adipose tissue, and circulates abundantly in human sera, potentially playing an important role in energy homeostasis and obesity-related inflammation.16,20 The crystal structure of the trimeric globular domain of C1QTNF5 was determined at 1.34 Å resolution by Tu and Palczewski,21 who also showed that wild-type C1QTNF5 can multimerize into a bouquet-like octadecameric assembly containing six trimers.22 Previous in vitro studies have demonstrated that the S163R mutant is not secreted, has the tendency to aggregate, and is retained primarily within the endoplasmic reticulum.17,23
Interestingly, C1QTNF5 is expressed as a dicistronic transcript with MFRP, encoding the membrane-type frizzled related protein.6,24 The complete open reading frame of C1QTNF5 is located within the 3′ untranslated region of the final exon of MFRP. The two proteins were shown to directly interact; however, the functional significance of this interaction is not currently understood.18,22,23 Mutations in the two genes are associated with distinct disease phenotypes in humans, with the dominant S163R mutation in C1QTNF5 leading to L-ORD, whereas mutations in MFRP cause autosomal recessive retinitis pigmentosa (RP), nanophthalmos, foveoschisis, and optic disc drusen.25–27
We have previously shown that subretinal delivery of a fast-acting AAV8-733YF vector expressing wild-type MFRP led to robust transgene expression at its normal RPE location on the apical membrane and microvilli, prevented retinal degeneration, and rescued photoreceptor function in the rd6 mouse model of RP.28 In this study, we used the recombinant AAV vector technology to examine the pattern of mutant S163R transgene expression and its pathological effects following specific RPE targeting. We show that AAV-expressed wild-type C1QTNF5 is secreted apically toward photoreceptor cells and outer limiting membrane (OLM). In contrast, AAV-expressed S163R mutant forms large spherical intracellular aggregates within the RPE cells of normal mice, and gradually accumulates as thick, widespread extracellular basal deposits, leading to local regions of RPE atrophy and retinal degeneration. These findings have important mechanistic and therapeutic implications for L-ORD and perhaps for AMD as well.
Methods
AAV Vector Production and Subretinal Injections
Self-complementary AAV2 quadruple capsid tyrosine mutant (Y272,444,500,730F) vectors (scAAV2quadYF) were used for packaging and expressing either human wild-type C1QTNF5 (hC1QTNF5WT-HA) or the S163R mutant (hC1QTNF5(S163R)-HA), driven by an RPE-selective human BEST1 promoter.29 Both wild-type and mutant S163R C1QTNF5 were tagged with hemagglutinin (HA) at their C-terminal end for subsequent detection by immunostaining. The quadruple capsid tyrosine mutant vector AAV2 containing point mutations of surface-exposed tyrosine residues was chosen for transgene delivery in all these experiments based on its higher transduction efficiency following a single injection, compared to wild-type AAV2.30 The viral vectors were packaged, purified, and titered according to previously reported methods.31 The vectors were then subretinally injected in adult C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME, USA) as previously described.32 Each eye received 1 μL vector at a titer of 1 × 1012 genome copies/mL, leaving the left eye as an untreated control. Animals were maintained on a 12-hour light and 12-hour dark cycle in the University of Florida Health Science Center Animal Care Services Facility (Gainesville, FL, USA). All experiments were approved by University of Florida Institutional Animal Care and Use Committees and conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Immunostaining and Histologic Analysis
Eyes were harvested, processed for paraffin embedding, and sectioned at a thickness of 4 μm, at the same retinal locations for treated and untreated eyes. For morphologic analysis, the sections were stained with hematoxylin and eosin, and visualized by light microscopy. For immunostaining, deparaffinized tissue sections were dewaxed in xylene and rehydrated in a graded series of ethanol, then incubated with 0.3% Triton X-100 for 15 minutes, followed by blocking with a solution of 2% albumin for 30 minutes. Hemagglutinin-tagged C1QTNF5 proteins in the AAV-treated eyes were detected by immunostaining with a high-affinity anti-HA–fluorescein antibody (3F10; Roche Diagnostics, Indianapolis, IN, USA). Some sections were also incubated with mouse MFRP affinity-purified polyclonal antibody (AF3445, R&D Systems, Minneapolis, MN, USA), followed by the Alexa Fluor 594 secondary antibody (Molecular Probes/Invitrogen, Eugene, OR, USA). Negative controls to check for any potential autofluorescence in AAV-expressed S163R-treated retinas were also performed by omitting the anti-HA antibody, and purposefully overexposing those images to detect any background autofluorescence. The mouse monoclonal antibody B6-30 against rod opsin33 was used to label the outer segments. Fluorescence micrographs at higher magnification were also acquired using a ×100 oil immersion objective. All sections were examined by fluorescence microscopy using a microscope (Axiophot; Carl Zeiss Microscopy, LLC, Thornwood, NY, USA). Cryosections for Oil red O staining to detect neutral lipids in the AAV-expressed S163R-treated eyes were also used. Frozen liver sections were used as positive control for the lipid staining. The liver was from a mouse that received intraperitoneal injections with carbon tetrachloride (CCl4) to cause hepatocyte damage and liver fibrosis.34 Cryosections were air dried at room temperature for 30 minutes and rinsed with distilled water. Sections were then placed in polyethylene glycol (PEG), stained with Oil red O (Abcam, Cambridge, UK), rinsed with 85% PEG, then with distilled water, before adding an aqueous mounting medium (Dako, Carpinteria, CA, USA). Images were acquired using brightfield microscopy.
Transmission Electron Microscopy
For ultrastructural analysis, the injected eyes containing S163R aggregates and basal deposits that were identified at the light level were further examined by transmission electron microscopy (TEM). Paraffin blocks were submitted to the Electron Microscopy Core Laboratory, Interdisciplinary Center for Biotechnology Research, University of Florida. Areas of interest containing the RPE–choroid interface were excised from the paraffin blocks and placed in 100% xylene overnight. The dewaxed tissues were rehydrated through a decreasing ethanol series, water washed, buffer washed, and fixed with Trumps (Electron Microscopy Sciences, Hatfield, PA, USA). Fixed tissues were processed with the aid of a laboratory microwave (Pelco BioWave Pro; Ted Pella, Redding, CA, USA). The samples were washed in 0.1 M cacodylate buffer pH 7.24, postfixed with 2% OsO4, water washed and dehydrated in a graded ethanol series 25%, 50%, 75%, 95%, 100%. Dehydrated tissues were infiltrated with 1:1 ethanol/LR White hard acrylic resin (Electron Microscopy Sciences, Hatfield, PA, USA) then embedded in 100% LR White resin and polymerized at 60°C for 48 hours. The cured resin blocks were hand-trimmed and prepared for ultramicrotomy. Semi-thick (500 nm) and ultra-thin sections (100–120 nm) were cut using a diamond knife. Semi-thick sections were dried onto a glass slide and stained with toluidine blue. Ultra-thin sections were collected on carbon-coated Formvar copper slotted grids, poststained with 2% aq. uranyl acetate, and Reynolds lead citrate. Sections were examined with a FEI Tecnai Spirit LaB6 at 120 kV (FEI, Hillsboro, OR, USA) and images acquired with a charge-coupled device camera (AMT XR41, 2k X 2k; Advanced Microscopy Techniques, Woburn, WA).
Western Blot Analysis
Whole eyecups from AAV-S163R–treated eyes (1 month post injection) were homogenized by sonication in RIPA buffer (Sigma-Aldrich Corp., St. Louis, MO, USA) containing a complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). After centrifugation, the supernatant was mixed with 2X Laemmli's sample buffer containing dithiothreitol (DTT) and subjected to SDS-PAGE. Protein extracts from AAV–wild-type C1QTNF5 or AAV-S163R–injected eyecups were also analyzed under nondenaturing conditions by sonicating the tissues in a sample buffer lacking the SDS and DTT, and loading the supernatants onto 4% to 15% gradient Mini Protean TGX precast polyacrylamide gels (Bio-Rad Laboratories, Inc., Hercules, CA, USA). After protein transfer onto polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA) and blocking by incubation for 1 hour in Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE, USA), the membranes were probed with a mouse monoclonal primary antibody against the HA tag (Covance, Gaithersburg, MD, USA). Signals were detected with the Odyssey infrared fluorescence imaging system (LI-COR Biosciences), using an anti-mouse antibody conjugated with CW800 dye (LI-COR Biosciences).
Electroretinographic Analysis and Fundoscopy
Treated C57BL/6 mice were dark-adapted (>12 hours) and anesthetized by intraperitoneal injection. Full-field ERGs were recorded as previously described,32 using a UTAS Visual Diagnostic System (LKC Technologies, Gaithersburg, MD, USA). Dark-adapted ERGs were elicited with 0.02, 0.2 and 2 scot cd·s·m−2 stimuli. Cone-isolated ERGs were elicited with a 25 phot-cd·s·m−2 (10 dB) stimulus on a rod-suppressing background light after a preadaptation period. Differences in b-wave maximum amplitudes between AAV hC1QTNF5(S163R)-HA–injected and uninjected contralateral left eyes were analyzed by Student's t-test for paired samples (GraphPad Prism 6.0; GraphPad Software, San Diego, CA, USA), and considered statistically significant if P < 0.05. Electroretinographic data are presented as mean ± SEM. The Micron III fundus imaging system (Phoenix Research Laboratories, Pleasanton, CA, USA) was used to obtain digital images of the central retina.
SD-OCT
Spectral-domain optical coherence tomography (SD-OCT) was performed at 9 months post injection using a high-resolution SD-OCT instrument (Bioptigen, Inc., Research Triangle Park, Durham, NC, USA) for image capture and to measure the thickness of the retina, as previously described.35 Neural retina thickness was determined by measuring the distance from the vitreal face of the ganglion cell layer to the retinal pigment epithelium apical side. Four measurements at the same distance from the optical nerve head were recorded from each eye and averaged. Statistical analysis was performed by paired t-test, with P < 0.05 considered statistically significant.
Results
Distribution of AAV-Expressed Wild-Type C1QTNF5 and S163R Mutant
AAV2quadYF vectors containing either hC1QTNF5WT-HA or hC1QTNF5(S163R)-HA transgenes were delivered to wild-type mouse eyes in order to compare the distribution patterns of the wild-type and S163R mutant following specific RPE targeting, and to document potential pathological effects. In order to avoid detecting endogenous C1QTNF5, a C-terminal HA tag was added for the reliable detection of transgene expression by immunostaining in each case. We determined that wild-type C1QTNF5 was secreted apically from the RPE, reaching the outer limiting membrane and interdigitating with photoreceptor cells inner segments (Fig. 1A). Adeno-associated viral-expressed C1QTNF5 appears to diffuse further, beyond the outer limiting membrane, being detectable between photoreceptor nuclei (Fig. 1B).
Figure 1.
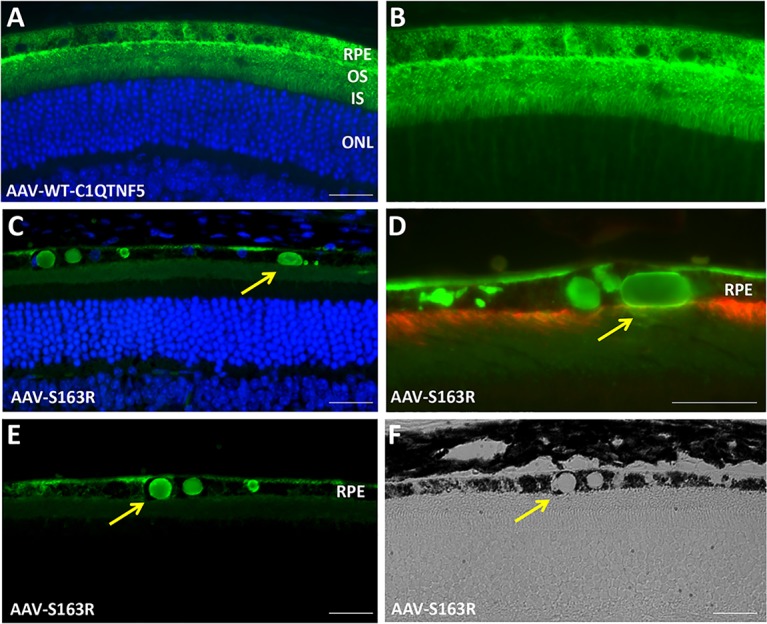
Detection of AAV-expressed wild-type C1QTNF5 and mutant S163R by immunofluorescence microscopy. (A) Vector-expressed wild-type C1QTNF5 (green) is present throughout the RPE, and apically secreted toward the OLM, where it interdigitates with photoreceptor inner segments. (B) Same image without DAPI nuclear stain, showing AAV-expressed wild-type C1QTNF5 diffusing beyond the OLM, between photoreceptor nuclei (2 months post injection). (C–E) Detection of AAV-expressed mutant S163R C1QTNF5 (green) by immunofluorescence microscopy. Round or oval-shaped S163R aggregates (arrows) are present throughout the RPE (4 months post injection). Note the continuous layer of S163R mutant protein deposit on the basal side of the RPE membrane. (D) Double-labeling with an anti-MFRP antibody (red) and anti-HA (green) following AAV-hC1QTNF5(S163R)-HA treatment. Note the reduced MFRP immunostaining in some areas containing large intracellular S163R aggregates (green), indicating loss of RPE microvilli (arrow). (F) Transparent spherical S163R aggregates as examined by brightfield light-microscopy (arrow). Scale bars: 30 μm. IS, inner segments; ONL, outer nuclear layer; OS, photoreceptor outer segments.
In stark contrast to vector expressed wild-type protein, AAV-expressed S163R is predominantly retained within RPE cells, where it forms large spherical or oval-shaped intracellular aggregates (Figs. 1C–E). In some regions containing the aggregates, RPE microvilli were missing, as revealed by double immunostaining with an anti-MFRP antibody, which detects the endogenous MFRP found abundantly across the entire length of RPE apical processes (Fig. 1D). Aggregates of S163R are transparent when examined under brightfield microscopy (Fig. 1F). Interestingly, S163R mutant protein was also secreted toward the basolateral side of the RPE, where it formed a continuous layer (Fig. 2). Sections from AAV-S163R–treated eyes were examined at higher magnification (×100) using differential interference contrast (DIC) microscopy (Fig. 2A) and merged with the fluorescent image (Fig. 2B) to provide a clear view of the S163R extracellular basal deposit and globular aggregates. Transmission electron microscopy was also used to examine the ultrastucture of the RPE-Bruch's membrane interface in S163R-injected eyes and confirmed that the S163R layer accumulates between the basement membrane of the RPE and its cell membrane, with large S163R aggregates being present inside the RPE cytoplasm (Figs. 2C, 2D).
Figure 2.
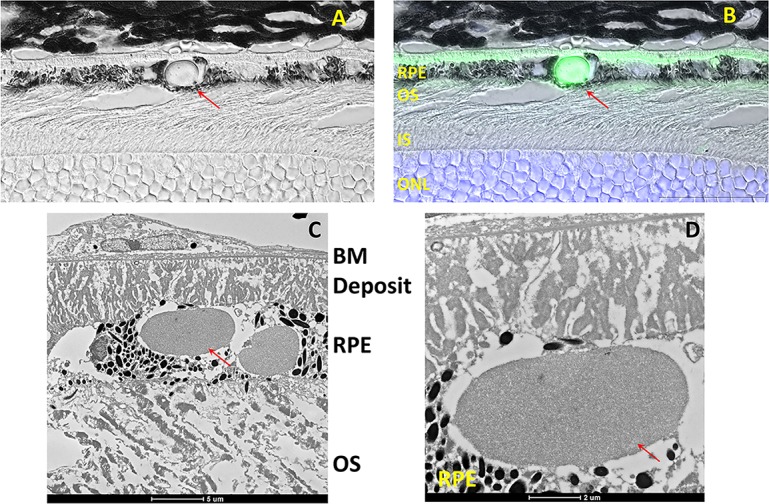
Examination of the S163R C1QTNF5 deposits at high magnification. (A) Image acquired using DIC microscopy at ×100 magnification. (B) Differential interference contrast image merged with the fluorescent image obtained following immunostaining with an anti-HA antibody. Note the intracellular globular S163R aggregate (red arrows) and the extracellular (green) basal RPE deposit. Scale bars: 25 μm. (C) Ultrastructural examination of the RPE/Bruch's membrane interface after AAV-S163R treatment using transmission electron microscopy. (D) Inset from (C) at higher magnification showing the area containing the intracellular and basal laminar deposits.
Staining the AAV-S163R–treated sections with hematoxylin and eosin (H & E stain) showed that the S163R aggregates appear eosinophilic (pink; Fig. 3). Aggregates of S163R were found throughout the RPE after a single subretinal delivery of hC1QTNF5(S163R)-HA vector at 4 months post injection. The retinal pigment epithelium appears vacuolated in many regions, although some of these may be where the aggregates were lost during tissue processing (Fig. 3, center and right panels).
Figure 3.
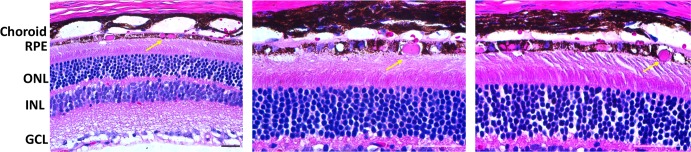
Histologic examination of AAV-hC1QTNF5(S163R)-HA–treated eyes by light microscopy at 4 months post injection. Staining with H & E of paraffin sections reveals the presence of eosinophilic (pink) aggregates (arrows) throughout the RPE cytoplasm of AAV-S163R–treated eyes. The choroid is detached from RPE (all panels) and photoreceptor outer segments have become disorganized (higher magnification images, middle and right). Scale bars: 20 μm.
Detection of S163R Deposits in Older Mice
By 9 months post injection, AAV-S163R–treated eyes display regions of RPE thinning and atrophy, as well as photoreceptor cell loss, particularly in those areas where thick, dome-shaped extracellular basal RPE deposits are present (Fig. 4B). In contrast, uninjected control eyes show normal RPE and retinal morphology (Fig. 4A). The basal RPE deposits consistently formed a continuous layer in every AAV-S163R–treated eye (Figs. 4C, 4D), and were determined to consist of S163R mutant protein by immunofluorescence following staining with the anti-HA antibody (Fig. 5A, Supplementary Fig. S1). We also tested for the presence of lipids following staining of cryosections with oil red O, and did not detect lipid accumulation in the AAV-S163R–injected eyes (Supplementary Fig. S1).
Figure 4.
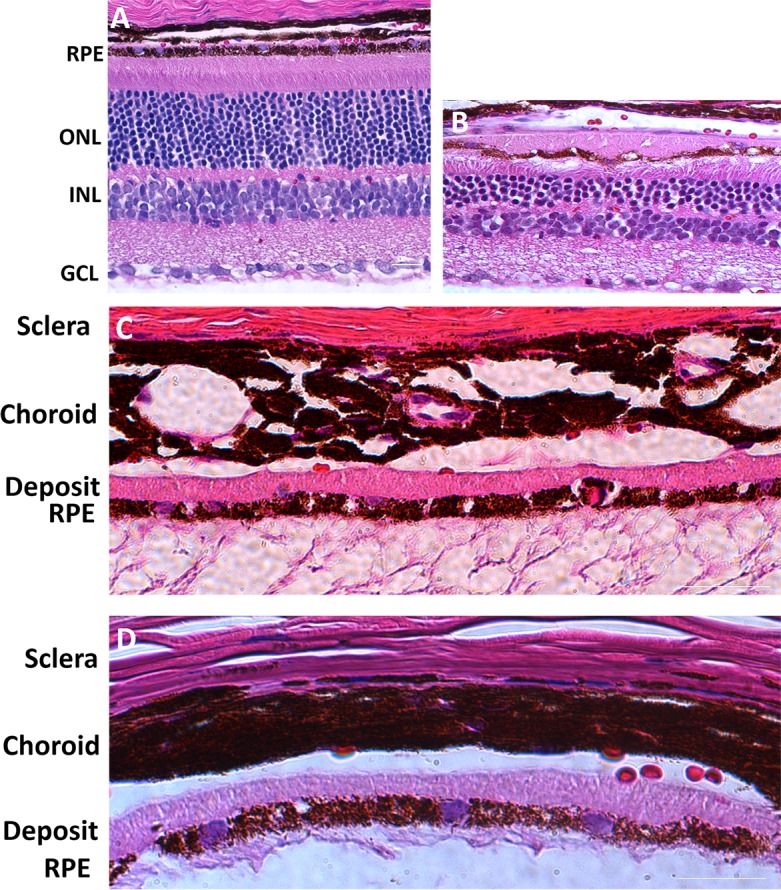
Histologic examination of AAV-hC1QTNF5(S163R)-HA–treated eyes by light microscopy at 9 months post injection. (A) Section stained with H & E of an untreated wild-type mouse eye showing normal RPE and retinal structure. (B) Section stained with H & E of an AAV-hC1QTNF5(S163R)-HA–injected eye. Note the thick deposit on the basal side of the RPE, the photoreceptor cell loss, and the RPE thinning and atrophy in contrast to the normal eye. (C, D) Higher magnification images showing the RPE and the eosinophilic (pink) basal deposits in two different eyes. Scale bars: 15 μm.
Figure 5.
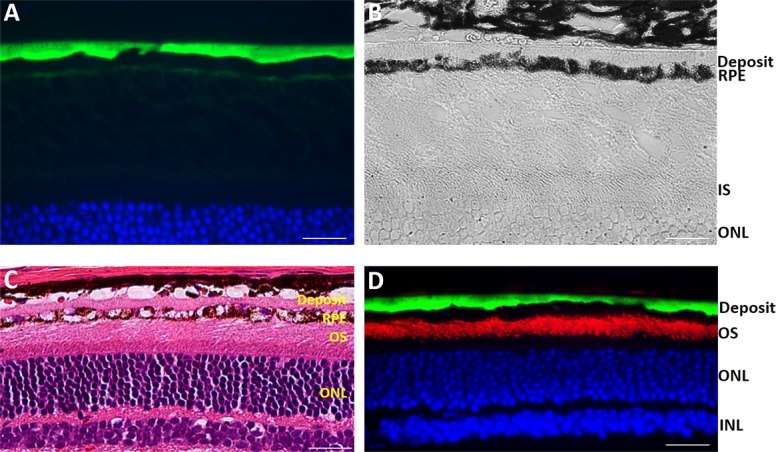
Examination of S163R expression at 9 months following AAV-hC1QTNF5(S163R)–HA delivery. (A) Detection of the AAV-expressed S163R by immunofluorescence. Note the thick, continuous S163R basal deposit, which appears transparent in the brightfield above the pigmented RPE (B). (C) Staining with H & E showing the thick basal deposit above a highly vacuolated, deteriorated RPE. (D) Double-labeling with an antibody against rod opsin showing the outer segments (red), and anti-HA (green) detecting the thick S163R deposit basal to the RPE. Scale bars: 20 μm.
The thick S163R deposits located between the Bruch's membrane and the deteriorating RPE layer appear transparent under brightfield microscopy (Fig. 5B) and pink following H & E staining (Fig. 5C). Interestingly, the spherical S163R C1QTNF5 aggregates are mostly absent at this stage, suggesting that the RPE may have developed a mechanism for their transport and deposition on their basal surface. The remaining photoreceptors retained their outer segments, as demonstrated by immunostaining with a rod opsin antibody (Fig. 5D). Low magnification images show that the S163R deposit above the RPE is continuous and widespread (Figs. 6A–C). The endogenous MFRP protein is expressed throughout the RPE apical membrane (Fig. 6C).
Figure 6.
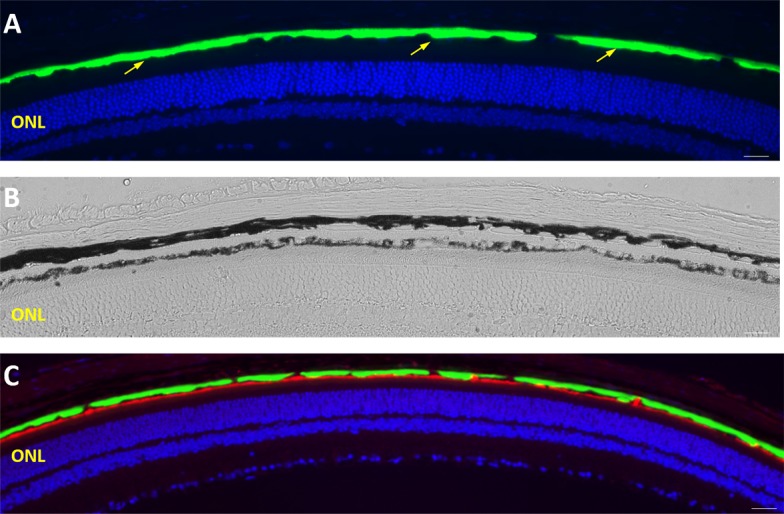
Widespread distribution of basal S163R deposits at 9 months following AAV-hC1QTNF5(S163R)–HA delivery. (A–C) Low magnification images of retinal sections showing continuous, widespread S163R expression (green) above the RPE by immunofluorescence (A, C). (B) Brightfield image showing the transparent basal S163R deposit between the choroid and the pigmented RPE. (C) The RPE apical membrane is shown in red as labeled by the anti-MFRP antibody.
Detection of AAV-Expressed S163R C1QTNF5 by Western Blot Analysis
We also evaluated the properties of AAV-expressed S163R mutant protein by Western blot analysis of whole eyecup protein extracts (Fig. 7). The adeno-associated viral-delivered mutant protein was detected primarily as a single immunoreactive band corresponding to the expected size of C1QTNF5 (slightly larger than 25 kDa) under denaturing and reducing conditions (Fig. 7A). We found S163R protein was present as high molecular weight aggregates when eyecups were sonicated in a buffer without SDS, a property consistent with results from previous in-vitro studies6,23 (Fig. 7B, first lane). In contrast, the AAV-expressed wild-type C1QTNF5 was detected as primarily a monomer, dimer, and trimer, as well as less abundant higher order oligomers in samples processed without SDS (Fig. 7B, second lane). In the presence of SDS, AAV-expressed wild-type C1QTNF5 was detected primarily as a strong monomeric immunoreactive band, and as less abundant signals corresponding to oligomeric and posttranslationally modified forms in a smeared appearance of immunoreactive bands (Fig. 7B, last lane).
Figure 7.
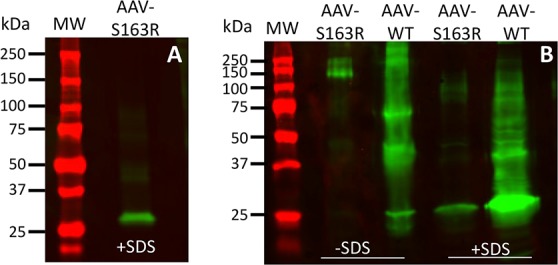
Western blot analysis of AAV-expressed S163R C1QTNF5. (A) Detection of AAV-expressed S163R mutant protein from whole eyecup extracts following SDS-PAGE separation and immunoblotting with an anti-HA antibody. (B) Detection of AAV-expressed S163R and wild-type proteins following resolution on nondenaturing gels. Note the distinct immunoreactive pattern of the S163R mutant compared to that of wild-type C1QTNF5. Prestained protein standards in kilodaltons are shown on the left.
Noninvasive Assessment of Pathological Effects
Noninvasive approaches were also used to analyze the structural and functional consequences of the mutant S163R expression in the RPE (Fig. 8). Full field ERG analysis was performed under scotopic dark-adapted, and photopic light adapted cone-mediated conditions to evaluate the effects of S163R protein on retinal function (Fig. 8A). There was a small but significant decrease in b-wave amplitudes in treated versus untreated mouse eyes by 2 months post injection under scotopic conditions at all light-intensities (Fig. 8A, left panel). For example, AAV-injected eyes had a rod-isolated b-wave amplitude (at 0.02 scot cd·s·m−2) of 238.5 ± 19.46 μV, which was 20% lower than the untreated controls, with a maximum b-wave amplitude average of 296.5 ± 26.12 μV (Average ± SEM, n = 13, P = 0.001). At 5 months post injection (Fig. 8A, right panel), the maximum ERG b-wave amplitudes reflecting the rod-mediated function (at 0.02 scot cd·s·m−2 flashes) further declined, being approximately 30% lower relative to control eyes (average of 134.3 ± 14.59 μV in AAV-treated versus 186 ± 9.02 μV in contralateral eyes, n = 5, P = 0.036). Maximum cone b-wave amplitudes recorded under photopic conditions were similar in treated and untreated eyes. Similar to other cases of RPE-based retinal degeneration, such as the rd6 model caused by lack of functional MFRP, cone-driven ERG amplitudes are initially less affected compared with that of rods, perhaps because cones can also rely on an alternate visual cycle using Müller cells.36,37
Figure 8.
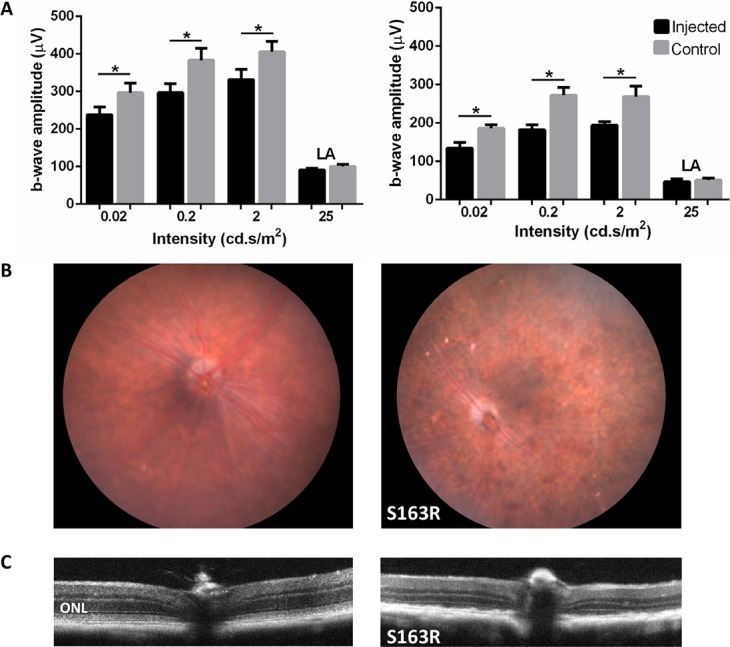
Evaluation of pathological effects following AAV-hC1QTNF5(S163R)–HA delivery using noninvasive approaches. (A) Comparison of retinal function in AAV-S163R-C1QTNF5–treated and untreated eyes following ERG analysis. Bar graphs show the maximum ERG b-wave amplitudes in AAV-S163R injected eyes (black) compared with untreated control eyes (gray) at 2 months (left panel) and 5 months (right panel) post injection. Maximum b-wave amplitudes in AAV-S163R injected eyes were significantly lower than untreated controls at all light intensities under scotopic conditions (*P < 0.05), and were similar under light-adapted conditions. (B) Fundus photographs show pigmentary abnormalities in AAV-S163R–injected eyes (right) in contrast to the normal untreated age-matched control (left). (C) Evaluation of AAV-S163R–treated eyes by OCT imaging. The OCT scan intersecting the optic nerve head shows the normal retinal laminar architecture in the untreated eye (left). The ONL thickness was markedly reduced in the AAV-S163R–treated eye (right) at 9 months post injection, with numerous irregular deposits visible.
Pathological changes in vector-treated eyes were also seen by fundoscopy and OCT (Figs. 8B, 8C). Eyes treated with AAV-S163R display an abnormal fundus with disperse pigmentary changes (Fig. 8B, right panel). By imaging with OCT, retinal thinning was seen in treated eyes by 9 months post injection, consistent with a loss of photoreceptor cells relative to untreated partner retinas (Fig. 8C). Comparison of retinal thicknesses showed that AAV-S163R treated eyes were 0.137 ± 0.021 mm thick, while untreated contralateral controls were 0.206 ± 0.014 mm (average ± SEM, n = 4, P = 0.01).
Discussion
Although L-ORD is a rare inherited disorder, some of its clinical features resemble AMD, a very common retinal degenerative disease that causes progressive loss of central vision that represents a major cause of blindness in the elderly.2,38,39 Specifically, both L-ORD and AMD exhibit age-related formation of drusen between the RPE and Bruch's membrane, a key predictor of eventual AMD.6 In spite of the identification of numerous mouse models that exhibit some aspects of AMD, none mimic the disease accurately due to the lack of an anatomical macula in rodents and the complex nature of the disease, which results from an incompletely understood interplay among multiple genetic and environmental factors.39–42 One feature that makes L-ORD a potentially informative model for AMD is its well-established monogenic etiology that leads to retinal pathology due to sub-RPE deposits that extend into the peripheral retina, causing widespread photoreceptor cell loss and RPE atrophy.6,7 Consequently, mouse models of L-ORD represent a novel platform for developing treatment strategies that may apply to key aspects of AMD as well.
Of the two knock-in mouse models of L-ORD generated to date, only one was found to display an ocular phenotype reflecting many of the early-stage characteristics of the disease, with the other model showing no abnormalities throughout its lifespan.43,44 Recombinant AAV vectors have several advantages over transgenic approaches, including the potential for quickly generating animal models, often with a faster time course of disease progression, controlling mutant protein expression using the appropriate vector serotype, promoter and titer, and producing graded levels of pathology, from early to late-stage disease phenotypes.45 Adeno-associated viral technology, combined with the use of an RPE-specific VMD2 (bestrophin) promoter, enabled us to examine the in vivo consequences of S163R C1QTNF5 mutant expression in wild-type mice. Our results show that S163R accumulates as intracellular aggregates within RPE cells, and gradually forms thick and widespread deposits on the basal side of the RPE. Interestingly, the S163R basal deposits are strikingly similar to those described in Efemp1 knock-in mice harboring the R345W mutation in the EFEMP1 protein, also known as fibulin-3.46,47 Efemp1 knock-in mice are a model for Malattia leventinese, also known as Doyne honeycomb retinal dystrophy, an autosomal dominant inherited macular degenerative disorder that shares many similarities with AMD.48,49 Less prominent basal laminar deposits were also observed by electron microscopic evaluation in a previously characterized knock-in Ctrp5+/− mouse model of L-ORD carrying the S163R mutation.43 The consequences of S163R C1QTNF5 overexpression were also examined in a previously described experiment using a lentiviral system.44 In that study, both photoreceptors and RPE appeared morphologically normal at 5 weeks following subretinal delivery in C57Bl6 mice. In addition to using a different delivery vector and promoter for transgene expression, most significantly, the postinjection analysis time was much longer in our current study, with the phenotypic effects becoming readily apparent by 4 months post injection. Similarly to the lentivirus study, we did not see apparent changes at earlier time points by morphologic examination of the eyes using light microscopy, supporting the idea that the phenotype is moderately slow to develop.
Expressed AAV wild-type C1QTNF5 displays a directional secretion from RPE, moving apically toward photoreceptor inner segments, and reaching the outer limiting membrane, a pattern similar to that of the endogenous protein.17,19 Its interaction with MFRP and its polarized secretion toward the photoreceptor layer suggests that wild-type C1QTNF5 plays a significant role on the apical side of the RPE. Consistent with this view, S163R aggregates clearly have a detrimental impact on the apical RPE membrane, as seen by RPE microvilli being absent or sparse in some regions of AAV-hC1QTNF5(S163R)–HA treated eyes. The accumulation of mutant protein intracellular globules with time and the abnormal distribution of S163R toward the basal side of the RPE, suggest its presence could interfere with not only the transport of nutrients—including oxygen and vitamin A from the choroid to the photoreceptor layer—but also secretion of waste materials from the RPE, thus causing degeneration of both RPE and photoreceptor cells.7 Previous studies demonstrated that sub-RPE deposits from L-ORD patients are rich in oil red O-positive lipids, protein, and calcium.7,13 However, we did not detect the presence of lipids following staining of cryosections from AAV-S163R–injected mouse eyes, suggesting that the pathogenesis of L-ORD is incompletely recapitulated in this AAV-based mouse model.
Many factors that alter the stratified extracellular matrix at the RPE/choroid interface could lead to drusen formation.50–52 Critically, C1QTNF5 interacts with complement factor H, a secreted glycoprotein that functions as a negative regulator of complement activation.53 Single-nucleotide polymorphisms in genes encoding CFH and related complement proteins have been associated with an increased risk of developing AMD.54–57 In addition, other systemic factors may also contribute to the disease pathology in patients, as C1QTNF5 was found to circulate in the human sera with broad interindividual variation.20 C1QTNF5 has a collagen domain (residues 30–98) containing 23 uninterrupted Gly-X-Y repeats,21 and this is perhaps the reason why both S163R aggregates and basal deposits appear pink when stained with hematoxylin and eosin, similar to other collagen-containing structures. It is interesting to note that in L-ORD patients collagen deposits are detected within the inner part of the thick drusen accumulating between the basolateral side of the RPE and the Bruch's membrane,7 suggesting that S163R deposits could be present at those sites in patients and act as a stimulus for drusen formation. By extension therefore, AAV-expressed S163R mutant, being misdirected toward the basal side of the RPE, may alter its normal binding to CFH, triggering complement activation over time, and thus accelerating the formation of the drusen seen in patients.58 Protein aggregation is considered a pathological trigger in many neurodegenerative disorders, including Alzheimer, Parkinson, and Huntington diseases.59,60 Future experiments aimed at enhancing the autophagic pathway will determine if the disease progression can be slowed by increasing the ability of the RPE to degrade mutant S163R efficiently.61 Another approach is the use of small interfering RNAs to inhibit mutant protein expression.62
In conclusion, we show that RPE-targeted expression of the S163R mutant of C1QTNF5 can generate a mouse model that displays pathological features similar to some of those found in patients with advanced L-ORD, including RPE thinning, RPE cell loss, and retinal degeneration. S163R mutant protein forms large, transparent, spherical aggregates throughout the RPE, and thick basal deposits, in contrast to the wild-type protein which is secreted in the opposite direction, toward the apical microvilli and photoreceptor cells. The mechanism leading to the formation of S163R basal deposits remains to be elucidated, but promises to yield valuable clues as to how to treat L-ORD and perhaps some aspects of AMD.
Supplementary Material
Acknowledgments
Supported by NIH Grants EY021721 and EY018331, and by Grants from Foundation Fighting Blindness, Research to Prevent Blindness, Inc., and Macula Vision Research Foundation.
Disclosure: A. Dinculescu, None; S.-H. Min, None; F.M. Dyka, None; W.-T. Deng, None; R.M. Stupay, None; V. Chiodo, None; W.C. Smith, None; W.W. Hauswirth, AGTC, Inc. (I, C)
References
- 1. Booij JC,, Baas DC,, Beisekeeva J,, Gorgels TG,, Bergen AA. The dynamic nature of Bruch's membrane. Prog Retin Eye Res. 2010. ; 29: 1–18. [DOI] [PubMed] [Google Scholar]
- 2. Lim LS,, Mitchell P,, Seddon JM,, Holz FG,, Wong TY. Age-related macular degeneration. Lancet. 2012. ; 379: 1728–1738. [DOI] [PubMed] [Google Scholar]
- 3. Bhutto I,, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch's membrane/choriocapillaris complex. Mol Aspects Med. 2012. ; 33: 295–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kay P,, Yang YC,, Paraoan L. Directional protein secretion by the retinal pigment epithelium: roles in retinal health and the development of age-related macular degeneration. J Cell Mol Med. 2013. ; 17: 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sparrow JR,, Hicks D,, Hamel CP. The retinal pigment epithelium in health and disease. Curr Mol Med. 2010. ; 10: 802–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hayward C,, Shu X,, Cideciyan AV,, et al. Mutation in a short-chain collagen gene, CTRP5, results in extracellular deposit formation in late-onset retinal degeneration: a genetic model for age-related macular degeneration. Hum Mol Genet. 2003. ; 12: 2657–2667. [DOI] [PubMed] [Google Scholar]
- 7. Kuntz CA,, Jacobson SG,, Cideciyan AV,, et al. Sub-retinal pigment epithelial deposits in a dominant late-onset retinal degeneration. Invest Ophthalmol Vis Sci. 1996. ; 37: 1772–1782. [PubMed] [Google Scholar]
- 8. Vincent A,, Munier FL,, Vandenhoven CC,, Wright T,, Westall CA,, Heon E. The characterization of retinal phenotype in a family with C1QTNF5-related late-onset retinal degeneration. Retina. 2012. ; 32: 1643–1651. [DOI] [PubMed] [Google Scholar]
- 9. Soumplis V,, Sergouniotis PI,, Robson AG,, et al. Phenotypic findings in C1QTNF5 retinopathy (late-onset retinal degeneration). Acta Ophthalmol. 2013; 91: e191–e195. [DOI] [PubMed] [Google Scholar]
- 10. Borooah S,, Collins C,, Wright A,, Dhillon B. Late-onset retinal macular degeneration: clinical insights into an inherited retinal degeneration. Br J Ophthalmol. 2009. ; 93: 284–289. [DOI] [PubMed] [Google Scholar]
- 11. Ayyagari R,, Mandal MN,, Karoukis AJ,, et al. Late-onset macular degeneration and long anterior lens zonules result from a CTRP5 gene mutation. Invest Ophthalmol Vis Sci. 2005. ; 46: 3363–3371. [DOI] [PubMed] [Google Scholar]
- 12. Subrayan V,, Morris B,, Armbrecht AM,, Wright AF,, Dhillon B. Long anterior lens zonules in late-onset retinal degeneration (L-ORD). Am J Ophthalmol. 2005. ; 140: 1127–1129. [DOI] [PubMed] [Google Scholar]
- 13. Milam AH,, Curcio CA,, Cideciyan AV,, et al. Dominant late-onset retinal degeneration with regional variation of sub-retinal pigment epithelium deposits, retinal function, and photoreceptor degeneration. Ophthalmology. 2000. ; 107: 2256–2266. [DOI] [PubMed] [Google Scholar]
- 14. Jacobson SG,, Cideciyan AV,, Sumaroka A,, Roman AJ,, Wright AF. Late-onset retinal degeneration caused by C1QTNF5 mutation: sub-retinal pigment epithelium deposits and visual consequences. JAMA Ophthalmol. 2014; 132: 1252–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seldin MM,, Tan SY,, Wong GW. Metabolic function of the CTRP family of hormones. Rev Endocr Metab Disord. 2014. ; 15: 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schaffler A,, Buechler C. CTRP family: linking immunity to metabolism. Trends Endocrinol Metab. 2012. ; 23: 194–204. [DOI] [PubMed] [Google Scholar]
- 17. Mandal MN,, Vasireddy V,, Reddy GB,, et al. CTRP5 is a membrane-associated and secretory protein in the RPE and ciliary body and the S163R mutation of CTRP5 impairs its secretion. Invest Ophthalmol Vis Sci. 2006. ; 47: 5505–5513. [DOI] [PubMed] [Google Scholar]
- 18. Mandal MN,, Vasireddy V,, Jablonski MM,, et al. Spatial and temporal expression of MFRP and its interaction with CTRP5. Invest Ophthalmol Vis Sci. 2006. ; 47: 5514–5521. [DOI] [PubMed] [Google Scholar]
- 19. Won J,, Smith RS,, Peachey NS,, et al. Membrane frizzled-related protein is necessary for the normal development and maintenance of photoreceptor outer segments. Vis Neurosci. 2008. ; 25: 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schmid A,, Kopp A,, Aslanidis C,, Wabitsch M,, Muller M,, Schaffler A. Regulation and function of C1Q/TNF-related protein-5 (CTRP-5) in the context of adipocyte biology. Exp Clin Endocrinol Diabetes. 2013. ; 121: 310–317. [DOI] [PubMed] [Google Scholar]
- 21. Tu X,, Palczewski K. Crystal structure of the globular domain of C1QTNF5: implications for late-onset retinal macular degeneration. J Struct Biol. 2012. ; 180: 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tu X,, Palczewski K. The macular degeneration-linked C1QTNF5 (S163) mutation causes higher-order structural rearrangements. J Struct Biol. 2014. ; 186: 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shu X,, Tulloch B,, Lennon A,, et al. Disease mechanisms in late-onset retinal macular degeneration associated with mutation in C1QTNF5. Hum Mol Genet. 2006. ; 15: 1680–1689. [DOI] [PubMed] [Google Scholar]
- 24. Kameya S,, Hawes NL,, Chang B,, Heckenlively JR,, Naggert JK,, Nishina PM. Mfrp a gene encoding a frizzled related protein, is mutated in the mouse retinal degeneration 6. Hum Mol Genet. 2002. ; 11: 1879–1886. [DOI] [PubMed] [Google Scholar]
- 25. Ayala-Ramirez R,, Graue-Wiechers F,, Robredo V,, Amato-Almanza M,, Horta-Diez I,, Zenteno JC. A new autosomal recessive syndrome consisting of posterior microphthalmos retinitis pigmentosa, foveoschisis, and optic disc drusen is caused by a MFRP gene mutation. Mol Vis. 2006. ; 12: 1483–1489. [PubMed] [Google Scholar]
- 26. Mukhopadhyay R,, Sergouniotis PI,, Mackay DS,, et al. A detailed phenotypic assessment of individuals affected by MFRP-related oculopathy. Mol Vis. 2010. ; 16: 540–548. [PMC free article] [PubMed] [Google Scholar]
- 27. Zenteno JC,, Buentello-Volante B,, Quiroz-Gonzalez MA,, Quiroz-Reyes MA. Compound heterozygosity for a novel and a recurrent MFRP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis. 2009. ; 15: 1794–1798. [PMC free article] [PubMed] [Google Scholar]
- 28. Dinculescu A,, Estreicher J,, Zenteno JC,, et al. Gene therapy for retinitis pigmentosa caused by MFRP mutations: human phenotype and preliminary proof of concept. Hum Gene Ther. 2012. ; 23: 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guziewicz KE,, Zangerl B,, Komaromy AM,, et al. Recombinant AAV-mediated BEST1 transfer to the retinal pigment epithelium: analysis of serotype-dependent retinal effects. PLoS One. 2013. ; 8: e75666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Petrs-Silva H,, Dinculescu A,, Li Q,, et al. Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol Ther. 2011. ; 19: 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jacobson SG,, Acland GM,, Aguirre GD,, et al. Safety of recombinant adeno-associated virus type 2-RPE65 vector delivered by ocular subretinal injection. Mol Ther. 2006. ; 13: 1074–1084. [DOI] [PubMed] [Google Scholar]
- 32. Pang JJ,, Boye SL,, Kumar A,, et al. AAV-mediated gene therapy for retinal degeneration in the rd10 mouse containing a recessive PDEbeta mutation. Invest Ophthalmol Vis Sci. 2008. ; 49: 4278–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adamus G,, Zam ZS,, Arendt A,, Palczewski K,, McDowell JH,, Hargrave PA. Anti-rhodopsin monoclonal antibodies of defined specificity: characterization and application. Vision Res. 1991. ; 31: 17–31. [DOI] [PubMed] [Google Scholar]
- 34. Pi L,, Robinson PM,, Jorgensen M,, et al. Connective tissue growth factor and integrin αvβ6: a new pair of regulators critical for ductular reaction and biliary fibrosis in mice. Hepatology. 2015. ; 61: 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mao H,, Seo SJ,, Biswal MR,, et al. Mitochondrial oxidative stress in the retinal pigment epithelium leads to localized retinal degeneration. Invest Ophthalmol Vis Sci. 2014. ; 55: 4613–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Engelhardt M,, Tosha C,, Lopes VS,, et al. Functional and morphological analysis of the subretinal injection of retinal pigment epithelium cells. Vis Neurosci. 2012. ; 29: 83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mata NL,, Radu RA,, Clemmons RC,, Travis GH. Isomerization and oxidation of vitamin a in cone-dominant retinas: a novel pathway for visual-pigment regeneration in daylight. Neuron. 2002. ; 36: 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Friedman DS,, O'Colmain BJ,, Munoz B,, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004. ; 122: 564–572. [DOI] [PubMed] [Google Scholar]
- 39. Fritsche LG,, Fariss RN,, Stambolian D,, Abecasis GR,, Curcio CA,, Swaroop A. Age-related macular degeneration: genetics and biology coming together. Annu Rev Genomics Hum Genet. 2014. ; 15: 151–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ambati J,, Fowler BJ. Mechanisms of age-related macular degeneration. Neuron. 2012. ; 75: 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fletcher EL,, Jobling AI,, Greferath U,, et al. Studying age-related macular degeneration using animal models. Optom Vis Sci. 2014. ; 91: 878–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Edwards AO,, Malek G. Molecular genetics of AMD and current animal models. Angiogenesis. 2007. ; 10: 119–132. [DOI] [PubMed] [Google Scholar]
- 43. Chavali VR,, Khan NW,, Cukras CA,, Bartsch DU,, Jablonski MM,, Ayyagari RA. CTRP5 gene S163R mutation knock-in mouse model for late-onset retinal degeneration. Hum Mol Genet. 2011. ; 20: 2000–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shu X,, Luhmann UF,, Aleman TS,, et al. Characterisation of a C1qtnf5 Ser163Arg knock-in mouse model of late-onset retinal macular degeneration. PLoS One. 2011. ; 6: e27433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Klein RL,, Dayton RD,, Tatom JB,, Diaczynsky CG,, Salvatore MF. Tau expression levels from various adeno-associated virus vector serotypes produce graded neurodegenerative disease states. Eur J Neurosci. 2008. ; 27: 1615–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fu L,, Garland D,, Yang Z,, et al. The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum Mol Genet. 2007. ; 16: 2411–2422. [DOI] [PubMed] [Google Scholar]
- 47. Marmorstein LY,, McLaughlin PJ,, Peachey NS,, Sasaki T,, Marmorstein AD. Formation and progression of sub-retinal pigment epithelium deposits in Efemp1 mutation knock-in mice: a model for the early pathogenic course of macular degeneration. Hum Mol Genet. 2007. ; 16: 2423–2432. [DOI] [PubMed] [Google Scholar]
- 48. Marmorstein LY,, Munier FL,, Arsenijevic Y,, et al. Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002. ; 99: 13067–13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stone EM,, Lotery AJ,, Munier FL,, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999. ; 22: 199–202. [DOI] [PubMed] [Google Scholar]
- 50. Mullins RF,, Russell SR,, Anderson DH,, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000. ; 14: 835–846. [PubMed] [Google Scholar]
- 51. Anderson DH,, Mullins RF,, Hageman GS,, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002. ; 134: 411–431. [DOI] [PubMed] [Google Scholar]
- 52. Gehrs KM,, Jackson JR,, Brown EN,, Allikmets R,, Hageman GS. Complement age-related macular degeneration and a vision of the future. Arch Ophthalmol. 2010. ; 128: 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shu XH,, Clark SJ,, Dodds AW,, et al. C1QTNF5, which is mutated in late-onset retinal macular degeneration, interacts with complement factor H. Mol Immunol. 2007. ; 44: 240–240. [Google Scholar]
- 54. Klein RJ,, Zeiss C,, Chew EY,, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005. ; 308: 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hageman GS,, Anderson DH,, Johnson LV,, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005. ; 102: 7227–7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Edwards AO,, Ritter R,, III,, Abel KJ,, Manning A,, Panhuysen C,, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005; 308: 421–424. [DOI] [PubMed] [Google Scholar]
- 57. Gold B,, Merriam JE,, Zernant J,, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006. ; 38: 458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scholl HP,, Charbel Issa P,, Walier M,, et al. Systemic complement activation in age-related macular degeneration. PLoS One. 2008. ; 3: e2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Irvine GB,, El-Agnaf OM,, Shankar GM,, Walsh DM. Protein aggregation in the brain: the molecular basis for Alzheimer's and Parkinson's diseases. Mol Med. 2008. ; 14: 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Weiss KR,, Kimura Y,, Lee WC,, Littleton JT. Huntingtin aggregation kinetics and their pathological role in a Drosophila Huntington's disease model. Genetics. 2012. ; 190: 581–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kaarniranta K,, Sinha D,, Blasiak J,, et al. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy. 2013. ; 9: 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chavali VR,, Vasireddy V,, Ayyagari R. Silencing the expression of CTRP5/C1QTNF5 and ELOVL4 genes by small interfering RNA. Adv Exp Med Biol. 2012. ; 723: 225–233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.