

Abstract
Aims/Introduction
A combination of multiple genetic and environmental factors contribute to the pathogenesis of type 2 diabetes. Copy number variations (CNVs) are associated with complex human diseases. However, CNVs can cause genotype deviation from the Hardy–Weinberg equilibrium (HWE). A genetic case–control association study in 216 Thai diabetic patients and 192 non-diabetic controls found that, after excluding genotyping errors, genotype distribution of calpain 10 (CAPN10) SNP44 (rs2975760) deviated from HWE. Here, we aimed to detect CNV within the CAPN10 SNP44 region.
Materials and Methods
CNV within the CAPN10 SNP44 region was detected using denaturing high-performance liquid chromatography, and the results confirmed by real-time quantitative polymerase chain reaction with SYBR Green I.
Results
Both methods successfully identified CNV in the CAPN10 SNP44 region, obtaining concordant results. Correction of genotype calling based on the status of identified CNVs showed that the CAPN10 SNP44 genotype is in good agreement with HWE (P > 0.05). However, no association between CNV genotypes and risk of type 2 diabetes was observed.
Conclusions
Identified CNVs for CAPN10 SNP44 genotypes lead to deviation from HWE. Furthermore, both denaturing high-performance liquid chromatography and real-time quantitative polymerase chain reaction are useful for detecting CNVs.
Keywords: CAPN10, Copy number variations, Denaturing high-performance liquid chromatography
Introduction
Type 2 diabetes is a complex disease that involves participation of several genetic and environmental factors1,2. Recent genetic research has identified several susceptibility genes. Discovery of type 2 diabetes genes is expected to aid development of novel therapeutic interventions or disease prevention. Calpain 10 (CAPN10), a type 2 diabetes susceptibility gene, was identified from a genome-wide scan. CAPN10 encodes an intracellular cysteine protease that plays a physiological role in insulin-mediated glucose metabolism and insulin production from pancreatic β-cells3,4. It is expressed in various tissues including pancreatic islets, adipose tissue, skeletal muscle and the liver3. CAPN10 is located on chromosome 2q37.3, and consists of 15 exons spanning 31 kb. Four single nucleotide polymorphisms (SNPs), SNP44 (rs2975760), SNP43 (rs3792267), Indel19 (rs3842570) and SNP63 (rs5030952), and/or their haplotypes have previously been associated with type 2 diabetes, insulin action and insulin secretion5,6. However, the impact of CAPN10 variants on type 2 diabetes had not been investigated in the Thai population; therefore, we previously examined the association between these markers and diabetes in a sample population. Genotype frequencies showed that Indel19 and SNP44 do not meet the assumption of the Hardy–Weinberg equilibrium (HWE; P < 0.05), and show increased homozygosity and decreased heterozygosity. Deviations from HWE commonly indicate genotyping errors, population stratification or some other artifact7. Another possibility currently being discussed in the literature to a greater extent is the presence of copy number variations (CNVs). CNVs correspond to relatively large genomic regions that are deleted (fewer than the normal number) or duplicated (more than the normal number) on certain chromosomes. It is now recognized that CNVs could be a major source of heritable variation among complex traits and can affect disease susceptibility8,9. In humans, over 14,478 CNV loci have been recorded, based on 89,427 different entries covering approximately one-third of the genome10. The large proportion of CNVs in the human genome indicates that numerous SNPs could lie within these regions. Furthermore, common deletions spanning SNP sites result in homozygosity excess and heterozygosity deficit, which can lead to deviation from HWE11,12. The extent of known CNVs covering the entire CAPN10 locus is catalogued in the Database of Genomic Variants. In our previous cohort study, we used denaturing high-performance liquid chromatography (DHPLC) to determine the presence of CNVs in the Indel19 marker region, and to ascertain whether these CNVs lead to genotype deviation from HWE13. In the present study, we used DHPLC to provide further evidence supporting the presence of CNVs in CAPN10 SNP44, and confirmed the results by real-time quantitative polymerase chain reaction (PCR) to ensure accuracy and reliability. In addition, we investigated the role of this CNV and the type 2 diabetes risk in our population.
Materials and Methods
Study Participants
A total of 216 patients with type 2 diabetes and 192 non-diabetic control subjects were included in the present study. Patients with type 2 diabetes were recruited from the Diabetic Clinic, Siriraj Hospital, Mahidol University, Bangkok, Thailand. Diabetes was defined according to the American Diabetes Association13. Non-diabetic controls were selected from participants at the health check-up center at the Department of Preventive and Social Medicine, Siriraj Hospital. Inclusion criteria were: (i) fasting plasma glucose <100 mg/dL (6.1 mmol/L); (ii) glycated hemoglobin A1c (HbA1c) <5.7%; (iii) no family history of type 2 diabetes in the first degree relatives; (iv) age ≥50 years; (v) no hypertension; and (vi) body mass index range 18.50–24.99 kg/m2. Study approval was granted from the institutional ethics committee. The patients and control participants provided informed consent before participating in the study.
PCR-Restriction Fragment Length Polymorphism Analysis
Genomic deoxyribonucleic acid (DNA) was extracted from peripheral blood leukocytes (obtained from ethylenediaminetetraacetic acid anticoagulated blood) using the standard phenol chloroform method. All participants were genotyped for SNP44 by PCR-restriction fragment length polymorphism (PCR-RFLP). The primers are listed in Table S1. PCR products were digested using MwoI restriction endonuclease (Fermentas, Vilnius, Lithuania). Digestion products were separated on 10% acrylamide and silver stained. Allele T was detected as a 183-, 118- and 74-bp fragments, and allele C as 183-, 102-, 74- and 16-bp fragments.
DHPLC Analysis
To detect CNVs in the CAPN10 SNP44 region by multiplex PCR and DHPLC, multiplex PCR was carried out using three primer pairs, specifically, co-amplified CAPN10 SNP44 region, cytotoxic T lymphocyte associated protein-4 (CTLA4, chromosome 2q) and dystrophin (DMD, chromosome X) genes. CTLA-4 and DMD were used as two and single copy (observed only in males) internal control genes, respectively. Primer sequences are summarized in Table S1. Multiplex PCR was carried out in a 25-μL total volume containing 125 ng genomic DNA; 4.5, 3.0 and 3.0 μmol/L of each primer for CAPN10 SNP44, DMD and CTLA-4, respectively, 2 mmol/L dNTPs, 0.5 units IMMOLASE™ DNA polymerase (Bioline Inc, London, UK), 2.5 mL Immobuffer and 1.5 mmol/L MgCl2, provided by the manufacturer. PCR for linear amplification was carried out in a T gradient Thermocycler (Biometra, Goettingen, Germany) with an initial denaturation step at 95°C for 10 min, followed by 27 cycles consisting of denaturation at 95°C for 30 s, annealing at 60°C for 30 s, extension at 72°C for 30 s and a final extension step at 72°C for 5 min. Multiplex PCR products were analyzed by gel electrophoresis through 2.5% LE-agarose and visualized after ethidium bromide staining. For quantification, 10 μL amplified products were processed through non-denaturing conditions at 50°C using the WAVE™ Nucleic Acid Fragment Analysis System (Transgenomic Inc., San Jose, CA, USA). The mobile phase was carried out in 0.1 mol/L triethylammonium acetate solution (buffer A). PCR products were then eluted using an acetonitrile gradient (49–58%). The gradient was created by mixing buffer A and 0.1 mol/L triethylammonium acetate solution in water, with 25% acetonitrile, pH 7 (buffer B). Each sample was then separated by DHPLC and quantified by fluorescence detection. Data were analyzed using appropriate software (Transgenomic Inc.). The results clearly show discrete chromatography profiles peaks: the x-axis represents elute time (min) and the y-axis represents fluorescence intensity. Relative peak intensity of each sample directly reflects copy number. For SNP44, the expected ratio of peak height compared with internal control genes should be 1 for samples without CNVs (genotypes T/T, T/C and C/C), 0.5 for samples with deletions and 1.5 for samples with duplications. Triplicate assays were carried out to check analysis accuracy.
Real-Time Quantitative PCR
Presence of CNVs was confirmed using specific primers for the target sequence (SNP44) and reference sequence (GAPDH), designed using Primer 3 (http://frodo.wi.mit.edu). Primer sequences and gene locations are listed in Table S2. Real-time quantitative PCR was carried out using the Light Cycler 480® system (Roche Diagnostics GmbH, Mannheim, Germany). GAPDH was simultaneously co-amplified with SNP44 and served as an internal standard control. Amplification reactions (20 μL) were carried out using 50 ng template DNA, 1xLightCycler® 480 SYBR Green I Masterkit (Roche), and 0.25 μmol/L of each forward and reverse primer. Thermal cycling was one cycle with predenaturation at 95°C for 10 min, followed by 45 cycles at 95°C for 30 s, 60°C for 30 s and 72°C for 30 s. In each assay, two normal control DNA and non-template controls were included. For data analysis, SNP44 gene copy number was determined by comparing the target SNP44 sequence to the reference (GAPDH). The relative gene copy number for each sample was determined using the comparative threshold cycle number (Ct) method. Each sample was run in triplicate in separate tubes to allow SNP44 quantification relative to GAPDH. Ct was determined for all PCR reactions. ΔCt represents the mean Ct value of each sample, and was calculated for SNP44 and GAPDH. By using a calibrator sample of normal control DNA, the gene copy number of unknown samples was estimated based on the following formula:
 |
Relative SNP44 copy number was determined using the formula 2 × 2(−ΔΔCT) as described, previously13.
Statistical Analysis
Genotype distribution was tested for deviation from HWE using the Chi square-test from a web-based program (http://ihg.gsf.de/cgi-bin/hw/hwal.pl). P-values for comparing cases and controls, and determining type 2 diabetes association were obtained using Pearson's chi square-test. Odds ratios (ORs) were calculated for one copy genotypes either (T/–) or (C/–) by comparing with the normal two copies genotype (TT + TC + CC). After excluding the identified CNV (1 copy), the CAPN10 SNP44 genotype was determined according to genetic models: TT vs TC vs CC (co-dominant model); (CC + TC) vs TT (dominant model); and CC vs (TC + TT; recessive model). For each genetic model, ORs were estimated by comparing each genotype with the other genotypes.
The linear regression model was used to analyze continuous variables adjusted for covariate variables of age, sex, body mass index and drugs where appropriate. P-values ≤0.05 were considered statistically significant. Statistical power calculation for SNP44 was carried out using the Power and Sample size program (PS program version 3.0; http://mc.vanderbilt.edu/wiki/Main/PowerSampleSize). To detect an association with the traits examined in the present study, 28.6% power (α = 0.05) was calculated.
Results
Clinical characteristics and laboratory parameters of the study participants are shown (Table1). Type 2 diabetic patients had higher body mass index, waist-to-hip ratio, waist circumference and blood pressure. Fasting plasma glucose, serum triglyceride, serum total cholesterol and serum low-density lipoprotein (LDL)-cholesterol levels were also significantly higher in diabetic patients. Non-diabetic controls had higher serum high-density (HDL)-cholesterol levels.
Table 1.
Clinical characteristics of study participants
| Clinical characteristics | Type 2 diabetes | Non-diabetic controls | P-value |
|---|---|---|---|
| n (male/female) | 202 (58/144) | 188 (53/135) | – |
| Age (years) | 53.75 ± 11.00 | 53.34 ± 9.09 | NS |
| Age at diagnosis (years) | 49.39 ± 10.87 | ND | ND |
| Weight (kg) | 68.57 ± 12.46 | 59.25 ± 9.89 | <0.001 |
| BMI (kg/m2) | 27.84 ± 4.95 | 23.91 ± 3.17 | <0.001 |
| Waist circumference (cm) | 87.99 ± 10.90 | 82.21 ± 9.37 | <0.001 |
| Hip circumference (cm) | 99.82 ± 9.73 | 95.62 ± 6.69 | <0.001 |
| Waist-to-hip ratio | 0.89 ± 0.07 | 0.86 ± 0.06 | <0.001 |
| Systolic BP (mmHg) | 132.13 ± 16.16 | 116.14 ± 15.12 | <0.001 |
| Diastolic BP (mmHg) | 81.42 ± 10.40 | 70.31 ± 9.90 | <0.001 |
| Fasting glucose (mmol/L) | 11.34 ± 5.09 | 4.88 ± 0.35 | <0.001 |
| HbA1c (%) | 8.34 ± 2.58 | 5.36 ± 0.26 | <0.001 |
| Total cholesterol (mmol/L) | 5.86 ± 1.19 | 5.32 ± 0.93 | <0.001 |
| Triglyceride (mmol/L) | 4.63 ± 2.33 | 2.68 ± 1.11 | <0.001 |
| LDL (mmol/L) | 3.70 ± 1.08 | 3.23 ± 0.86 | <0.001 |
| HDL (mmol/L) | 1.27 ± 0.31 | 1.58 ± 0.42 | <0.001 |
Data are shown as mean ± standard deviation. P-values compare clinical and laboratory data between 202 type 2 diabetes patients and 188 non-diabetic controls with completely available data, using either Mann–Whitney U-test or Student's unpaired t-test. BMI, body mass index; BP, blood pressure; HbA1c, glycated hemoglobin A1c; HDL, high-density lipoprotein; LDL, low-density lipoprotein; ND, not determined; NS, not significant.
We previously investigated the association of CAPN10 Indel19 and SNP44 with type 2 diabetes. The genotypes obtained for these SNPs showed deviation from HWE (Table S2). We initially examined CAPN10 Indel19, and successfully confirmed the presence of CNVs in the CAPN10 region by DHPLC13. Here, using the same cohort comprising 216 type 2 diabetes patients and 192 non-diabetic controls, we further examined the presence of CNVs in the SNP44 region using DHPLC, and confirmed the results by real-time quantitative PCR with SYBR Green I. First, we carried out DHPLC analysis. CAPN10 SNP44 chromatograms clearly showed the different patterns between male and female individuals harboring two copies or one copy of the variant (Figure1). Relative CAPN10 SNP44 copy number was determined from DHPLC peak heights of amplified products by comparing with the reference internal control genes (DMD and CTLA-4). Chromatogram peak heights from individuals carrying two normal copies of CAPN10 SNP44 were equal to CTLA-4 peak heights (Figure1a,b). Conversely, chromatogram peak heights reflecting heterozygous deletion (1 copy) of SNP44 were only half CTLA-4 peak heights (Figure1a,b).
Figure 1.
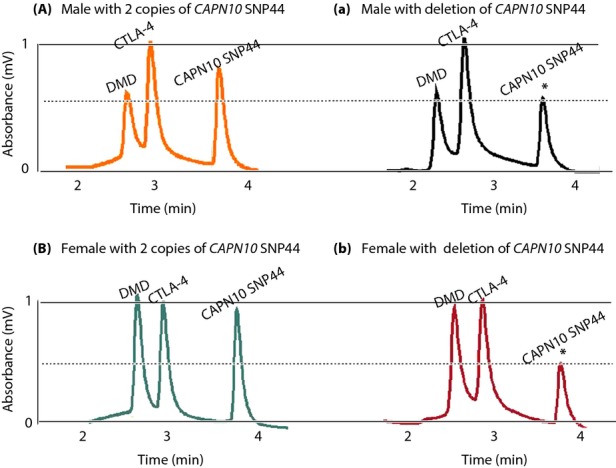
Denaturing high-performance liquid chromatography chromatogram of CAPN10 SNP44 copy number in male and female individuals. Each panel includes genotypes with three peaks corresponding to polymerase chain reaction amplification of CAPN10 SNP44, DMD and CTLA-4, respectively, obtained from (A) a normal male control or (B) female with two copies of CAPN10 SNP44. (a) Male and (b) female individuals with gene deletions are shown. Peaks corresponding to deletions are shown with an asterisk on top of the peak.
We carried out real-time quantitative PCR using SYBR Green in the same cohort to confirm our DHPLC results. PCR amplification plots of normal control samples showed nearly identical Ct values for CAPN10 SNP44, compared with GAPDH (Figure2a). Individuals with CAPN10 SNP44 deletions showed increased Ct values of nearly 0.5 cycles compared with GAPDH (Figure2b). The copy number status of CAPN10 SNP44 among non-diabetic controls and type 2 diabetes participants is shown (Figure2c). The results obtained from the two methods were blinded for CNV calling genetic results, and the concordance rate between them was 98%. We next investigated the relationship between CAPN10 SNP44 gene copy number and the risk of type 2 diabetes. There was no difference in the one copy genotype frequency either (T/–) or (C/–), when compared between case and control groups (Table2). After excluding CNVs, re-analysis showed that SNP44 genotypes fitted well within HWE in both non-diabetic controls and type 2 diabetes patients (P = 0.742 and 0.771, respectively; Table3). We then re-examined the association, finding that CAPN10 SNP44 was not associated with type 2 diabetes under co-dominant, dominant or recessive models (Table2). We also found no association between clinical characteristics and laboratory parameters of the studied participants and CNV genotypes (Tables S3 and S4).
Figure 2.
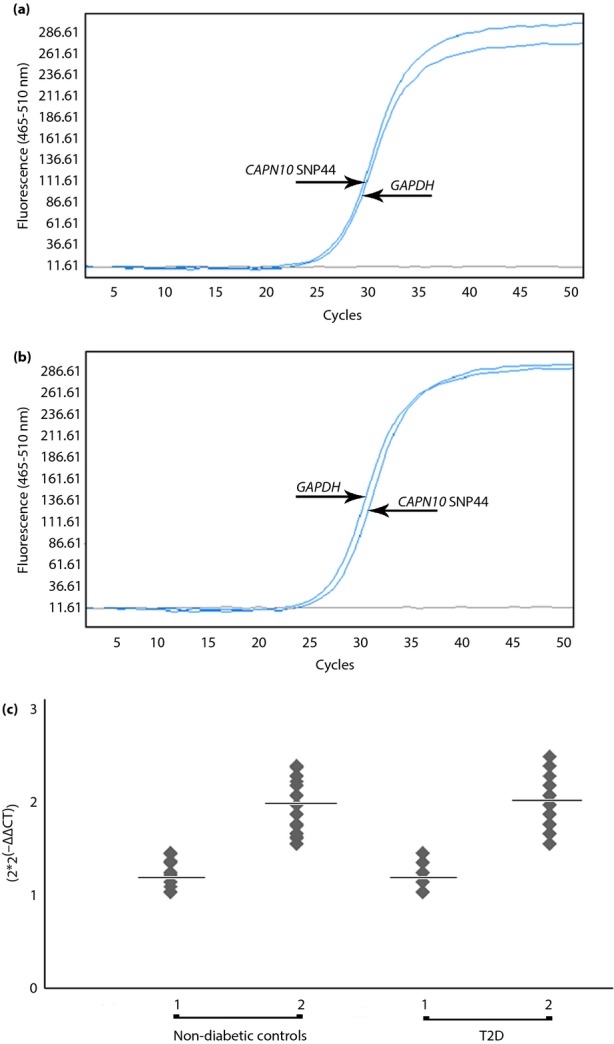
Amplification plots of CAPN10 SNP44 copy number status determined by real-time quantitative polymerase chain reaction, including (a) a normal control of CAPN10 SNP44, and (b) an individual with a gene deletion of gene. Quantification analysis of CAPN10 SNP44 copy number in non-diabetic controls and diabetic patients. In each group, copy numbers are shown as one copy or two copies of genes (x-axis). Analysis of gene copy number was carried out using the formula 2 × 2−ΔΔCt (y-axis). (c) The copy number mean after 2 × 2−ΔΔCt calculation in each group is represented by the horizontal line. T2D, type 2 diabetes.
Table 2.
Genotype distribution and association analysis of CAPN10 SNP44 copy number variation genotypes with type 2 diabetes risk
| Model | Genotype | Genotype frequency (%)† | P-value | OR (95% CI) | |
|---|---|---|---|---|---|
| Control (n = 192) | Case (n = 216) | ||||
| 1 copy | |||||
| T/– | 64 (33.33) | 83 (38.42) | 0.313* | 1.23 (0.81–1.86) | |
| C/– | 8 (4.17) | 7 (3.24) | 0.732* | 0.83 (0.29–2.36) | |
| 2 copies | |||||
| Co-dominant | TT | 88 (73.33) | 99 (78.57) | 0.776 | 1 (reference) |
| TC | 29 (24.17) | 25 (19.84) | 0.77 (0.42–1.41) | ||
| CC | 3 (2.50) | 2 (1.59) | 0.59 (0.10–3.63) | ||
| Dominant | TT | 88 (73.33) | 99 (45.83) | 0.336 | 1 (reference) |
| TC + CC | 31 (25.83) | 27 (21.43) | 0.75 (0.42–1.35) | ||
| Recessive | TT + TC | 117 (97.50) | 124 (98.41) | 0.956 | 1 (reference) |
| CC | 3 (2.50) | 2 (1.59) | 0.59 (0.09–3.62) |
*P-value assessed by comparing either (T/–) or (C/–) genotype and (TT + TC + CC) genotype between cases and controls. †CAPN10 SNP44 was genotyped by denaturing high-performance liquid chromatography and real-time quantitative polymerase chain reaction. CI, confidence interval; OR, odds ratio.
Table 3.
CAPN10 SNP44 genotype distribution with or without consideration of identified copy number variations
| Group (n) | Observed (expected) genotype frequency without consideration of CNVs | P HWE * | Group (n) | Observed (expected) genotype frequency with consideration of CNVs | P HWE ** | ||||
|---|---|---|---|---|---|---|---|---|---|
| T/T | T/C | C/C | T/T | T/C | C/C | ||||
| Case (n = 216) | 182 (175.14) | 25 (38.72) | 9 (2.14) | 1.91 × 10–7 | Case (n = 126) | 99 (98.67) | 25 (25.66) | 2 (1.67) | 0.771 |
| Controls (n = 192) | 152 (144.39) | 29 (44.23) | 11 (3.39) | 1.83 × 10–7 | Controls (n = 120) | 88 (87.55) | 29 (29.90) | 3 (2.55) | 0.742 |
P-value reflects Hardy Weinberg Equilibrium (HWE) before* and after** consideration of identified copy number variations (CNVs).
Discussion
Genetic association studies assess the association between phenotypic traits and genetic variants within a population. Genotyping data from case–control studies must first be tested for deviation from HWE to better understand the genetic characteristics of the population. The HWE test is carried out using the Chi square testing procedure to compare observed with expected genotype frequencies within a population. The genotype frequencies should be in agreement with HWE in cases and controls. Various reasons for deviation from HWE have been proposed, including natural selection, non-random mating, mutation and migration14. However, in humans, these factors seldom occur in the general population. A significant deviation might indicate genotyping errors and sampling bias, which, by far, are the most common sources of deviation15. An additional theoretical concern is that the result of a genetic association study could be spurious if the genotype distribution in controls does not fulfil HWE. Data must therefore be analyzed carefully for all the aforementioned reasons.
In our previous report, we investigated the association of the CAPN10 variants, Indel19 and SNP44, with type 2 diabetes in the Thai population. Genotype distribution of these SNPs significantly deviated from HWE, with homozygotes excess and reduced heterozygotes in the same direction (Nattachet P, Kanjana C, Watip T, Yenchitsomanus PT, unpublished data). We then re-genotyped these SNPs using different methods, obtaining concordant results, and carefully selected case and control subjects according to our inclusion criteria. There is evidence that structural DNA variations, such as CNVs, influence genomic variability and risk of human diseases. Furthermore, recent studies have shown that the frequency of SNPs residing in CNV regions (CNVRs) might deviate from HWE16. CAPN10 variant genotypes are reported to deviate from HWE in several ethnic groups; for example, Korean, Spanish, British and Finnish17–19. However, these studies did not adequately describe why deviation of genotypes from HWE was observed. To address this, we used DHPLC and showed that CNV presence in the CAPN10 Indel19 region leads to deviation of genotypes from HWE. In the present study, our aim was to ratify the presence of CNVs in the CAPN10 SNP44 region using two independent techniques, DHPLC and real-time quantitative PCR, which increases the accuracy and reliability of our experimental results. Both methods produced highly concordant outcomes, and were equally effective in CNV detection in the CAPN10 SNP44 region.
DHPLC is powerful, simple, time saving and cost-effective, enabling CNV detection in the gene of interest. However, some limitations were proposed in our previous study13. Therefore, we also examined CAPN10 SNP44 genotyping results obtained by DHPLC using real-time quantitative PCR. This method is suitable for CNV detection in targeted regions. Two detection chemistries are generally used for this technique: the double-stranded DNA-intercalating agent, SYBR Green I dye, or sequence-specific TaqMan probes. We used SYBR Green I, because it is relatively simple and inexpensive compared with TaqMan probes. In addition, a previous study showed that gene copy number analysis using DHPLC and real-time quantitative PCR are compatible20.
The Database of Genomic Variants contains CNV information from many ethnic groups; for example, Caucasian, African-American and East Asian. The CNVs reported in Database of Genomic Variants to cover CAPN10 were shown by Redon et al.21, Wang et al.22 Jakobsson et al.23 and Dogan et al.24 (Figure S1). Several studies have attempted to identify CNVs specific for certain populations, such as European, Korean and Chinese25–27. Nevertheless, investigation of CNVs in the Thai population is very limited. We were not able to accurately extend the CNV breakpoints, which could partly be explained by the fact that only CAPN10 Indel19 and SNP44 were investigated. It is also possible that the CNVs identified in the present study were Thai-specific and differentially distributed among other populations. Future work should clarify this issue.
Type 2 diabetes is a metabolic disorder characterized by insulin resistance and relative insulin deficiency1. Most current studies attempting to discover genes causing type 2 diabetes focus only on SNPs as genetic markers, whereas studies using CNVs as newly discovered genetic markers are relatively scarce. Recently, reports have shown that several CNVRs are significantly associated with type 2 diabetes risk in various ethnic groups28,29. The present study failed to find an association between the CAPN10 CNV genotype and diabetes or any clinical parameter, possibly because of the small sample size preventing detection of a significant association. Nevertheless, association of CAPN10 CNVs with type 2 diabetes has not yet been reported. A further study using a larger sample size might be required to establish an association between these CNVs and disease risk in Thais. The present study adds to accumulating evidence that CNVRs might affect precise genotype calling of SNPs, leading to disagreement of the HWE assumption. Additionally, combining DHPLC and real-time quantitative PCR will confirm CNV presence in genetic studies, particularly when SNPs are not in perfect HWE.
In conclusion, the present study shows that genotype distribution of certain SNPs, that display deviation from HWE, might result from CNVs in proximity to these SNP markers. Furthermore, using different genotyping methods is crucial to confirm the presence of CNVs in a genomic region of interest. Investigations regarding precise locations, (including breakpoints), together with molecular characteristics of these CNVs are required to obtain more insight into the role of CNVs in pathogenesis of type 2 diabetes.
Acknowledgments
This study was supported by the Faculty of Medicine Siriraj Hospital, Mahidol University, and a Mahidol University research grant (to Napatawn Banchuin and NP). We are deeply grateful to all participants who voluntarily enrolled in this study. We thank the staff members of Siriraj Molecular Diabetes Research Group (SiMDRG), Division of Endocrinology and Metabolism, and the Department of Medicine Siriraj Hospital for their kind assistance. PY is a Senior Research Scholar of the Thailand Research Fund (TRF).
Disclosure
The authors declare no conflict of interest.
Supporting Information
Polymerase chain reaction primer sequences for denaturing high-performance liquid chromatography and real-time quantitative polymerase chain reaction analysis.
Table S2 | CAPN10 SNP44 genotype distribution obtained by Polymerase chain reaction-restriction fragment length polymorphism analysis.
Table S3 | Association between CAPN10 SNP44 copy number variations and clinical parameters in patients.
Table S4 | Association between CAPN10 SNP44 copy number variations and clinical parameters in controls.
Figure S1 | Copy number variations location harboring CAPN10 obtained from the Database of Genomic Variants (DGV).
References
- Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365:1333–1346. doi: 10.1016/S0140-6736(05)61032-X. [DOI] [PubMed] [Google Scholar]
- Busch CP, Hegele RA. Genetic determinants of type 2 diabetes mellitus. Clin Genet. 2001;60:243–254. doi: 10.1034/j.1399-0004.2001.600401.x. [DOI] [PubMed] [Google Scholar]
- Horikawa Y, Oda N, Cox NJ, et al. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet. 2000;26:163–175. doi: 10.1038/79876. [DOI] [PubMed] [Google Scholar]
- Harris F, Biswas S, Singh J, et al. Calpains and their multiple roles in diabetes mellitus. Ann N Y Acad Sci. 2006;1084:452–480. doi: 10.1196/annals.1372.011. [DOI] [PubMed] [Google Scholar]
- Kang ES, Kim HJ, Nam M, et al. A novel 111/121 diplotype in the Calpain-10 gene is associated with type 2 diabetes. J Hum Genet. 2006;51:629–633. doi: 10.1007/s10038-006-0410-9. [DOI] [PubMed] [Google Scholar]
- Tsuchiya T, Schwarz PE, Bosque-Plata LD, et al. Association of the calpain-10 gene with type 2 diabetes in Europeans: results of pooled and meta-analyses. Mol Genet Metab. 2006;89:174–184. doi: 10.1016/j.ymgme.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Hosking L, Lumsden S, Lewis K, et al. Detection of genotyping errors by Hardy-Weinberg equilibrium testing. Eur J Hum Genet. 2004;12:395–399. doi: 10.1038/sj.ejhg.5201164. [DOI] [PubMed] [Google Scholar]
- Freeman JL, Perry GH, Feuk L, et al. Copy number variation: new insights in genome diversity. Genome Res. 2006;16:949–961. doi: 10.1101/gr.3677206. [DOI] [PubMed] [Google Scholar]
- Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007;8:639–646. doi: 10.1038/nrg2149. [DOI] [PubMed] [Google Scholar]
- Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Hadnott TN, Perry GH, et al. Common deletion polymorphisms in the human genome. Nat Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- Bae JS, Cheong HS, Kim JO, et al. Identification of SNP markers for common CNV regions and association analysis of risk of subarachnoid aneurysmal hemorrhage in Japanese population. Biochem Biophys Res Commun. 2008;373:593–596. doi: 10.1016/j.bbrc.2008.06.083. [DOI] [PubMed] [Google Scholar]
- Plengvidhya N, Chanprasert K, Tangjittipokin W, et al. Identification of copy number variation of CAPN10 in Thais with type 2 diabetes by multiplex PCR and denaturing high performance liquid chromatography (DHPLC) Gene. 2012;506:383–386. doi: 10.1016/j.gene.2012.06.094. [DOI] [PubMed] [Google Scholar]
- Salanti G, Amountza G, Ntzani EE, et al. Hardy-Weinberg equilibrium in genetic association studies: an empirical evaluation of reporting, deviations, and power. Eur J Hum Genet. 2005;13:840–848. doi: 10.1038/sj.ejhg.5201410. [DOI] [PubMed] [Google Scholar]
- Cox DG, Kraft P. Quantification of the power of Hardy–Weinberg equilibrium testing to detect genotyping error. Hum Hered. 2006;61:10–14. doi: 10.1159/000091787. [DOI] [PubMed] [Google Scholar]
- Lee S, Kasif S, Weng Z, et al. Quantitative analysis of single nucleotide polymorphisms within copy number variation. PLoS ONE. 2008;3:e3906. doi: 10.1371/journal.pone.0003906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orho-Melander M, Klannemark M, Svensson MK, et al. Variants in the calpain-10 gene predispose to insulin resistance and elevated free fatty acid levels. Diabetes. 2002;51:2658–2664. doi: 10.2337/diabetes.51.8.2658. [DOI] [PubMed] [Google Scholar]
- Saez ME, Gonzalez-Sanchez JL, Ramirez-Lorca R, et al. The CAPN10 gene is associated with insulin resistance phenotypes in the Spanish population. PLoS ONE. 2008;3:e2953. doi: 10.1371/journal.pone.0002953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn S, Evans JC, White C, et al. Variation in the calpain-10 gene affects blood glucose levels in the British population. Diabetes. 2002;51:247–250. doi: 10.2337/diabetes.51.1.247. [DOI] [PubMed] [Google Scholar]
- Su YN, Hung CC, Li H, et al. Quantitative analysis of SMN1 and SMN2 genes based on DHPLC: a highly efficient and reliable carrier-screening test. Hum Mutat. 2005;25:460–467. doi: 10.1002/humu.20160. [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KK, deLeeuw RJ, Dosanjh NS, et al. A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet. 2007;80:91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson M, Scholz SW, Scheet P, et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- Dogan H, Can H, Otu HH. Whole genome sequence of a Turkish individual. PLoS ONE. 2014;9:e85233. doi: 10.1371/journal.pone.0085233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Hayward C, Wright AF, et al. Copy number variation across European populations. PLoS ONE. 2011;6:e23087. doi: 10.1371/journal.pone.0023087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim SH, Kim TM, Hu HJ, et al. Copy number variations in East-Asian population and their evolutionary and functional implications. Hum Mol Genet. 2010;19:1001–1008. doi: 10.1093/hmg/ddp564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou H, Li S, Yang Y, et al. A map of copy number variations in Chinese populations. PLoS ONE. 2011;6:e27341. doi: 10.1371/journal.pone.0027341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae JS, Cheong HS, Kim JH, et al. The genetic effect of copy number variations on the risk of type 2 diabetes in a Korean population. PLoS ONE. 2011;6:e19091. doi: 10.1371/journal.pone.0019091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon JP, Shim SM, Nam HY, et al. Copy number variation at leptin receptor gene locus associated with metabolic traits and the risk of type 2 diabetes mellitus. BMC Genom. 2010;11:426. doi: 10.1186/1471-2164-11-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Polymerase chain reaction primer sequences for denaturing high-performance liquid chromatography and real-time quantitative polymerase chain reaction analysis.
Table S2 | CAPN10 SNP44 genotype distribution obtained by Polymerase chain reaction-restriction fragment length polymorphism analysis.
Table S3 | Association between CAPN10 SNP44 copy number variations and clinical parameters in patients.
Table S4 | Association between CAPN10 SNP44 copy number variations and clinical parameters in controls.
Figure S1 | Copy number variations location harboring CAPN10 obtained from the Database of Genomic Variants (DGV).