

Abstract
We describe a case of type B insulin resistance syndrome associated with systemic lupus erythematosus (SLE) that was refractory to rituximab and successfully treated with a combination of oral glucocorticoids and cyclosporine. Prior to treatment, insulin resistance was severe, and application of a hyperinsulinemic euglycemic clamp was not possible despite the continuous intravenous infusion of insulin at a maximum rate of 9.0 mU/kg/min. The addition of cyclosporine to oral glucocorticoid therapy resulted in remission of insulin resistance. The combination of oral prednisolone and cyclosporine might be effective in treating type B insulin resistance syndrome, particularly in rituximab-resistant cases. However, nephrotoxicity is a particular concern for patients receiving long-term cyclosporine therapy.
Keywords: Cyclosporine, Systemic lupus erythematosus, Type B insulin resistance syndrome
Introduction
Type B insulin resistance syndrome, a rare form of insulin-resistant diabetes due to the presence of anti-insulin receptor antibodies, is associated with collagen vascular diseases in most cases, such as systemic lupus erythematosus (SLE), scleroderma, Sjögren's syndrome, or occasionally manifests as paraneoplastic syndrome of malignancy1–4. Patients with this syndrome usually present with hyperglycemia that is tolerant to oral antidiabetic agents or high doses of insulin3. The pathogenesis of this syndrome involves the interaction of anti-insulin receptor antibodies and cell surface insulin receptors1–4, with the former blocking the binding of insulin to the latter1,2. This frequent association of type B insulin resistance syndrome with collagen vascular diseases aggravates the clinical situation due to the presence of anti-insulin antibodies that prevent treatment of hyperglycemia, even with large doses of insulin.
Here, we describe a male patient with type B insulin resistance syndrome associated with SLE who was treated with rituximab followed by oral glucocorticoids and cyclosporine. We also review and discuss the effectiveness of cyclosporine in the treatment of this rare syndrome.
Case report
A 60-year-old man presented to his primary care physician with complaints of thirst, polyuria, weight loss, and dizziness. Past medical history was unremarkable, except for previous surgery for appendicitis. He was not taking any medication and had no family history of diabetes. At the clinic, he was tentatively diagnosed with poorly controlled type 2 diabetes mellitus with a blood glucose level of 419 mg/dL and glycated hemoglobin (HbA1c) level of 12.3%. Despite treatment with calorie restriction (1,600 kcal/day), metformin (500 mg/day), and a high dose of intensive insulin therapy (approximately 100 U/day), hyperglycemia did not improve.
The patient was referred to our hospital for a detailed examination. He appeared thin and weak despite intensive insulin treatment with a body weight of 53.8 kg (body mass index: 20.0 kg/m2) and had a height of 164 cm. His skin turgor was diminished and reflected dehydration, but acanthosis nigricans, a defining characteristic of severe insulin resistance, was not clinically apparent. Further, he exhibited ataxic gait and photosensitivity to sunlight. Findings of X-ray and ECG studies were normal. Radiological studies, including thoracic abdominal CT or MR imaging with contrast enhancement, revealed no malignant tumors. Brain MR imaging findings were also normal.
Laboratory results are shown in Table1. Hematological abnormalities including thrombocytopenia and lymphopenia were detected. The titer of anti-nuclear antibodies (ANA) was high (1:2,560) with a speckled staining pattern. Further, serum samples were negative for anti-double strand DNA antibodies and positive for anti-Smith antibodies. Fasting capillary glucose levels ranged from 200 to 250 mg/dL, and postprandial levels ranged from 300 to 400 mg/dL. Notably, anti-insulin antibodies were negative, but anti-insulin receptor antibodies were positive. Severe insulin resistance was detected based on highly elevated serum and urine C-peptide levels. We attempted to apply a hyperinsulinemic euglycemic clamp to assess and quantify the degree of insulin resistance (Figure1). However, this was not possible due to the patient's persistent, severe insulin resistance, despite the infusion of a high dose of intravenous insulin up to 9.0 mU/kg/min (Figure1). The patient exhibited photosensitivity and hematological abnormalities (thrombocytopenia and lymphopenia). In addition, the presence of anti-nuclear antibodies and anti-Smith antibodies—which is suggestive of systemic lupus erythematosus (SLE)—was observed along with extreme insulin resistance and anti-insulin receptor antibodies. We therefore diagnosed the patient with type B insulin resistance syndrome associated with SLE, based on the American College of Rheumatology criteria5. Although not apparent at that time, discoid rashes that spread to the scalp and truck and oral ulcers that developed later in the clinical course indicated SLE. Lupus disease activity was further indicated by a high Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) score, including thrombocytopenia, lymphopenia, low complement levels, high erythrocyte sedimentation rate, and oral ulcers. Given that creatinine clearance was normal for the patient's age and that body weight and urinalysis were normal, except for mild microalbuminuria, lupus nephritis did not appear active at that time.
Table 1.
Laboratory results
| Test | Results | Reference range |
|---|---|---|
| White cell count | 4.76 × 103 | 2.97–9.13 × 103 (per mm3) |
| Lymphocyte | 0.618 × 103 | 0.7–3.5 × 103 (per mm3) |
| Hemoglobin | 10.2 | 12.9–17.4 (g/dL) |
| Hematocrit | 30.6 | 38.6–50.9 (%) |
| Platelets | 6.3 × 104 | 14.3–33.5 × 104 (per mm3) |
| Total protein | 6.8 | 6.8–8.3 (g/dL) |
| Albumin | 3.0 | 4.2–5.1 (g/dL) |
| BUN | 12 | 9–22 (mg/dL) |
| Creatinine | 0.65 | 0.6–1.0 (mg/dL) |
| Na | 138 | 136–145 (mmol/L) |
| K | 4.0 | 3.4–4.5 (mmol/L) |
| Cl | 108 | 100–108 (mmol/L) |
| CRP | 0.11 | 0–0.1 (mg/dL) |
| ESR | 40 | 2–10 (mm/h) |
| Urine protein | (±) | Negative |
| Urine ketones | (-) | Negative |
| Glucose (fasting) | 210 | 70–110 (mg/dL) |
| Glucose (postprandial) | 318 | 70–140 (mg/dL) |
| HbA1c | 13.5 | 4.6–6.2 (%) |
| C peptide (fasting) | 5.7 | 0.6–1.8 (ng/mL) |
| C peptide (*postprandial) | 9.6 | 0–4.0 (ng/mL) |
| Urine C peptide | 394.8 | 20–155 (μg/day) |
| C3 | 15 | 86–160 (mg/dL) |
| C4 | 2.0 | 17–45 (mg/dL) |
| CH50 | <2.0 | 29–48 (U/mL) |
| IgG | 2572 | 870–1700 (mg/dL) |
| IgM | 138 | 35–220 (mg/dL) |
| IgA | 384 | 110–410 (mg/dL) |
| Rheumatoid Factor | 1 | 0–10 (IU/mL) |
| Anti-nuclear antibody | 1: 2,560, speckled pattern | <1:40 |
| Anti-dsDNA antibody | Negative | |
| Anti-Sm antibody | Positive | |
| Anti-SSA antibody | Positive | |
| Anti-SSB antibody | Negative | |
| Anti-Scl-70 antibody | Positive | |
| Anti-RNP antibody | Positive | |
| Anti-Jo1 antibody | Negative | |
| Anti-centromere antibody | Negative | |
| Anti-TG antibody | Negative | |
| Anti-TPO antibody | Positive | |
| Anti-IA2 antibody | Positive | |
| Anti-GAD antibody | Negative | |
| Anti-insulin antibody | Negative | |
| Anti-insulin receptor antibody | Positive |
All data are obtained during intensive insulin therapy.
Postprandial C peptide were measured 2 h after meal.
HbA1c, glycated hemoglobin; BUN, blood urea nitrogen; CRP, C reactive protein; ESR, erythrocyte sedimentation rate; Ig, immunoglobulin; dsDNA, double stranded DNA; Sm, Smith antigen; SSA, Ro/SSA antigen; SSB, La/SSB antigen; Scl-70, DNA-topoisomerase 1; RNP, ribonucleoprotein; TG, thyroglobulin; TPO, thyroid peroxidase; IA2, insulinoma associated protein; GAD, glutamic acid decarboxylase.
Figure 1.

Pre-treatment hyperinsulinemic euglycemic clamp. Hyperinsulinemic euglycemic clamp was applied to assess and quantify the degree of resistance to exogenous insulin. Euglycemia was not attainable due to the presence of severe insulin resistance despite the continuous infusion of intravenous insulin at a maximum dose of 9.0 mU/kg/min. Blood glucose levels (BG, red line) showed a linear decrease that was not completely related to the insulin infusion rate (IIR, green columns).
We first treated the patient with the anti CD-20 monoclonal antibody rituximab, based on a previous case report6. As rituximab was not approved for use in treating SLE by social health insurance in Japan, written informed consent was obtained from the patient and his family before treatment. The use of rituximab in this treatment protocol was approved by the Ethics Committee of Shinshu University and conformed to the provisions of the Declaration of Helsinki. Rituximab therapy was initiated at a dose of 375 mg/m2 of body surface area as an intravenous infusion once a week for 4 weeks, as previously described6. In addition to rituximab, oral prednisolone (20 mg) was also administered to suppress the immune response (Figure2). Unfortunately, the patient's hyperglycemia worsened following the initiation of treatment, most likely due to the adverse effects of glucocorticoids. In addition, severe subcutaneous bacterial infection of the scrotum occurred, and heavy proteinuria developed. We therefore performed a renal biopsy to establish a definite diagnosis. Renal pathology revealed mildly expanded mesangium regions and strong positive immunostaining of C1q and C3c, suggesting diffuse mesangial proliferative nephritis (class II according to World Health Organization classification).
Figure 2.
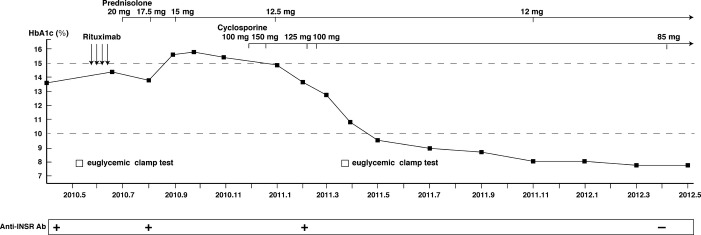
Schematic illustration of glucose metabolism. Rituximab therapy was initiated at a dose of 375 mg per square meter of body surface area as an intravenous infusion once a week for 4 weeks. Five months after the last rituximab infusion, severe insulin resistance showed no improvement. Anti-insulin receptor antibodies declined to an undetectable level following the addition of cyclosporine.
Following treatment of severe infection with broad-spectrum antibiotics, we added cyclosporine (starting dose of 100 mg/day, adjusted to 150 mg/day depending on trough levels) to the prednisolone therapy to increase immunosuppression and treat severe proteinuria. Trough levels of cyclosporine were monitored and maintained between 100 and 150 ng/mL for the induction of cyclosporine, and between 50 and 100 ng/mL thereafter. Surprisingly, a month after the addition of cyclosporine to the treatment regimen, the patient began to experience fasting hypoglycemia. We therefore gradually tapered the dose of insulin. We again attempted to apply a hyperinsulinemic euglycemic clamp to assess the degree of insulin resistance after treatment and obtained a mean glucose infusion rate (M value) of approximately 2.69 mg/kg/min, which suggested that insulin resistance was ameliorated. Since the addition of cyclosporine to oral prednisolone, relatively good glycemic control has been maintained, and anti-insulin receptor antibodies have declined to an undetectable level over approximately 1.5 years (Figure2). However, although the platelet number and the complement levels gradually returned to near-normal levels after the addition of cyclosporine, suggesting a suppression of lupus disease activity, the patient's renal function progressively declined (Figure3), and heavy proteinuria had an insufficient response to immunosuppressive therapy. Indeed, nephrotic proteinuria (>3.5 g/g creatinine) was ameliorated, with protein levels decreased to approximately 2.2 g/g creatinine (mean value during follow up period after the addition of cyclosporine), and this degree of proteinuria was maintained thereafter.
Figure 3.
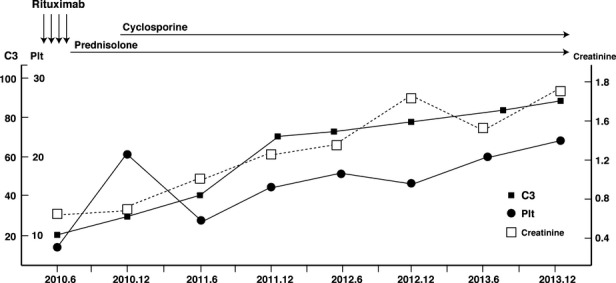
Schematic illustration of renal function and lupus activity. Platelet number (Plt) and complement (C3) level gradually returned to near normal after the addition of cyclosporine, suggesting suppression of lupus disease activity. The renal function of the patient gradually declined over the course of immunosuppressive therapy. Vertical axis indicates a scale of serum creatinine, C3, and platelet number. Normal range is as follows: serum creatinine: 0.63–1.05 mg/dL; C3: 86–160 mg/dL; and platelet number: 14.3–33.3 × 104, respectively. Note that the time scale is different from that of Figure2.
Discussion
Our treatment regimen referenced a case report from King's College Hospital in London that cited the successful treatment of type B insulin resistance syndrome with rituximab6. Given that the extreme insulin resistance observed in patients with this syndrome is caused by the presence of anti-insulin receptor antibodies, reducing the antibody titer with rituximab might be a reasonable approach. Nevertheless, in the present case, rituximab therapy was ineffective.
Previously reported treatments for type B insulin resistance syndrome include recombinant human insulin-like growth factor-1 (IGF-1), plasmapheresis, immunoglobulin G (IgG), and various combinations of immunosuppressants2–4,6–10. Based on more than 30 years of expert experience, Malek et al.3 of the NIH notably advocated the use of combination therapy with rituximab, pulse steroids, and immunosuppressive drugs, including cyclophosphamide, cyclosporine, and azathioprine. In addition, Zhang et al.10 also reported a case that was successfully treated with a combination of intravenous cyclophosphamide, methylprednisolone, and IgG. Unfortunately, these reports of highly efficient protocols for treating type B insulin resistance syndrome with fewer adverse effects were not available when our treatment began in April 2010. However, given that the rarity of this disease prevents researchers from conducting randomized placebo-controlled trials, as discussed by Malek et al.3, identifying the most effective course of treatment is difficult10. In this context, our case report may provide an alternative therapeutic regimen, particularly for use in rituximab-resistant cases.
The calcineurin inhibitor cyclosporine reduces the transcriptional activation of various inflammatory cytokines. In the present case, the addition of cyclosporine to maintenance glucocorticoid therapy led to remission of insulin resistance. Although some case reports have described the use of cyclosporine in combination with other immunosuppressants3,7,8, we consider the combination of cyclosporine and oral glucocorticoids to be a novel approach. Whether the success of our treatment was due to reinforced immunosuppression or effects specific to cyclosporine remains to be elucidated. Our rituximab therapy aimed to achieve remission not of lupus nephritis but of type B insulin resistance syndrome. Following the addition of cyclosporine, the platelet number and complement level gradually returned to near normal, and the SLEDAI score also improved, suggesting a suppression of lupus disease activity. However, the patient's renal function declined during the course of treatment with immunosuppressants. The degree of proteinuria also decreased somewhat after the treatment, albeit not sufficiently, we speculated that this renal impairment was the additive result of both the nephrotoxicity of cyclosporine and influence of chronic active lupus nephritis. Given that nephrotoxicity is a particular concern in patients receiving long-term cyclosporine therapy, the short-term use of cyclosporine might negate this problem. However, as Malek et al.3 described, whether or not type B insulin resistance syndrome recurs once immunosuppression is terminated is unknown. In addition, whether or not our patient should be on cyclosporine to maintain remission of insulin resistance is also unknown. Further study is needed to identify which patients should be on long-term maintenance immunosuppressive therapy to prevent recurrence.
In conclusion, although caution is required due to nephrotoxicity, cyclosporine might contribute to remission of type B insulin resistance syndrome associated with SLE in rituximab-resistant cases.
Acknowledgments
We thank Suguru Mishina of Nikkiso Co., Ltd., for providing technical assistance with the hyperinsulinemic euglycemic glucose clamp.
Disclosure
The authors declare no conflict of interest.
References
- Flier JS, Kahn CR, Roth J, et al. Antibodies that impair insulin receptor binding in an unusual diabetic syndrome with severe insulin resistance. Science. 1975;190:63–65. doi: 10.1126/science.170678. [DOI] [PubMed] [Google Scholar]
- Kahn CR, Flier JS, Bar RS, et al. The syndromes of insulin resistance and acanthosis nigricans. Insulin-receptor disorders in man. N Engl J Med. 1976;294:739–745. doi: 10.1056/NEJM197604012941401. [DOI] [PubMed] [Google Scholar]
- Malek R, Chong AY, Lupsa BC, et al. Treatment of type B insulin resistance: a novel approach to reduce insulin receptor autoantibodies. J Clin Endocrinolo Metab. 2010;95:3641–3647. doi: 10.1210/jc.2010-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arioglu E, Andewelt A, Diabo C, et al. Clinical course of the syndrome of autoantibodies to the insulin receptor (type B insulin resistance): a 28-year perspective. Medicine (Baltimore) 2002;81:87–100. doi: 10.1097/00005792-200203000-00001. [DOI] [PubMed] [Google Scholar]
- Petri M, Orbai AM, Alarcón GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64:2677–2686. doi: 10.1002/art.34473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll AP, Thomas S, Mufti GJ. Rituximab therapy for the type B syndrome of severe insulin resistance. N Engl J Med. 2004;350:310–311. doi: 10.1056/NEJM200401153500324. [DOI] [PubMed] [Google Scholar]
- Eriksson JW, Bremell T, Eliasson B, et al. Successful treatment with plasmapharesis, cyclophosphamide, and cyclosporine in type B syndrome of insulin resistance. Diabetes Care. 1998;21:1217–1220. doi: 10.2337/diacare.21.8.1217. [DOI] [PubMed] [Google Scholar]
- Kawashiri SY, Kawakami A, Fujikawa K, et al. Type B insulin resistance complicated with systemic lupus erythematosus. Intern Med. 2010;49:487–490. doi: 10.2169/internalmedicine.49.2746. [DOI] [PubMed] [Google Scholar]
- Tran HA, Reeves GE. Treatment of type B insulin resistance with immunoglobulin: novel use of an old therapy. Med J Aust. 2009;190:168. doi: 10.5694/j.1326-5377.2009.tb02335.x. [DOI] [PubMed] [Google Scholar]
- Zhang S, Wang G, Wang J. Type B insulin resistance syndrome induced by systemic lupus erythematosus and successfully treated with intravenous immunoglobulin: case report and systematic review. Clin Rheumatol. 2013;32:181–188. doi: 10.1007/s10067-012-2098-x. [DOI] [PubMed] [Google Scholar]