

Abstract
IMPORTANCE
For patients with X-linked retinitis pigmentosa and clinicians alike, phenotypic variability can be challenging because it complicates counseling regarding patients’ likely visual prognosis.
OBJECTIVE
To evaluate the clinical findings from patients with X-linked retinitis pigmentosa with 13 distinct RPGR mutations and assess for phenotypic concordance or variability.
DESIGN
Retrospective medical record review of data collected from 1985 to 2011.
SETTING
Kellogg Eye Center, University of Michigan.
PATIENTS
A total of 42 patients with X-linked retinitis pigmentosa with mutations in RPGR. Age at first visit ranged from 4 to 53 years, with follow-up ranging from 1 to 11 visits (median follow-up time, 5.5 years; range, 1.4-32.7 years, for 23 patients with >1 visit).
MAIN OUTCOMES AND MEASURES
Clinical data assessed for concordance included visual acuity (VA), Goldmann visual fields (GVFs), and full-field electroretinography (ERG). Electroretinography phenotype (cone-rod vs rod-cone dysfunction) was defined by the extent of photopic vs scotopic abnormality. Qualitative GVF phenotype was determined by the GVF pattern, where central or peripheral loss suggested cone or rod dysfunction, respectively. Goldmann visual fields were also quantified and compared among patients.
RESULTS
Each mutation was detected in 2 or more related or unrelated patients. Five mutations in 11 patients displayed strong concordance of VA, while 4 mutations in 16 patients revealed moderate concordance of VA. A definitive cone-rod or rod-cone ERG pattern consistent among patients was found in 6 of 13 mutations (46.2%); the remaining mutations were characterized by patients demonstrating both phenotypes or who had limited data or nonrecordable ERG values. Concordant GVF phenotypes (7 rod-cone pattern vs 4 cone-rod pattern) were seen in 11 of 13 mutations (84.6%). All 6 mutations displaying a constant ERG pattern within the mutation group revealed a GVF phenotype consistent with the ERG findings.
CONCLUSIONS AND RELEVANCE
While VA and ERG phenotypes are concordant in only some patients carrying identical mutations, assessment of GVF phenotypes revealed stronger phenotypic conservation. Phenotypic concordance is important for establishing proper counseling of patients diagnosed as having X-linked retinitis pigmentosa, as well as for establishing accurate patient selection and efficacy monitoring in therapeutic trials.
Mutations in the retinitis pigmentosa GTPase regulator gene (RPGR) and RP2 are responsible for most cases of X-linked retinitis pigmentosa (XLRP), which is generally more severe than retinal diseases involving other patterns of inheritance.1,2 Disease from RPGR mutations is characterized by significant phenotypic variability. Studies into the etiology of this variability have been complicated by the elusive physiological function of RPGR,3-9 as well as multiple isoforms resulting from alternative splicing.10 Most phenotype studies have focused on comparing disease severity between mutations in the 2 predominant retinal RPGR isoforms3,11-14 and suggested that mutations in exon open reading frame 15 (ORF15) exhibit lesser disease severity compared with those in exons 1-14.12,13 In a study of 156 patients with RPGR mutations, Sharon et al13 used multiple regression analysis to illustrate that patients with ORF15 mutations tended to have larger visual field areas and higher electroretinographic (ERG) amplitudes at comparable ages than patients with mutations in exons 1-14. Fahim et al12 recently reported increased phenotypic variability and reduced severity in a study of 98 patients with RPGR-ORF15 mutations.
While mutations in different regions of the same gene can differentially affect the ultimate protein outcome and result in different phenotypes,6,14 it is more difficult to explain variability among patients with the same mutation.15-19 The most dramatic example of this was demonstrated in a case report of dizygotic twins with an ORF15 mutation.20 One patient had a clear cone-rod dysfunction, color vision deficits, macular atrophy, minimal peripheral constriction, and a cone-rod pattern on ERG, while his sibling exhibited foveal sparing, severe peripheral field loss, and a nonrecordable (NR) ERG. Furthermore, Fahim et al reported phenotypic variability in large families with the same mutations.
For patients and clinicians alike, phenotypic variability can be challenging because it complicates counseling of patients regarding their likely visual prognosis. Furthermore, as new therapies are developed, variability in clinical course makes patient selection for clinical trials more challenging and clouds the assessment of treatment outcomes. In this study, we investigated phenotypic variability in a cohort of 42 patients with 13 distinct RPGR mutations by assessing different visual function parameters.
Methods
Mutation Screening
DNA from lymphocytes of patients was extracted using Qiagen blood DNA purification kits (Qiagen). For RPGR mutational screening, exons were amplified using polymerase chain reaction using published primers.21,22 All mutations were originally identified in our laboratory with the exception of exon 2-3 deletion, which was first reported by another laboratory and then confirmed by our laboratory (for polymerase chain reaction conditions, see the eFigure in Supplement).
Patient Selection and Phenotype Analysis
A retrospective review was performed of 42 male patients with XLRP carrying mutations in RPGR who were seen at the Kellogg Eye Center, University of Michigan. All patients provided informed consent and the study was approved by the University of Michigan institutional review board. Medical records were reviewed for the following clinical features: visual acuity (VA), funduscopic appearance on color fundus photographs and in the medical record, standardized full-field ERG data, and Goldmann visual field (GVF) data. Clinical findings were recorded for each patient visit; however, not all outcome measures were available at all patient visits. Patients with the same mutation were grouped together, resulting in groups representing 13 distinct mutations.
Concordance in this study was defined as the similarity of clinical ophthalmic parameters between patients at comparable age. By assessing concordance among patients with the same mutation, we evaluated whether a certain mutation resulted in a similar phenotype in all patients.
Visual Acuity Concordance
Snellen VA values were converted to a logMAR scale before an average was obtained for both eyes. Because this was a retrospective study, the best available VA was recorded (whether correction or pinhole was used). Three brackets reflecting severity of VA were created as follows: 20/20 to 20/70 (relatively good acuity), 20/80 to 20/200 (moderate to limited acuity), and less than 20/200 (limited acuity). We compared the VA of patients in each mutation group who were closest in age and less than 7 years apart from each other. We tallied the percentage of age groupings within each mutation group that exhibited the same VA severity category. Based on this analysis, mutations were described as exhibiting strong (75%-100%), moderate (26%-74%), or low (0%-25%) Snellen VA concordance.
Because of possible testing unreliability in young patients (those who improved in VA with age), we excluded from this analysis VA measurements from patients younger than the age of 5 years and visits that occurred after the initiation of experimental treatments (eg, ciliary neurotrophic factor trial). We were unable to analyze 2 mutations for VA concordance because the available data were from patients who were greater than 7 years apart in age at visits. In addition to using average VA, this analysis was done by selecting all right or all left eyes.
Visual Acuity Progression
Progression of disease (worsening of vision) was assessed by plotting logMAR VA (average of 2 eyes) vs age at every visit, and best-fit lines were generated by least-squares regression analysis for each mutation group. Visual acuity measured from individuals younger than 5 years and those occurring after initiation on an experimental treatment trial were not included. Overall VA deterioration for all patients in our cohort was assessed by least-squares and polynomial regression analyses.
Full-Field Electroretinography Concordance
Abnormal photopic and scotopic B-wave amplitudes were expressed as the percentage of the respective mean values de rived from control subjects. Patients whose scotopic deficiency was more severe (defined as a lower percentage of mean) were considered to have rod-cone dysfunction, while patients with a worse photopic deficiency were categorized as having cone-rod dysfunction.23 In addition, ERG phenotype was confirmed by assessment of the relative abnormalities in photopic vs scotopic B-wave implicit times by determining the percentage increase over 2 standard deviations above normal mean. Each mutation group was assessed for concordance, with concordance defined as consistent ERG phenotypes between patients in a group. We defined patients younger than 10 years of age with NR scotopic and photopic parameters as having a rod-cone phenotype based on our clinical experience. Older patients with NR ERG parameters were left unclassified because it is difficult to determine the ERG pheno-type in this situation. If a mutation group contained multiple older patients with NR ERG values, the mutation group was not assessed for concordance.
Goldmann Visual Field Concordance
Patients were grouped into specific GVF phenotypes based on each patient's pattern of visual field loss. Central loss over time with relative preservation of peripheral vision indicated a cone-rod GVF phenotype; peripheral visual field loss with the smaller target isopters showing greater peripheral constriction suggested a rod-cone phenotype (Figure 1). A tight ring scotoma with absence of smaller isopters centrally was interpreted as a cone-rod GVF phenotype.23 Concordance of GVF pheno-type was assessed within each mutation group and compared with the respective ERG phenotype, if available. Total planimetric areas for the I4e isopter were quantified in Adobe Photoshop CS3 in square degrees and square millimeters for all patients. We did not correct for cartographic distortion, given that, to our knowledge, most previous longitudinal studies of GVF progression have not included such corrections. Quantified areas were compared between patients with the same mutation who were fewer than 6 years apart in age and exhibited the same qualitative GVF phenotype.
Figure 1. Examples of Goldmann Visual Field (GVF) Phenotypes and Phenotypic Concordance and Discordance.

Illustrations show an example of discordant GVF phenotypes in 2 cousins with the same mutation (p.Glu746fs) (A, cone-rod phenotype and B, rod-cone phenotype). The rod-cone phenotypes show an example of GVF phenotypic concordance in 2 brothers with the same mutation (exon 2-3 deletion) (C and D). Isopters shown in this figure include IV4e (red), III4e (green), and I4e (black).
Results
Patient Demographic Data and Mutational Findings
For all 42 patients, age at first visit ranged from 4 to 53 years, with follow-up ranging from 1 to 11 visits (median follow-up time, 5.5 years; range, 1.4-32.7 years, for 23 patients with >1 visit). Mutations are numbered from 1 to 13 as shown; 10 mutations occurred in RPGR-ORF15 and 3 in RPGR–exons 1-14 (Figure 2 and Table 1). Each mutation was identified in 2 or more related or unrelated patients. Three mutations were novel (exon 2-3 deletion, p.Glu922fs, and p.Glu1014fs), while the others had been previously reported.13,22,24-29 Individuals included for comparison in 8 of the mutations were related, while the remaining mutations were identified in at least 2 families.
Figure 2. Graphical Representation of Mutations in RPGR Identified in Our Cohort.
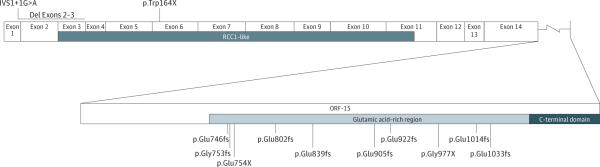
Ten of the identified mutations were located in ORF15. RCC1 indicates regulator of chromosome condensation 1.
Table 1.
Characteristics of RPGR Mutations and VA Concordance in a Mutation Group
| Patient No. | Nucleotide | Mutationa | Exon | Domain | No. of Patients (n = 42) | No. of Families (n = 24) | VA Concordanceb |
|---|---|---|---|---|---|---|---|
| 1 | c.28 + 1 G>A | Splice24 | 1 | NA | 2 | 1 | Indeterminate |
| 2 | Exon 2-3 deletion | 2-3 | RCC1 | 2 | 1 | Strong | |
| 3 | c.492 G>A | p.Trp164X25 | 6 | RCC1 | 4 | 1 | Strong |
| 4 | c.2236_2237 del GA | p.Glu746fs26 | ORF15 | GAR | 4 | 3 | Moderate |
| 5 | c.2257_2260 del GGAG | p.Gly753fs27 | ORF15 | GAR | 4 | 2 | Strong |
| 6 | c.2260 C>T | p.Glu754X28 | ORF15 | GAR | 2 | 1 | Weak |
| 7 | c.2405_2406 del AG | p.Glu802fs28 | ORF15 | GAR | 4 | 4 | Moderate |
| 8 | c.2516_2520 del AAGGG | p.Glu839fs13 | ORF15 | GAR | 7 | 4 | Moderate |
| 9 | c.2714_2715 del AA | p.Glu905fs13 | ORF15 | GAR | 4 | 2 | Weak |
| 10 | c.2764 del G | p.Glu922fs | ORF15 | GAR | 3 | 1 | Strong |
| 11 | c.2929 G>T | p.Glu977X29 | ORF15 | GAR | 2 | 1 | Moderate |
| 12 | c.3039_3040 del GG | p.Glu1014fs | ORF15 | GAR | 2 | 1 | Strong |
| 13 | c.3096_3097 del GG | p.Glu1033fs22 | ORF15 | GAR | 2 | 2 | Indeterminate |
Abbreviations: GAR, glutamic acid rich; NA, not available; ORF15, open reading frame 15; RCC1, regulator of chromosome condensation 1-like; VA, visual acuity.
Previously reported mutations are referenced. Mutations 2, 9, and 11 are novel.
Visual acuity concordance of each mutation using mean logMAR VA at each visit.
Visual Acuity Concordance
Five distinct mutations in 11 patients from 5 families displayed strong concordance of VA with age (Table 1). Four mutations in 16 patients from 11 families demonstrated moderate concordance of VA. Two mutations in 6 patients from 3 families exhibited low concordance, while the remaining 2 mutations could not be assessed for concordance because of a large difference in age of the patients. These findings are summarized in Table 1. Similar results were obtained if all right or all left eyes were selected for this analysis.
Visual Acuity Deterioration
Visual acuity data from each visit were plotted with respect to age for 40 patients and showed a tendency to worsen with age (best-fit slope: 0.015 logMAR units/y; R2 = 0.48 with least-squares regression and R2 = 0.55 with polynomial regression; Figure 3A and B, respectively). The R2 values of these analyses did not improve with setting a floor of analysis at higher age (eg, >20 years) to account for a possible latent phase of disease. Individually, 10 mutations appeared to worsen with age using least-squares regression analysis (Table 2). Whether considered individually or together, lines of best fit for most mutations exhibited low R2 values. Mutations 5, 7, and 10 revealed higher R2 values compared with other mutations. Three mutations (p.Glu905fs, p.Glu922fs, and p.Glu1014fs) exhibited negative sloping lines for VA progression, suggesting an improvement in VA with age. However, these 3 mutations included patients with visits at younger than 10 years of age.
Figure 3. Overall Longitudinal LogMAR Visual Acuity (VA) Deterioration.
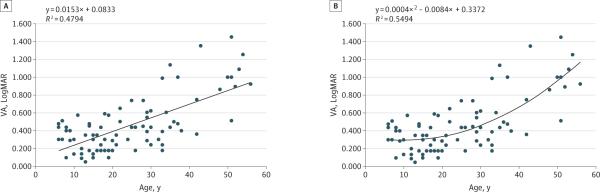
Average logMAR VA from all visits from 40 patients was plotted against age, which showed that VA tended to worsen with age. A, Least-squares regression analysis with a slope reflecting VA deterioration with time (in logMAR units/y) and corresponding R2 coefficient. B, Polynomial regression analysis with corresponding R2 coefficient. Visual acuity measurements from patients younger than age 5 years or those taken from patients after initiation of experimental therapies were excluded. The R2 values for these calculations did not improve with setting a floor of analysis at higher ages (eg, excluding data points at younger than age 20 years).
Table 2.
Longitudinal Visual Acuity Deterioration by Mutationa
| Mutation | Slope | R 2 |
|---|---|---|
| 1 | 0.0032 | 1.0000 |
| 2 | 0.0091 | 0.1739 |
| 3 | 0.0111 | 0.3602 |
| 4 | 0.0176 | 0.4217 |
| 5 | 0.0175 | 0.7391 |
| 6 | 0.0727 | 0.3370 |
| 7 | 0.0127 | 0.7977 |
| 8 | 0.0015 | 0.0053 |
| 9 | −0.0065 | 0.0923 |
| 10 | −0.0285 | 0.7817 |
| 11 | 0.0619 | 0.6558 |
| 12 | −0.0455 | 0.6942 |
| 13 | 0.0196 | 1.0000 |
| All | 0.0153 | 0.4794 |
LogMAR visual acuity was plotted against age for all patients in each mutation. Slope of visual acuity decline (in logMAR units/y) and an R2 value were calculated for every mutation using least-squares regression analysis and are shown.
Electroretinography Concordance
Electroretinography phenotypes were assigned to all patients with available ERG data (Table 3). Eleven patients displayed rod-cone dysfunction, while 12 exhibited cone-rod dysfunction. Patients with 9 of 13 mutations had adequate ERG data to categorize a definitive phenotype. The remaining had unavailable or NR ERGs, precluding categorization for each mutation as a whole. Of the 9 mutations with definable phenotypes, 6 displayed phenotypic concordance, while 3 did not (Table 3). The 3 that did not exhibit concordance included patients with both cone-rod and rod-cone ERG dysfunction. The p.Glu839fs mutation was identified in a sibling pair (cases 8D and 8E) whose ERG phenotypes were contrasting. Thirteen of 14 patients with adequate implicit time data for phenotypic categorization exhibited the same ERG phenotype whether amplitude or implicit time was compared. Patients with mutations located at the 3′-end of ORF15 all exhibited a cone-rod ERG phenotype.
Table 3.
ERG and GVF Phenotypes of Each Patient and Assessment of Phenotypic Concordance in Each Mutation
| No. (%) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| VA |
OD ERGa |
OS ERGa |
GVF Concordance | |||||||
| Case ID/Age, y | OD | OS | Photopic B-wave | Scotopic B-wave | Photopic B-wave | Scotopic B-wave | ERG Phenotype | ERG-Concordance | GVF Phenotype | |
| 1A/6 | 20/40 | 20/40 | NA | NA | NA | NA | Unknown | Unknown | Rod-cone | Yes |
| 1B/21 | 20/50 | 20/40 | 13 (3.6) | NR | 6 (3.0) | NR | Rod-cone | Rod-cone | ||
| 2A/9 | 20/50 | 20/50 | NR | NR | NR | NR | Rod-cone | Yes | Rod-cone | Yes |
| 2B/21 | 20/30 | 20/40 | NR | NR | NR | NR | Unknown | Rod-cone | ||
| 3A/12 | 20/40 | 20/25 | 28 (16.9) | 32 (11.7) | 13 (7.8) | 38 (13.9) | Rod-cone | Yes | Rod-cone | Yes |
| 3B/15 | 20/40 | 20/40 | 56 (33.7) | 60 (21.9) | 52 (31.3) | 56 (20.4) | Rod-cone | Rod-cone | ||
| 3C/37 | 20/CF | 20/200 | NR | NR | NR | NR | Unknown | Rod-cone | ||
| 3D/51 | 20/70 | 20/60 | NA | NA | NA | NA | Unknown | Unknown | ||
| 4A/24 | 20/40 | 20/40 | NA | NA | NA | NA | Unknown | No | Rod-cone | No |
| 4B/30 | 20/100 | 20/70 | NR | NR | NR | NR | Unknown | Unknown | ||
| 4C/33 | 20/160 | 20/250 | 25 (14.8) | NR | 12 (7.1) | NR | Rod-cone | Rod-cone | ||
| 4D/52 | 20/70 | 20/400 | NR | 80 (21.3) | NR | 100 (26.7) | Cone-rod | Cone-rod | ||
| 5A/7 | 20/60 | 20/70 | NA | 10 (7.3) | NA | NA | Unknown | Unknown | Rod-cone | Yes |
| 5B/33 | 20/40 | 20/30 | NA | NA | NA | NA | Unknown | Rod-cone | ||
| 5C/48 | 20/30 | 20/30 | <5 (3.0) | 30 (10.9) | 20 (12.0) | 30 (10.9) | Rod-cone | Rod-cone | ||
| 5D/48 | 20/300 | 20/70 | NR | NR | NR | NR | Unknown | Rod-cone | ||
| 6A/34 | 20/80 | 20/80 | 15 (9.0) | 35 (12.8) | 19 (11.4) | 21 (7.7) | Cone-rod | Yes | Cone-rod | Yes |
| 6B/42 | 20/200 | 20/100 | NR | 31 (11.3) | NR | 53 (19.3) | Cone-rod | Cone-rod | ||
| 7A/7 | 20/40 | 20/40 | 4.3 (2.5) | 95.3 (25.4) | 13.1 (7.8) | 40.2 (10.7) | Cone-rod | No | Rod-cone | Yes |
| 7B/22 | 20/80 | 20/50 | 8 (4.8) | NR | 8 (4.8) | NR | Rod-cone | Rod-cone | ||
| 7C/29 | 20/80 | 20/80 | NR | NR | NR | NR | Unknown | Rod-cone | ||
| 7D/15 | 20/60 | 20/60 | 9 (5.5) | NR | 14 (8.6) | NR | Rod-cone | Rod-cone | ||
| 8A/7 | 20/40 | 20/40 | 20 (12.0) | 108 (39.4) | 22 (13.3) | 96 (35.0) | Cone-rod | No | Rod-cone | Yes |
| 8B/19 | 20/25 | 20/25 | NA | NA | NA | NA | Unknown | Rod-cone | ||
| 8C/5 | 20/150 | 20/100 | NA | NA | NA | NA | Unknown | Rod-cone | ||
| 8D/11 | 20/80 | 20/70 | 9.2 (5.5) | NR | 12.6 (7.6) | NR | Rod-cone | Rod-cone | ||
| 8E/15 | 20/50 | 20/30 | 10 (6.0) | 60 (21.9) | 10 (6.0) | 60 (29.2) | Cone-rod | Rod-cone | ||
| 8F/28 | 20/63 | 20/50 | NR | NR | NR | NR | Unknown | Rod-cone | ||
| 8G/29 | 20/50 | 20/100 | NR | NR | NR | NR | Unknown | Rod-cone | ||
| 9A/22 | 20/40 | 20/200 | NA | 100 (36.5) | NA | NA | Unknown | Unknown | Cone-rod | No |
| 9B/26 | 20/60 | 20/50 | NR | NR | NR | NR | Unknown | Cone-rod | ||
| 9C/35 | 20/60 | 20/50 | NR | NA | NR | NA | Unknown | Cone-rod | ||
| 9D/6 | 20/60 | 20/60 | 16 (9.6) | NR | 13 (7.8) | NR | Rod-cone | Rod-cone | ||
| 10A/6 | 20/60 | 20/50 | 3.6 (2.2) | NR | <2 (1.2) | NR | Rod-cone | Unknown | Rod-cone | Yes |
| 10B/16 | 20/30 | 20/30 | NA | NA | NA | NA | Unknown | Rod-cone | ||
| 10C/45 | LPO | LPO | NA | NA | NA | NA | Unknown | Unknown | ||
| 11A/26 | 20/30 | 20/30 | 16 (9.6) | 260 (94.9) | 18 (10.8) | 260 (87.5) | Cone-rod | Yes | Cone-rod | Yes |
| 11B/33 | 20/30 | 20/30 | 5 (3.0) | 90 (32.8) | 4 (2.4) | 90 (20.8) | Cone-rod | Cone-rod | ||
| 12A/8 | 20/50 | 20/50 | 13 (7.8) | 82 (29.9) | 19 (11.4) | 92 (33.6) | Cone-rod | Yes | Cone-rod | Yes |
| 12B/10 | 20/30 | 20/25 | 74 (44.6) | 174 (63.5) | 68 (41.0) | 87 (43.8) | Cone-rod | Cone-rod | ||
| 12B/13 | 20/25 | 20/20 | 41 (24.7) | 97 (35.4) | 32 (19.3) | 78 (25.2) | Cone-rod | Cone-rod | ||
| 13A/52 | 20/500 | 20/500 | 6 (3.7) | NR | 9 (5.5) | 21 (7.5) | Cone-rod | Yes | Cone-rod | Yes |
| 13B/35 | 20/300 | 20/250 | 6 (3.7) | 75 (26.8) | 5 (3.1) | 82 (32.8) | Cone-rod | Cone-rod | ||
Abbreviations: CF, count fingers; ERG, electroretinography; GVF, Goldmann visual field; LPO, lightperception only; NA, notavailable; NR, nonrecordable; OD, right eye; OS, left eye; VA, visual acuity.
Mean full-field ERG parameters: 166 for photopic B-wave and 274 for scotopic B-wave.
Goldmann Visual Field Concordance
Visual field phenotypes were determined for all patients with available data (Table 3). Overall, 12 patients (mean, 29.8 years; median, 33.0 years) exhibited a cone-rod pattern of field loss, while 27 (mean, 19.6 years; median, 16.0 years) exhibited a rod-cone pattern. Eleven of 13 mutations (84.6%) displayed concordant GVF phenotypes (7 rod-cone vs 4 cone-rod), while 2 mutations exhibited both GVF phenotypes. All 6 mutations displaying a concordant ERG pattern revealed a GVF phenotype consistent with the ERG. Patients with mutations at the 3′-end of ORF15 consistently exhibited a cone-rod GVF pheno-type. Examples of GVF discordance and concordance are shown in Figure 1. All phenotypic concordance data are summarized in Figure 4. Limited quantified GVF data were available for comparison between patients with the same mutation, given large differences in age. However, patients who had a cone-rod GVF phenotype tended to exhibit larger preserved areas than those with rod-cone phenotypes. Furthermore, patients who exhibited the same qualitative phenotype tended to exhibit similar quantified total areas (Table 4). Selected examples of quantified visual fields are shown in Figure 5.
Figure 4. Summary of Phenotypic Concordance in Visual Acuity (VA), Electroretinography (ERG), and Goldmann Visual Field (GVF) Data.
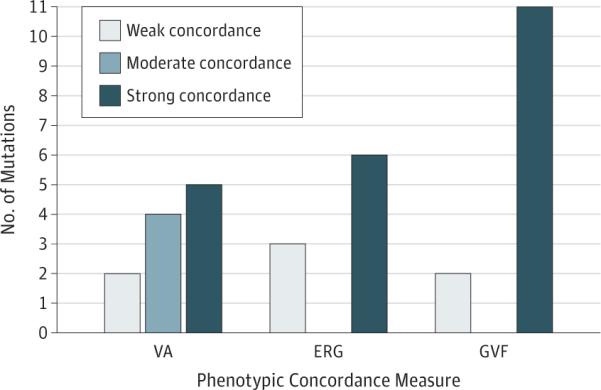
Numbers of mutations exhibiting strong, moderate, or weak concordance for each modality are shown. Phenotypic concordance was highest when assessed by comparing GVF phenotypes between patients.
Table 4.
Comparison of Quantified Goldmann Visual Fields in Patients With the Same RPGR Mutations and the Same Qualitative Phenotypea
| Mutation No. | Patient Age, y | Phenotype | I4e Area, deg2 | I4e Area, mm2 | % Normal I4e Area | % Difference |
|---|---|---|---|---|---|---|
| 2 | 13 | Rod-cone | 98.8 | 142.3 | 0.80 | 0.13 |
| 14 | 83.1 | 119.7 | 0.67 | |||
| 4 | 24 | Rod-cone | 54.3 | 78.2 | 0.44 | 0.02 |
| 30 | 56.8 | 81.8 | 0.46 | |||
| 5 | 24 | Rod-cone | 93.3 | 134.3 | 0.76 | 2.04 |
| 27 | 292.5 | 421.2 | 2.80 | |||
| 5 | 27 | Rod-cone | 292.5 | 421.2 | 2.80 | 0.09 |
| 29 | 354.5 | 510.5 | 2.89 | |||
| 7 | 23 | Cone-rod | 326.9 | 470.8 | 2.66 | 2.47 |
| 29 | 23.8 | 34.3 | 0.19 | |||
| 8 | 19 | Rod-cone | 138.3 | 199.2 | 1.13 | 1.10 |
| 20 | 37.4 | 53.8 | 0.30 | |||
| 12 | 10 | Cone-rod | 4541.6 | 6539.9 | 36.99 | 22.13 |
| 10 | 7258.1 | 10 451.8 | 59.12 | |||
Goldmann visual fields from patients with the same mutations who were within 6 years of age at time of testing were quantified using Adobe Photoshop CS3. Normal total I4e area was determined by calculating the average total I4e areas from 10 normal eyes (12 276 deg2 or 17678.35 mm2) to determine percentage of normal in patients. Only patients with the same mutations who were deemed to exhibit the same qualitative phenotype were compared. Data from all right eyes are shown. These comparisons show that areas are similar in patients who display the same qualitative Goldmann visual field phenotypes and have the same mutations.
Figure 5. Comparison of Quantified Goldmann Visual Fields for Select RPGR Mutations.

Visual fields from patients with the same mutations who were within 6 years of age at time of testing were quantified using Adobe Photoshop CS3. Normal total I4e area was determined by calculating the average total I4e from 10 normal eyes to determine the percentage of normal in patients. A and B represent 2 siblings (exon 2-3 deletion) both exhibiting a rod-cone phenotype. The percentage difference between the 2 I4e areas was 0.13%. C and D also represent 2 siblings with a rod-cone phenotype (p.Gly753fs), whose percentage difference was 0.09%. E and F represent 2 siblings with a mutation near the 3′-end of RPGR (p.Glu1014fs) who both exhibit a cone-rod Goldmann visual field phenotype. Their quantified percentage difference was larger at 22.1%, but both patients displayed relatively large visual fields with the I4e isopter.
Discussion
Genotype-phenotype relationships for XLRP caused by mutations in RPGR present a unique challenge for clinicians because of phenotypic variability reported in patients with the same mutation, the reasons for which are poorly understood. Further studies are needed to elucidate the possible causes, which may include genetic modifiers, epigenetic differences, environmental influences, or random effects. Phenotypic concordance is important to establish before therapeutic trials for appropriate patient selection and given that assessments of treatment efficacy would be more accurate if the expected natural disease progression were understood. Because of the high numbers of patients with XLRP with documented mutations in our retinal dystrophy clinic, we explored this question in 42 patients with 13 distinct mutations. We explored data from multiple patients in each mutation to gain a better understanding of the phenotypic manifestations of each mutation.
Our study provided interesting insights into XLRP phenotypes. It is crucial to determine whether patients exhibit a cone-rod or rod-cone pattern of retinal degeneration because it helps in determining disease prognosis and management. Our finding of strong phenotypic concordance of GVF phenotype in 11 of 13 mutations was striking. Encouragingly, patients also exhibited quantitative concordance when their qualitative phenotype was similar. In contrast to the global reflection of photoreceptor function provided by full-field ERG testing, GVF provides a geographic indication of local photoreceptor dysfunction or degeneration. Given the distribution of cones and rods in the retina,30 patterns of loss facilitate phenotypic categorization of which type of photoreceptor is likely to be more severely affected. Grover et al31 and Heckenlively23 have previously described different patterns of field loss in retinitis pigmentosa. The concordance revealed with this modality suggests that studies assessing phenotypic conservation within mutations would benefit from GVF findings, which are often underappreciated clinically. This is especially important when full-field ERG testing reveals globally depressed cone and rod function, as observed in many of our patients.
Interestingly, we found that all patients with available data in the 3 non-ORF15 mutation groups (which were located proximally in the RPGR gene: splice mutant, exon 2-3 deletion, and p.Trp164X) exhibited a rod-cone pattern of degeneration using both ERG and GVF at a relatively young age. The high level of concordance in VA, ERG, and GVF reflects a severe rod-cone phenotype likely resulting from putative null-protein products in all 3 mutations. Although involving only 8 patients, the fact that the least severe ERG was found in a 15-year-old subject is a strong indication of the necessity of the RPGR protein in retinal function and photoreceptor survival.
Our cohort of patients with mutations in ORF15 was significantly larger. It is important to note that all 10 of these mutations were either frame-shift or nonsense mutations and would likely also prove catastrophic. These 34 patients exhibited much more heterogeneity with respect to VA, ERG, and GVF. Specifically, patients exhibited varying degrees of cone-rod or rod-cone disease severity spanning a broader range of ages. This variability in ORF15 mutations as a whole is similar to past reports.12,13 When evaluating mutations for phenotypic conservation using our analytic framework, all patients in the 3 mutation groups closest to the C-terminal domain (p.Glu977X, p.Glu1014fs, and p.Glu1033fs) consistently exhibited a cone-rod ERG pattern of degeneration, as well as a cone-rod GVF pattern. This finding is consistent with the finding of mutations at the 3′-end of RPGR in X-linked cone-rod dystrophy.22,32-35
Before beginning this study, we accepted multiple caveats of working with retrospective data in assessing pheno typic concordance. For example, we understood the complexities of comparing ophthalmic parameters measured at different times or by different operators, although standard testing methods were used for all patients. Furthermore, studies have shown similar variability in GVF testing.36 Limitations such as these and inadequate/incomplete follow-up and missing data in many patients limited a more complete analysis. We attempted to overcome some of these by analyzing only mutations with adequate data for each analysis, comparing VA from patients who were closest in age, and by excluding visits from patients having interventions and from very young patients with unreliable testing.
Importantly, phenotypic concordance in our cohort was more commonly observed when mutations were in the same family. However, many mutations found in different families exhibited VA, ERG, and GVF concordance. A separate analysis did not show a relationship between coefficients of relatedness and concordance in any parameter, given that patients who were very distant exhibited phenotypic concordance as well.
Assessment of VA deterioration within mutations was complicated because some patients (most often those younger than the age of 10 years) actually improved with respect to VA with time. This is most likely secondary to improved patient testing acumen with age and the absence of a true best-corrected VA recording in some instances. While these retrospective results are important, more complete follow-up, ideally in a prospective study of VA over time, would be most useful in determining the natural course of VA deterioration for each mutation.
Electroretinography phenotypes are traditionally determined by the relative deterioration in cone vs rod function as reflected by percentage loss compared with the respective means.23 A major problem encountered in our studies and other studies is the fact that many patients present with NR ERG values across all parameters. While useful as a marker of early panretinal dysfunction and disease severity, it precludes categorization in our analytic framework. Furthermore, some patients who have been lost to follow-up had unavailable ERG data. This limited the number of mutations available for comparison. Despite these limitations, 6 of 9 mutations displayed ERG phenotypic concordance. The 3 discordant mutations displayed low (often both less than 10% of mean) or NR ERG values in both rods and cones, which made it difficult to categorize the pattern of photoreceptor degeneration.
It is possible that long-term trends in deterioration of cone or rod function may lend themselves to better phenotypic categorization. However, many patients often only received ERG testing at the beginning of their assessment. Only 1 patient (patient 12B in Table 3) in our cohort received ERG testing twice, at ages 10 and 13 years. He was characterized as exhibiting cone-rod dysfunction at both these points, given consistently lower photopic amplitudes. However, the degree of decline in both rod and cone parameters at these 2 points were comparable, suggesting that both are severely affected quite early. In such situations where the decline in ERG function is similar in both classes of photoreceptors, the ERG phenotypes as described in this study are unhelpful phenotypic classifications given the non-specific degeneration.
Phenotypic concordance is important to establish for proper counseling of patients diagnosed as having XLRP, as well as for accurate patient selection and efficacy monitoring in therapeutic trials. In this context, it is an important finding that assessment of GVF exhibits striking concordance of phenotypes both qualitatively and quantitatively in different RPGR mutations.
Supplementary Material
Acknowledgments
Funding/Support: This research was supported by the Foundation Fighting Blindness and the National Eye Institute (Core Center for Vision Research grant EY007003).
Footnotes
Supplemental content at jamaophthalmology.com
Author Contributions: Mr Zahid, Dr Khan, and Ms Branham contributed equally to this study.
Conflict of Interest Disclosures: None reported.
REFERENCES
- 1.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 2.Sandberg MA, Rosner B, Weigel-DiFranco C, Dryja TP, Berson EL. Disease course of patients with X-linked retinitis pigmentosa due to RPGR gene mutations. Invest Ophthalmol Vis Sci. 2007;48(3):1298–1304. doi: 10.1167/iovs.06-0971. [DOI] [PubMed] [Google Scholar]
- 3.Hong DH, Pawlyk B, Sokolov M, et al. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest Ophthalmol Vis Sci. 2003;44(6):2413–2421. doi: 10.1167/iovs.02-1206. [DOI] [PubMed] [Google Scholar]
- 4.Hong DH, Yue G, Adamian M, Li T. Retinitis pigmentosa GTPase regulator (RPGRr)-interacting protein is stably associated with the photoreceptor ciliary axoneme and anchors RPGR to the connecting cilium. J Biol Chem. 2001;276(15):12091–12099. doi: 10.1074/jbc.M009351200. [DOI] [PubMed] [Google Scholar]
- 5.Hosch J, Lorenz B, Stieger K. RPGR: role in the photoreceptor cilium, human retinal disease, and gene therapy. Ophthalmic Genet. 2011;32(1):1–11. doi: 10.3109/13816810.2010.535889. [DOI] [PubMed] [Google Scholar]
- 6.Murga-Zamalloa CA, Atkins SJ, Peranen J, Swaroop A, Khanna H. Interaction of retinitis pigmentosa GTPase regulator (RPGR) with RAB8A GTPase: implications for cilia dysfunction and photoreceptor degeneration. Hum Mol Genet. 2010;19(18):3591–3598. doi: 10.1093/hmg/ddq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murga-Zamalloa CA, Desai NJ, Hildebrandt F, Khanna H. Interaction of ciliary disease protein retinitis pigmentosa GTPase regulator with nephronophthisis-associated proteins in mammalian retinas. Mol Vis. 2010;16:1373–1381. [PMC free article] [PubMed] [Google Scholar]
- 8.Shu X, Fry AM, Tulloch B, et al. RPGR ORF15 isoform co-localizes with RPGRIP1 at centrioles and basal bodies and interacts with nucleophosmin. Hum Mol Genet. 2005;14(9):1183–1197. doi: 10.1093/hmg/ddi129. [DOI] [PubMed] [Google Scholar]
- 9.Gakovic M, Shu X, Kasioulis I, Carpanini S, Moraga I, Wright AF. The role of RPGR in cilia formation and actin stability. Hum Mol Genet. 2011;20(24):4840–4850. doi: 10.1093/hmg/ddr423. [DOI] [PubMed] [Google Scholar]
- 10.Murga-Zamalloa C, Swaroop A, Khanna H. Multiprotein complexes of Retinitis Pigmentosa GTPase regulator (RPGR), a ciliary protein mutated in X-linked Retinitis Pigmentosa (XLRP). Adv Exp Med Biol. 2010;664:105–114. doi: 10.1007/978-1-4419-1399-9_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He S, Parapuram SK, Hurd TW, et al. Retinitis pigmentosa GTPase regulator (RPGR) protein isoforms in mammalian retina: insights into X-linked retinitis pigmentosa and associated ciliopathies. Vision Res. 2008;48(3):366–376. doi: 10.1016/j.visres.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fahim AT, Bowne SJ, Sullivan LS, et al. Allelic heterogeneity and genetic modifier loci contribute to clinical variation in males with X-linked retinitis pigmentosa due to RPGR mutations. PLoS One. 2011;6(8):e23021. doi: 10.1371/journal.pone.0023021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharon D, Sandberg MA, Rabe VW, Stillberger M, Dryja TP, Berson EL. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am J Hum Genet. 2003;73(5):1131–1146. doi: 10.1086/379379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh AK, Murga-Zamalloa CA, Chan L, Hitchcock PF, Swaroop A, Khanna H. Human retinopathy-associated ciliary protein retinitis pigmentosa GTPase regulator mediates cilia-dependent vertebrate development. Hum Mol Genet. 2010;19(1):90–98. doi: 10.1093/hmg/ddp469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Maskari A, O'grady A, Pal B, McKibbin M. Phenotypic progression in X-linked retinitis pigmentosa secondary to a novel mutation in the RPGR gene. Eye (Lond) 2009;23(3):519–521. doi: 10.1038/eye.2008.427. [DOI] [PubMed] [Google Scholar]
- 16.Andréasson S, Ponjavic V, Abrahamson M, et al. Phenotypes in three Swedish families with X-linked retinitis pigmentosa caused by different mutations in the RPGR gene. Am J Ophthalmol. 1997;124(1):95–102. doi: 10.1016/s0002-9394(14)71649-6. [DOI] [PubMed] [Google Scholar]
- 17.Jin ZB, Liu XQ, Hayakawa M, Murakami A, Nao-i N. Mutational analysis of RPGR and RP2 genes in Japanese patients with retinitis pigmentosa: identification of four mutations. Mol Vis. 2006;12:1167–1174. [PubMed] [Google Scholar]
- 18.Koenekoop RK, Loyer M, Hand CK, et al. Novel RPGR mutations with distinct retinitis pigmentosa phenotypes in French-Canadian families. Am J Ophthalmol. 2003;136(4):678–687. doi: 10.1016/s0002-9394(03)00331-3. [DOI] [PubMed] [Google Scholar]
- 19.Wu DM, Khanna H, Atmaca-Sonmez P, et al. Long-term follow-up of a family with dominant X-linked retinitis pigmentosa. Eye (Lond) 2010;24(5):764–774. doi: 10.1038/eye.2009.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walia S, Fishman GA, Swaroop A, et al. Discordant phenotypes in fraternal twins having an identical mutation in exon ORF15 of the RPGR gene. Arch Ophthalmol. 2008;126(3):379–384. doi: 10.1001/archophthalmol.2007.72. [DOI] [PubMed] [Google Scholar]
- 21.Meindl A, Dry K, Herrmann K, et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet. 1996;13(1):35–42. doi: 10.1038/ng0596-35. [DOI] [PubMed] [Google Scholar]
- 22.Demirci FY, Rigatti BW, Wen G, et al. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet. 2002;70(4):1049–1053. doi: 10.1086/339620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heckenlively JR. Retinitis Pigmentosa. Lippincott; Philadelphia, PA: 1988. [Google Scholar]
- 24.Zito I, Thiselton DL, Gorin MB, et al. Identification of novel RPGR (retinitis pigmentosa GTPase regulator) mutations in a subset of X-linked retinitis pigmentosa families segregating with the RP3 locus. Hum Genet. 1999;105(1-2):57–62. doi: 10.1007/s004399900110. [DOI] [PubMed] [Google Scholar]
- 25.Buraczynska M, Wu W, Fujita R, et al. Spectrum of mutations in the RPGR gene that are identified in 20% of families with X-linked retinitis pigmentosa. Am J Hum Genet. 1997;61(6):1287–1292. doi: 10.1086/301646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vervoort R, Lennon A, Bird AC, et al. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. 2000;25(4):462–466. doi: 10.1038/78182. [DOI] [PubMed] [Google Scholar]
- 27.Bader I, Brandau O, Achatz H, et al. X-linked retinitis pigmentosa: RPGR mutations in most families with definite X linkage and clustering of mutations in a short sequence stretch of exon ORF15. Invest Ophthalmol Vis Sci. 2003;44(4):1458–1463. doi: 10.1167/iovs.02-0605. [DOI] [PubMed] [Google Scholar]
- 28.Breuer DK, Yashar BM, Filippova E, et al. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002;70(6):1545–1554. doi: 10.1086/340848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ayyagari R, Demirci FY, Liu J, et al. X-linked recessive atrophic macular degeneration from RPGR mutation. Genomics. 2002;80(2):166–171. doi: 10.1006/geno.2002.6815. [DOI] [PubMed] [Google Scholar]
- 30.Kaufman PL, Alm A. Adler's Physiology of the Eye. 10th ed. Mosby; St. Louis, MO: 2002. [Google Scholar]
- 31.Grover S, Fishman GA, Brown J., Jr Patterns of visual field progression in patients with retinitis pigmentosa. Ophthalmology. 1998;105(6):1069–1075. doi: 10.1016/S0161-6420(98)96009-2. [DOI] [PubMed] [Google Scholar]
- 32.Mears AJ, Hiriyanna S, Vervoort R, et al. Remapping of the RP15 locus for X-linked cone-rod degeneration to Xp11.4-p21.1, and identification of a de novo insertion in the RPGR exon ORF15. Am J Hum Genet. 2000;67(4):1000–1003. doi: 10.1086/303091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergen AA, Pinckers AJ. Localization of a novel X-linked progressive cone dystrophy gene to Xq27: evidence for genetic heterogeneity. Am J Hum Genet. 1997;60(6):1468–1473. doi: 10.1086/515458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jalkanen R, Demirci FY, Tyynismaa H, et al. A new genetic locus for X linked progressive cone-rod dystrophy. J Med Genet. 2003;40(6):418–423. doi: 10.1136/jmg.40.6.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ebenezer ND, Michaelides M, Jenkins SA, et al. Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Invest Ophthalmol Vis Sci. 2005;46(6):1891–1898. doi: 10.1167/iovs.04-1482. [DOI] [PubMed] [Google Scholar]
- 36.Bittner AK, Iftikhar MH, Dagnelie G. Test-retest, within-visit variability of Goldmann visual fields in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2011;52(11):8042–8046. doi: 10.1167/iovs.11-8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.