

Abstract
The constitutive androstane receptor (CAR) and peroxisome proliferator-activated receptor gamma coactivator-1α (PGC1α) are master regulators of drug metabolism and gluconeogenesis, respectively. In supporting the cross talk between drug metabolism and energy metabolism, activation of CAR has been shown to suppress hepatic gluconeogenesis and ameliorate hyperglycemia in vivo, but the underlying molecular mechanism remains elusive. In this study, we demonstrated that CAR suppressed hepatic gluconeogenic gene expression through posttranslational regulation of the subcellular localization and degradation of PGC1α. Activated CAR translocated into the nucleus and served as an adaptor protein to recruit PGC1α to the Cullin1 E3 ligase complex for ubiquitination. The interaction between CAR and PGC1α also led to their sequestration within the promyelocytic leukemia protein-nuclear bodies, where PGC1α and CAR subsequently underwent proteasomal degradation. Taken together, our findings revealed an unexpected function of CAR in recruiting an E3 ligase and targeting the gluconeogenic activity of PGC1α. Both drug metabolism and gluconeogenesis are energy-demanding processes. The negative regulation of PGC1α by CAR may represent a cellular adaptive mechanism to accommodate energy-restricted conditions.
CAR was initially recognized as a xenobiotic receptor that senses foreign chemicals and transcriptionally regulates the expression of drug metabolizing enzymes and transporters in the liver (1). More recent studies suggested that constitutive androstane receptor (CAR) can also restore glucose homeostasis under diabetic conditions. We and others showed that activation of CAR suppressed hepatic gluconeogenic gene expression and glucose production, and ameliorated hyperglycemia in genetic (ob/ob) and diet-induced obese mice (2, 3), as well as in a mouse model of gestational diabetes (4). A potential metabolic benefit of CAR activation in human glucose metabolism has been suggested by several clinical reports showing that administration of phenobarbital, a prototypical CAR activator, decreased plasma glucose levels and improved insulin sensitivity in diabetic patients (5–7).
Although the role of CAR in the cross talk between xenobiotic metabolism and glucose homeostasis has been recognized, few studies have probed into the underlying molecular mechanisms. It has been suggested that CAR may prevent the recruitment of the forkhead box protein O1 (FoxO1) to the gluconeogenic gene promoters (8). CAR may also interfere with the hepatocyte nuclear factor 4α (HNF4α)-mediated gluconeogenesis by competing for the direct repeat spaced by one nucleotide (DR1) type binding motif and its coactivators (9). However, due to the limited evidence and mostly in vitro nature of the aforementioned studies, the molecular pathway that underlies the CAR-mediated suppression of gluconeogenesis in physiological context remained to be characterized.
PPARγ coactivator-1α (PGC1α) is an inducible transcriptional coactivator that plays an essential role in regulating energy metabolism. During fasting, glucagon and cAMP induce the hepatic expression of PGC1α by activating the transcription factor cAMP-response element-binding protein, which in turn induces the expression of phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase), 2 rate-limiting gluconeogenic enzymes, by coactivating transcriptional factors such as HNF4α and glucocorticoid receptor (10). The hepatic expression of PGC1α is robustly elevated in obese mice to a level comparable with the fasting state, contributing to the hyperglycemia and obesity-related prediabetic symptoms (11).
The promyelocytic leukemia protein-nuclear bodies (PML-NBs) are macromolecular nuclear structures distributed in discrete nuclear foci. Dynamic orchestrations of PML-NBs constantly sequestrate and release transcriptional factors/coactivators and mediate their posttranslational modifications in response to cellular stresses (12). PML-NBs have been implicated in the regulation of diverse cellular functions, including the induction of apoptosis and cellular senescence, inhibition of proliferation, maintenance of genomic stability and antiviral responses. Recent evidence suggests that PML also participates in glucose and lipid metabolism. PML regulates fatty acid oxidation, which is essential for hematopoietic stem cells maintenance and cancer cell survival (13, 14). PML ablation in mice leads to accelerated fatty acid metabolism, abnormal glucose metabolism, and insulin resistance (15).
In the present work, we discovered a posttranslational mechanism by which CAR suppresses the gluconeogenic activity of PGC1α. Upon ligand activation, CAR translocates from cytoplasm into the nucleus where it recruits PGC1α to the Cullin1 E3 ligase complex for ubiquitination. The interaction between CAR and PGC1α also triggers the sequestration of both proteins into PML-NBs, which is required for the degradation of PGC1α and suppression of gluconeogenesis both in vitro and in vivo. Interestingly, CAR can inhibit the gluconeogenic activity of PGC1α independent of its traditional transcriptional activity.
Materials and Methods
Animals
C57BL/6j mice were purchased from The Jackson Laboratory. Pml−/− mice (16) were purchased from the National Cancer Institute Mouse Repository. CAR−/− mice were previously described (17). For the fasting-refeeding experiment, mice were subjected to overnight fasting (16 h), followed by 12 hours of refeeding before killing. For high-fat diet (HFD) feeding, mice were fed with HFD (TD.06414) from Harlan. When necessary, mice were ip injected with 1,4-Bis-[2-(3,5 dichloropyridyloxy)]benzene (TCPOBOP) in Dimethyl sulfoxide (DMSO) (0.25 mg/kg) once per week. The ip glucose tolerance test (IPGTT) was performed after overnight fasting. The mice were ip injected with 2 g/kg glucose, and blood glucose levels were measured at the time points of 0, 15, 30, 60, 90, and 120 minutes. All mice were housed in a pathogen-free animal facility under a standard 12-hour light, 12-hour dark cycle with free access to food and water. The use of mice in this study complied with all relevant federal guidelines and institutional policies.
Plasmids, cell transfection, and reporter assay
pCMV-HA-CAR (WT, D8, D30, coactivator-binding mutant [CBM], CC/AA, and L346F), pCMV-Flag-PGC1α, pCMV-PPARα, pCMV-HA-PPARγ, pCMV-FXR, pCMV-LXR, pCMV-HNF4α, pCMV-HA/Flag-Cullin1 (WT, N terminus of Cullin1 [Cul1NT], CullNT_Mut), pCMV-HA/Flag-Skp1 (WT, Mut), pCMV-Flag-Rbx1, Gal4-PGC1α (1–400), and pCMV-PML/Myc-PML were cloned using standard molecular cloning techniques. pcDNA-Flag-PGC1α (plasmid 1026) (18), Gal4-PGC1α (plasmid 8892) (19), and pcDNA3-DN-CUL1-FLAG (plasmid 15818) (20) were purchased from Addgene. The G6Pase-luciferase reporter plasmid (21) was a gift from Dr Richard M. O'Brien (Department of Molecular Physiology and Biophysics, Vanderbilt University Medical School). pcDNA-CAR (CC/AA) (9) was a gift from Dr Jongsook Kim Kemper (Department of Molecular and Integrative Physiology, University of Illinois at Urbana-Champaign). HEK293T, HepG2, and Hepa1-6 cells were obtained from ATCC. Transient transfections were performed with the TransIT-LT1 Transfection Reagent (Mirus). Cells were harvested and measured for luciferase and β-galactosidase (β-gal) activities 24 hours after transfection. Transfection efficiency was normalized against β-gal activity derived from the cotransfected pCMX-β-gal plasmid.
Adenovirus, lentivirus, and stable cell line
Flag-tagged mouse CAR adenovirus (Ad-Flag-CAR) was generated by using the AdEasy Adenoviral Vector System from Life Technologies. HA-tagged PGC1α adenovirus (Ad-HA-PGC1α) was made using shuttle vector pAd-Track HA-PGC1α (plasmid 14427) (22) from Addgene. Ad-PGC1α RNAi (23) and scrambled RNA interference (RNAi) adenoviruses were gifts from Dr Marc R. Montminy (Salk Institute, La Jolla, CA). The sequences for scrambled RNAi and PGC1α RNAi are 5′-GGCATTACAGTATCGATCAGA-3′ and 5′-GGTGGATTGAAGTGGTGTAGA-3′, respectively. Mouse PML RNAi lentivirus was generated using pLKO.1-mPML RNAi vector from Open Biosystems. pLKO.1-nonspecific RNAi vector, packaging vector psPAX2 and envelope vector pMD2.G were from Addgene. PML RNAi knockdown Hepa1-6 cells were selected by puromycin (5–10 μg/mL) for 2 weeks after the lentiviral infection.
Western blotting and coimmunoprecipitation (coIP)
For Western blot analysis, cells were lysed in ice-cold radioimmunoprecipitation assay buffer containing a protease inhibitor cocktail from Roche. Primary antibodies used include anti-HA (C29F4) and anti-HNF4α (C11F12) from Cell Signaling, anti-Flag (M2) and anti-Myc (M4439) from Sigma, anti-PML (clone 36.1-104) from Millipore, and anti-PGC1α (H300) and anti-CAR (M-150) from Santa Cruz Biotechnology, Inc. For coIP, cells were lysed in immunoprecipitation buffer (150mM NaCl, 50mM Tris-HCl [pH 7.5], and 1% Nonidet P-40) supplemented with a protease-inhibitor cocktail. The lysates were precleared by incubation with protein G magnetic beads and incubated with primary antibody overnight at 4°C, followed by incubation with protein G magnetic beads for 1 hour at room temperature. Protein G beads were then washed 5 times with ice-cold immunoprecipitation buffer, eluted with protein loading buffer, and analyzed by Western blotting.
Chromatin immunoprecipitation (ChIP) assay
Primary mouse hepatocytes were infected with Ad-HA-PGC1α and/or Ad-Flag-CAR (Multiplicity of infection = 5) for 48 hours and fixed in 1% formaldehyde for 15 minutes in room temperature. Nuclear extracts were sonicated and aliquot of sheared chromatin (equivalent of 2 × 105 cells) was immunoprecipitated with anti-HA, anti-HNF4α, or normal rabbit IgG. Immunoprecipitated chromatin was decross-linked, ethanol precipitated, and quantified by quantitative real-time PCR. Recoveries were calculated as the percentage of input. For the mG6Pase promoter, we used the next primers: mG6Pase_-300 bp_F, 5′-GCTGTTTTTGTGTGCCTGTT-3′ and mG6Pase_-300 bp_R, 5′-TGCTATCAGTCTGTGCCTTG-3′; and mG6Pase _-3000 bp_F, 5′-CAGTGCTCCCAGAGTTCCTC-3′ and mG6Pase_-3000 bp_R, 5′-TGAGGAGCAGGGCTGTCTGT-3′. For the mPepck promoter, we used the next primers: mPepck_-300 bp_F, 5′-GGCCTCCCAACATTCATTAAC-3′ and mPepck_-300 bp_R, 5′-CGCCCTCCTTGCTTTAAATA-3′; and mPepck_-3000 bp_F, 5′-TCCAGCATACACAGAGGATCA-3′ and mPepck_-3000 bp_R, 5′-TGCAGTCCAGCTAATGCAAC-3′.
In vitro ubiquitination assay
The in vitro ubiquitination assay was performed based on a previously described protocol (24). The CAR associated E3 complex was purified from the 293T cells transfected with HA-Flag-CAR and HA-Cullin1/Skp1/Rbx1 with the anti-Flag M2 affinity gel from Sigma (St Louis, MO). The recombinant Flag-PGC1 was purified from the 293T cells transfected with Flag-PGC1 with the anti-Flag M2 affinity gel. For the in vitro ubiquitination reaction, the purified E3 complex and PGC1 were incubated in the presence of ATP, ubiquitin, and E1/E2 (UbcH5a and UbcH3) from a Ubiquitination Assay kit from Enzo Life Sciences. The reactions were terminated by adding Western blotting loading buffer, and the products were resolved on SDS-PAGE.
Mouse primary hepatocyte isolation and culture
Mouse primary hepatocytes were isolated from 8- to 12-week-old male wild type (WT), CAR−/−, or Pml−/− mice. Briefly, the liver was first perfused with Hanks' buffered salt solution containing 0.5mM EGTA, 0.1M HEPES at 5 mL/min for 5–10 minutes and then perfused with L-15 medium containing 1.8mM CaCl2, 0.1M HEPES, 20-μg/mL liberase TM (Roche). After perfusion, the dissociated hepatocytes were filtered through 50-μm pore mesh and collected by centrifugation at 400 rpm for 4 minutes at 4°C. Hepatocytes were seeded onto type 1 collagen-coated dishes or slides in William E medium containing 5% fetal bovine serum, 1μM dexamethasone, and 100nM insulin. The hepatocytes were maintained with medium (HepatoZYME-SFM supplemented with 100nM dexamethasone, 100nM insulin, and 0.2% BSA) the next day. For forskolin (FSK) treatment, primary hepatocytes were changed to maintenance medium without hormones for 6 hours before treating with FSK (10μM) and/or TCPOBOP (500nM) for mRNA analysis, or in 1-mL gluconeogenic medium (glucose-free DMEM, 20mM sodium lactate, 2mM sodium pyruvate, and 0.5% BSA) for hepatic glucose production assay. Medium glucose concentration was measured using a glucose oxidase assay kit (GAGO20-1KT) from Sigma (St Louis, MO).
Immunofluorescence and confocal microscopy
Primary mouse hepatocytes or cell lines were grown on slides and treated when necessary. Cells were fixed in 4% paraformaldehyde for 15 minutes in room temperature followed by blocking with PBS containing 5% donkey serum and 0.3% Triton X-100 for 30 minutes. Slides were incubated in diluted primary antibody overnight at 4°C followed by incubation with fluorochrome-conjugated secondary antibody for 2 hours at room temperature in the dark. Slides were mounted and scanned using confocal microscopy to obtain images.
Quantitative real-time PCR
Total RNA was isolated using the TRIzol reagent from Invitrogen. Reverse transcription was performed with random hexamer primers and Superscript RT III enzyme from Invitrogen. SYBR Green-based real-time PCR was performed with the ABI 7300 Real-Time PCR System. Data were normalized against internal control cyclophillin.
Statistical analysis
All results were presented as mean ± SD. Statistical significance between groups was determined using an unpaired 2-tailed Student's t test, with P < .05 considered statistically significantly.
Results
CAR suppresses gluconeogenic gene expression through inhibiting the PGC1α activity
Activation of CAR has been shown to suppress hepatic gluconeogenesis and ameliorate hyperglycemia in animal models and human patients (2, 3, 5–7). In searching for the mechanism by which CAR inhibits gluconeogenesis, we noticed the inhibitory effect of the CAR agonist TCPOBOP on hepatic gluconeogenic gene expression was most dramatic in HFD-fed mice (Supplemental Figure 1), suggesting that CAR might have targeted a HFD-inducible factor in the liver. One such candidate factor is PGC1α, whose expression is markedly elevated in diabetic conditions (Supplemental Figure 1) (11, 25). To directly evaluate the effect of CAR on PGC1α activity, we found that CAR efficiently suppressed the PGC1α-responsive activation of the G6Pase-luciferase reporter gene (Figure 1A). The inhibition was obvious in the absence of an exogenously added ligand, and was enhanced by the addition of TCPOBOP. In primary mouse hepatocytes, FSK treatment increased the expression of G6Pase and Pepck mainly via the cAMP-response element-binding protein-mediated induction of PGC1α (10). We showed that treatment with TCPOBOP suppressed the FSK-responsive induction of G6Pase and Pepck without affecting the expression of PGC1α (Figure 1B, left panel), and this effect was abolished in hepatocytes isolated from the CAR−/− mice (Figure 1B, right panel). The inhibition of FSK-responsive induction of G6Pase and PEPCK was also observed in primary human hepatocytes treated with 6-(4-Chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime, a human CAR-specific agonist (Figure 1C). The inhibitory effect of TCPOBOP was PGC1α dependent, because the inhibition of both the gluconeogenic gene expression (Figure 1D) and glucose production (Figure 1E) was attenuated by PGC1α knockdown. The efficiency of PGC1α RNAi knockdown was validated by real-time PCR (Supplemental Figure 2A). To directly test whether CAR activation inhibited PGC1α, we overexpressed PGC1α in primary mouse hepatocytes using adenovirus. Overexpression of PGC1α was sufficient to induce the expression of G6Pase and Pepck as expected, which was attenuated in cells coinfected with the CAR expressing adenovirus and treated with TCPOBOP (Figure 1F). The adenoviral overexpression of PGC1α was validated by real-time PCR (Supplemental Figure 2B). At the functional level, CAR activation inhibited PGC1α-induced glucose production in primary mouse hepatocytes (Figure 1G). In the loss-of-function model, we showed the chow diet-fed CAR−/− mice had elevated basal expression and compromised fasting-responsive induction of G6Pase and Pepck (Figure 1H). In addition, the hepatic expression of CAR fluctuated in response to fasting, refeeding and HFD feeding, mirroring the pattern of PGC1α (Figure 1I), suggesting that CAR may be coregulated with PGC1α and suppresses its activity to fine-tune hepatic glucose homeostasis.
Figure 1.

CAR suppresses gluconeogenic gene expression through inhibiting PGC1α activity. A, Cotransfection of CAR inhibited the PGC1α-responsive activation of the G6Pase luciferase reporter gene in 293T cells. B, Mouse primary hepatocytes isolated from WT (left panel) or CAR null (right panel) mice were treated with TCPOBOP (TC) (500nM) or DMSO for 12 hours before treatment of FSK (10μM) for 2 hours. The gene expression was measured by real-time PCR. C, Human primary hepatocytes were treated with 6-(4-Chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (1μM) for 12 hours, followed by a FSK (10μM) treatment for 2 hours. The gene expression was measured by real-time PCR. D, Primary hepatocyte from WT mice infected with Ad-scramble RNAi or Ad-PGC1α RNAi for 48 hours was treated with TC (500nM) or DMSO for 12 hours before treatment of FSK (10μM) for 2 hours. The gene expression was measured by real-time PCR. E, Mouse primary hepatocytes were pretreated with TC (500nM) overnight in the maintenance medium. Glucose production was measured after incubation with FSK (10μM) with or without TC (500nM) in the glucose-free medium for 4 hours. F and G, Primary hepatocytes from CAR null mice were infected with Ad-GFP, Ad-CAR, or Ad-PGC1α and treated with or without TC (500nM) for 12 hours before measuring the mRNA expression of G6Pase and Pepck (F) and glucose production (G). H, Hepatic expression of G6Pase and Pepck in fed, overnight fasted (16 h), and refed (12 h) WT and CAR null mice. n = 5 for each group. I, Expression of CAR and PGC1α in mouse liver during the fasting-refed transition (left panel, n = 4 for each group) and upon a 12-week HFD feeding (right panel, n = 5 for each group). *, P < .05; **, P < .01.
CAR reduces the recruitment of PGC1α to the gluconeogenic gene promoters and causes redistribution of PGC1α to PML-NBs
We then used ChIP assay to determine whether CAR activation reduced the recruitment of PGC1α to the promoters of gluconeogenic genes. Indeed, treatment of primary mouse hepatocytes with TCPOBOP suppressed the recruitment of PGC1α to the proximal promoter regions of G6Pase (−250 bp) and Pepck (−300 bp) genes that harbor the HNF4α and FoxO1 binding sites (21) without affecting the nonspecific binding of PGC1α to the distal promoter regions (Figure 2A). The inhibition appeared to be PGC1α specific, because the recruitment of HNF4α to the G6Pase and Pepck gene promoters was not affected in the same cells (Figure 2B). A direct interaction between CAR and PGC1α had been reported (26, 27), which was verified by our coIP experiment (Supplemental Figure 3). However, a simple “coactivator quenching” model in which CAR competes with other transcriptional factors for the binding of PGC1α was unlikely the underlying mechanism, because among a panel of nuclear receptors known to interact with and coactivated by PGC1α, only CAR showed inhibition of the PGC1α activity (Supplemental Figure 4). Instead, we found CAR activation induced a dramatic redistribution of nuclear PGC1α. Treatment of primary mouse hepatocytes with TCPOBOP triggered the translocation of CAR from cytoplasm to nucleus to concentrate at the spot-like subnuclear loci, which turned out to be PML-NBs, a multiprotein subnuclear structures, as confirmed by their colocalization with the PML protein (12) (Figure 2, C and D). A similar pattern of CAR-responsive redistribution of PGC1α and CAR to the PML-NBs was observed in the human hepatoma HepG2 cells cotransfected with CAR and PGC1α (Supplemental Figure 5). The interaction between CAR and PGC1α was required for their targeting to the PML-NBs, because transfection of CAR or PGC1α alone resulted in an even distribution of both proteins in the nucleus (Supplemental Figure 5). PML is the essential component of the PML-NBs, in which PML multimerizes to function as a critical scaffold for the assembly of the entire complex (28). We then hypothesized that redistribution of CAR and PGC1α may have been mediated through the interaction between the CAR-PGC1α complex and PML. Indeed, coIP assay showed that transfection of PGC1α or CAR alone in 293T cells resulted in little interaction with PML, whereas coexpression of both proteins significantly enhanced their association with PML (Figure 2E), which was consistent with the immunofluorescence results (Supplemental Figure 5). These results suggested that the formation of CAR-PGC1α complex was a prerequisite for their interaction with PML.
Figure 2.

CAR reduces the recruitment of PGC1α to the gluconeogenic gene promoters and causes redistribution of PGC1α to PML-NBs. A and B, Mouse primary hepatocytes were infected with Ad-HA-PGC1α and/or Ad-CAR for 48 hours, in the absence or presence of TCPOBOP (TC) (500nM). PGC1α (A) and HNF4α (B) ChIPs were facilitated by using anti-HA and anti-HNF4α antibodies, respectively (**, P < .01; N.S., not significant). C and D, Mouse primary hepatocytes were infected with Ad-PGC1α and/or Ad-CAR for 48 hours, in the absence or presence of TC (500nM) before immunofluorescent detection of CAR and PGC1α (C) or PML and PGC1α (D). E, 293T cells were cotransfected with Myc-PML, Flag-PGC1α, and HA-CAR before subjecting to immunoprecipitation and immunoblotting as indicated.
CAR promotes ubiquitination-proteasomal degradation of PGC1α, which is required for gluconeogenic suppression
We noticed that treatment with TCPOBOP dramatically reduced the protein level of both CAR and PGC1α (Figure 2E), suggesting a ligand-dependent degradation of the CAR-PGC1α complex upon their redistribution to the PML-NBs. Indeed, interaction of the CAR-PGC1α complex to PML was associated with increased ubiquitination of both CAR and PGC1α, and their degradation was triggered by TCPOBOP (Figure 3A). In vivo treatment of HFD-fed mice with TCPOBOP significantly reduced the protein level of PGC1α, CAR, and PML in the liver (Figure 3B). Treatment of cells with the proteasome inhibitor MG132 inhibited the degradation of both PGC1α and CAR, with PGC1α enriched in the insoluble pellet fraction (Figure 3C). These results were consistent with the notion that PGC1α has a tendency to form insoluble protein aggregates when poly-ubiquitinated (29). The effect of proteasome inhibitor suggested the degradation of CAR and PGC1α was achieved through the ubiquitination-proteasome pathway. Because the formation of CAR-PGC1α complex was the prerequisite for their interaction with PML and subsequent degradation, we reasoned the binding between CAR and PGC1α was necessary and sufficient to trigger the cascade. Indeed, mutation of CAR at the 2 conserved amino acid residues within H12/AF2 (E355A) and H3 (K187A) (CBM) that constitute the hydrophobic cleft for the binding of coregulators (30) disrupted the interaction between CAR and PGC1α (Figure 3D), which in turn abolished the CAR-induced ubiquitination and degradation of PGC1α (Figure 3, E and F). At the functional level, the suppressive effect of CAR on the PGC1α-responsive activation of the G6Pase-lucifarase reporter gene was nearly abolished by the CBM mutations (Figure 3G).
Figure 3.

CAR promotes ubiquitination-proteasomal degradation of PGC1α, which is required for gluconeogenic suppression. A, The same samples as in Figure 2E were used to detect the polyubiquitination of PGC1α, CAR, and PML with a ubiquitin antibody. B, Mice fed with HFD were treated with TCPOBOP (0.2 mg/kg, once per week) or vehicle for 12 weeks. Total liver lysates were analyzed by immunoblotting for the detection of endogenous PGC1α, CAR, and PML (n = 4 for each group). The protein expression level was quantified by densitometry (*, P < .05). C, 293T cells were cotransfected with Myc-PML, Flag-PGC1α, and HA-CAR and treated with TC (500nM) or MG132 (10μM) before subjecting to Western blotting. Proteins in both the soluble and insoluble fractions were analyzed. D and E, 293T cells were cotransfected with Flag-PGC1α, HA-tagged WT CAR, or CAR CBM mutant, followed by immunoprecipitation and immunoblotting to evaluate their interaction (D), and their polyubiquitination was detected by immunoblotting with an ubiquitin antibody (E). “Nonspecific” in D denotes the nonspecific band detected by the PGC1 antibody, which was used as a loading control. F, 293T cells were cotransfected with Flag-PGC1α, HA-tagged WT CAR, or CAR CBM mutant and treated with or without TC (500nM). The expression of PGC1α and CAR was measured by Western blotting. Nonspecific, nonspecific band detected by the PGC1 antibody, which was used as a loading control. G, The suppressive effects of WT CAR or CAR CBM mutant on PGC1α activity were evaluated by G6Pase luciferase reporter assay in 293T cells.
PML-NBs are required for CAR to induce PGC1α degradation and suppress gluconeogenic gene expression
The association between redistribution of CAR and PGC1α to the PML-NBs and their degradation prompted us to determine whether PML-NBs were required for CAR-induced PGC1α degradation and inhibition of gluconeogenesis. In Hepa1-6 cells stably expressing PML shRNA, the formation of PML-NBs was disrupted as expected, and PGC1α remained evenly distributed in the nucleus when cotransfected with CAR (Figure 4A, top panel). The inhibitory effect of CAR on PGC1α-responsive activation of the G6Pase-lucifarease reporter gene was attenuated in PML knockdown cells (Figure 4A, bottom panel). In contrast, overexpression of PML facilitated PML-NB formation and targeting of PGC1α to PML-NBs (Figure 4B, top panel). Interestingly, overexpression of PML had a marginal effect in enhancing CAR-induced suppression of PGC1α activity (Figure 4B, bottom panel), likely due to the abundance of the endogenous PML. In primary hepatocytes isolated from the Pml−/− mice, CAR activation failed to induce the subnuclear redistribution of PGC1α (Figure 4C) and inhibition of the FSK-responsive induction of gluconeogenic genes (Figure 4D). Moreover, treatment of primary mouse hepatocytes with As2O3, a PML degrading chemical (31), abolished the TCPOBOP-responsive degradation of PGC1α and CAR (Figure 4E). In vivo treatment of HFD-fed Pml−/− mice with TCPOBOP failed to reduce the protein level of PGC1α and CAR (Figure 4F) and suppress the gluconeogenic gene expression (Figure 4G). Moreover, the benefit of TCPOBOP in improving the performance of the IPGTT in WT mice was abolished in the Pml−/− mice (Figure 4H). Interestingly, CAR-induced PGC1α ubiquitination seemed to be PML independent, because the PGC1α ubiquitination was intact in the presence of As2O3 (Supplemental Figure 6), suggesting that PML-NBs might not be required for the ubiquitination of PGC1α but were indispensable for its degradation.
Figure 4.

PML-NBs are required for CAR to induce PGC1α degradation and suppress gluconeogenic gene expression. A, Immunofluorescent detection of PML and PGC1α in stable PML-knockdown Hepa1–6 cells generated by infecting cell with lenti-scramble or lenti-shPML and cotransfected with CAR and PGC1α (top panel). The effect of PML-knockdown on CAR-responsive inhibition of PGC1α activity was measured by the G6Pase luciferase reporter gene assay (bottom panel). B, Immunofluorescent detection of PML and PGC1α in Hepa1–6 cells cotransfected with CAR, PGC1α, and PML or empty vector (top panel). The effect of PML overexpression on CAR-responsive inhibition of PGC1α activity was measured by the G6Pase luciferase reporter gene assay (bottom panel). C, Primary hepatocytes isolated from WT and Pml−/− mice were infected with Ad-HA-PGC1α and Ad-Flag-CAR and treated with DMSO or TC (500nM) for 48 hours before immunofluorescent detection of PGC1α and PML. D, Primary hepatocytes from Pml−/− mice were pretreated with TC (500nM) or DMSO for 12 hours before FSK (10μM) treatment for 2 hours. Gene expression was measured by real-time PCR. E, 293T cells were cotransfected with Flag-PGC1α and HA-CAR and treated with TC (500nM) and A2O3 (10μM) as indicated. “Nonspecific” denotes the nonspecific band detected by the PGC1 antibody, which was used as a loading control. F and G, Pml−/− mice and their heterozygous littermates were fed with HFD for 4 weeks (n = 4 for each group), followed by treatment with TC (0.25 mg/kg, once per week) or vehicle for additional 2 weeks. Total liver lysates were subjected to immunoblotting for the detection of endogenous PGC1α and CAR (left panel). The quantification of the results is shown (right panel) (F). The same mice in were used to measure the expression of Pepck and G6Pase (G). H, Six-week-old WT and Pml−/− mice (n = 3–4 for each group) were fed with HFD for 2 weeks before receiving weekly TCPOBOP injections (0.25 mg/kg body weight) for 4 weeks while the HFD feeding continued. IPGTT was performed 2 days after the final dose of TCPOBOP. *, P < .05; **, P < .01; n.s., statistically not significant.
CAR recruits the Cullin1 E3 ligase to promote the ubiquitination of PGC1α
In understanding the mechanism by which CAR promotes PGC1α ubiquitination, we hypothesized that CAR may serve as an adaptor protein to present PGC1α to an E3 ligase for ubiquitination and subsequent degradation. Among a panel of E3 ubiquitin ligases, CAR showed ligand-independent interaction with Cullin1 (Figure 5A), the major structural scaffold protein of the Skp1-Cullin1-F box protein (SCF) complex (32). The interaction between CAR and Cullin1 complex was further supported by the coIP of CAR with Rbx1 and Skp1, another 2 core components of the Cullin1 complex (Figure 5B). It has been reported that Rbx1 and Cullin1 form a catalytic core complex that recruits a cognate E2 ubiquitin conjugating enzyme, and Skp1 serves as an adaptor to bring the F-box protein together with a specific substrate and Cullin1/Rbx1/E2 in the neighborhood (33). We showed that CAR interacted with the Cul1NT (Figure 5C). Interestingly, CAR retained its interaction with the mutant Cul1NT harboring mutations (Y46A/T47A/Y50A) that abolished the binding of Cullin1 to Skp1 (Figure 5C and Supplemental Figure 7). Furthermore, CAR interacted with both the WT Skp1 and its Cullin1-binding deficient mutant (Figure 5D and Supplemental Figure 7). Taken together, our results suggested that CAR formed a unique complex with Cullin1, which was mediated through both Cullin1 and Skp1. We also showed that PGC1α was coIP with Cullin1 in the presence of CAR in a ligand independent manner (Figure 5E), indicating that CAR is an adaptor protein that bridges PGC1α and the Cullin1 E3 complex. We then performed in vitro ubiquitination to directly demonstrate that the CAR-recruited E3 complex is capable of catalyzing PGC1α ubiquitination. The CAR-containing Cullin1 E3 ligase and PGC1α were expressed in cells and purified by immunoprecipitation (Supplemental Figure 8). Incubation of purified PGC1α with CAR-containing E3 ligase resulted in an increased PGC1α poly-ubiquitination (Figure 5F). In vivo ubiquitination assay showed that CAR was able to induce the ubiquitination of PGC1α, which was largely abolished by the cotransfection of a dominant-negative Cullin1 (DN-Cul1) (Figure 5G). At the functional level, cotransfection of DN-Cul1 (Figure 5H, left panel), or treatment with the Cullin1 inhibitor MLN4924 (Figure 5H, right panel) (34) largely abolished the inhibition of PGC1α activity by CAR in G6Pase luciferase reporter gene assays. These results collectively suggested that CAR functions as an adaptor protein to present PGC1α to the Cullin1 E3 complex for ubiquitination (Figure 5I).
Figure 5.

CAR recruits the Cullin1 E3 ligase to promote the ubiquitination of PGC1α. A, 293T cells were cotransfected with Flag-Cullin1 and HA-CAR and treated with or without TC (500nM) as indicated before being subjected to immunoprecipitation and immunoblotting. B, 293T cells were cotransfected with HA-CAR and Flag-Skp1 or Flag-Rbx1 as indicated before being subjected to immunoprecipitation and immunoblotting. The asterisk next to the top band denotes the heavy chain of IgG. C, 293T cells were cotransfected with HA-CAR and Flag-Cul1NT WT or mutant (Y46A/T47A/Y50A) as indicated before being subjected to immunoprecipitation and immunoblotting. D, 293T cells were cotransfected with HA-CAR and Flag-Skp1 WT or mutant (N108K, Y109N) as indicated before being subjected to immunoprecipitation and immunoblotting. E, 293T cells were cotransfected with HA-Cullin1, HA-CAR, and Flag-PGC1α and treated with or without TC (500nM) as indicated before being subjected to immunoprecipitation and immunoblotting. F, In vitro ubiquitination of PGC1α by CAR-containing Cullin1/SCF E3 ligase complex. CAR-containing Cullin1/SCF E3 complex was purified and mixed with purified PGC1α (see Supplemental Figure 8 for details) in the presence of E1, E2, ubiquitin, and ATP. G, 293T cells were cotransfected with HA-ubiquitin (Ub), HA-CAR, Flag-PGC1α, and DN-Cul1, followed by immunoprecipitation and immunoblotting as indicated. *, P < .05. H, Cotransfection of DN-Cul1 (left panel) or treatment with SCF inhibitor MLN4924 (right panel) abolished the inhibition of PGC1α activity by CAR in G6Pase reporter gene assay (*, P < .05). I, Proposed formation of CAR-associated E3 ligase and mechanism of CAR-mediated ubiquitination of PGC1α.
CAR-mediated inhibition of PGC1α requires active AF2 domain but is independent of DNA-binding
Our results suggested that the interaction between CAR and PGC1α was sufficient to induce PGC1α ubiquitination, but the subsequent PGC1α degradation (Figure 3C) and maximum inhibition of PGC1α activity required the presence of CAR ligand (Figure 1A). To determine whether the ligand-bound conformation of CAR was necessary to trigger PGC1α degradation, we generated 2 CAR mutants with the H12/AF2 deletion (D8) and H11-H12 deletion (D30), respectively. Both D8 and D30 mutants retained their ability to interact with PGC1α and PML (Supplemental Figure 9A), to induce the redistribution of PGC1α to PML-NBs (Supplemental Figure 9B), and to trigger PGC1α ubiquitination (Supplemental Figure 9C). However, both mutants failed to induce PGC1α and CAR degradation in the presence of TCPOBOP (Figure 6A), or to suppress PGC1α-responsive activation of the G6Pase luciferase reporter gene (Figure 6B). We also generated the L346F mutant of CAR that was reported to stabilize the AF2 helix in the active conformation and mimic the TCPOBOP-bound receptor (35). Compared with the WT CAR, transfection of the L346F mutant reduced the basal protein level of the cotransfected PGC1α, and TCPOBOP had less effect in promoting PGC1α and CAR degradation (Figure 6C). The proteasome inhibitor lactacystin was able to stabilize the PGC1α and CAR proteins regardless of the mutation of CAR (Figure 6C). These results suggested that the L346F mutation destabilized both CAR and PGC1α by enhancing their proteasome-mediated degradation. In the G6Pase luciferase reporter gene assay, the L346F mutant was more efficient than WT CAR in inhibiting PGC1α activity, and this inhibition cannot be further enhanced by TCPOBOP (Figure 6D).
Figure 6.

CAR-mediated inhibition of PGC1α requires active AF2 domain but is independent of DNA-binding. A, Cotransfection of PGC1α and WT CAR or D8 and D30 in 293T cells with or without TC (500nM) treatment for 24 hours, followed by immunoblotting to detect the protein level of PGC1α and CAR. B, The suppressive effect of CAR D8 and D30 on PGC1α activity was measured by G6Pase luciferase reporter assay. C, Cotransfection of PGC1α and WT CAR or L346F mutant in 293T cells with or without TC treatment (500nM, 24 h), followed by immunoblotting to detect the protein level of PGC1α and CAR. “Nonspecific” denotes the nonspecific band detected by the PGC1 antibody, which was used as a loading control. D, The suppressive effect of CAR L346F mutant on PGC1α activity was measured by G6Pase luciferase reporter assay. E, The transcriptional activity of WT CAR or CC/AA was measured by using the tk-PBRE reporter assay. PBRE, phenobarbital-response element. F, HepG2 cells were cotransfected with PGC1α, WT CAR or CC/AA, followed by immunofluorescent detection of PGC1α, CAR, and the endogenous PML. G, Cotransfection of PGC1α and WT CAR or CC/AA in 293T cells with or without TC treatment, followed by immunoblotting to detect PGC1α and CAR. H, The suppressive effect of WT CAR or CC/AA on PGC1α activity was measured by G6Pase luciferase reporter assay.
CAR is a DNA-binding transcriptional factor. We then used the CC/AA DNA-binding deficient mutant of CAR (9) to determine whether the transcriptional targets of CAR are required for its inhibition of PGC1α. The CC/AA mutant failed to transactivate its reporter gene as expected (Figure 6E). To our surprise, we found the CC/AA mutant retained its ability to induce the redistribution of PGC1α to PML-NBs (Figure 6F) and degradation of PGC1α (Figure 6G), and to suppress the PGC1α-responsive activation of the G6Pase luciferase reporter gene (Figure 6H).
Discussion
As summarized in Figure 7, our results revealed a novel molecular mechanism by which CAR inhibits gluconeogenesis by posttranslationally antagonizing PGC1α, a key gluconeogenic transcriptional factor. In this model, after ligand activation, CAR translocates from the cytoplasm into the nucleus to serve as an adaptor protein to present PGC1α to the Cullin1 E3 ligase for ubiquitination. The interaction between PML and the CAR-PGC1α complex lead to the redistribution of PGC1α and CAR to the PML-NBs, where the degradation of PGC1α occurs. As a result, CAR activation reduces the recruitment of PGC1α to the gluconeogenic gene promoters and suppresses hepatic glucose production.
Figure 7.
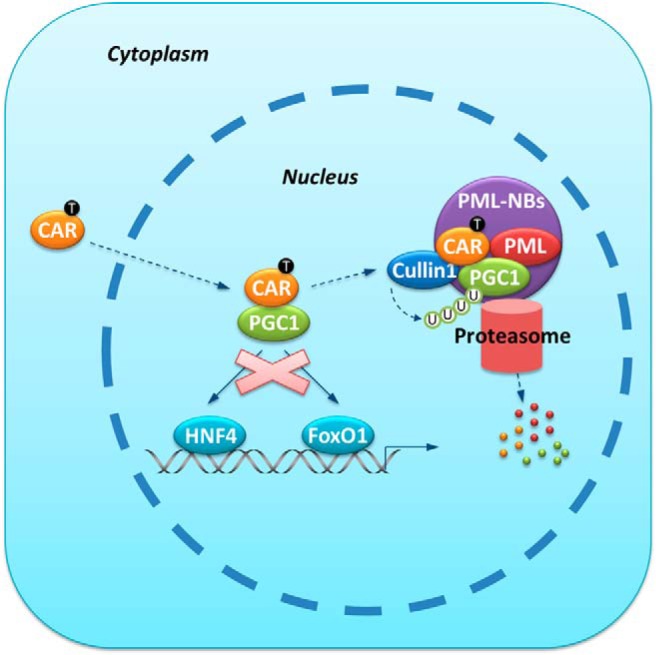
Mechanism of CAR-mediated suppression of hepatic gluconeogenesis. Hepatic gluconeogenic gene expression is facilitated by PGC1α-mediated coactivation of transcription factors such as HNF4 and FoxO1 during fasting or under the diabetic condition. When bound by its ligand TCPOBOP, CAR enters the nucleus and physically interacts with PGC1α. The CAR-PGC1α interaction results in the targeting of both proteins to the PML-NBs, where CAR-associated Cullin1 E3 ligase modifies PGC1α with poly-ubiquitin chain and promotes the proteasomal degradation of PGC1α. The gluconeogenic gene expression is inhibited due to the compromised availability of the PGC1α protein.
One of our most interesting findings is the recruitment of the Cullin1 E3 ligase complex by CAR. CAR is previously known as a master regulator of xenobiotic metabolism through its transcriptional regulation of drug metabolizing enzymes and transporters. To our knowledge, the recruitment of the Cullin1 E3 ligase complex and subsequent ubiquitination of PGC1α is the first example that CAR controls the protein turnover of a gluconeogenic transcriptional factor, which may have accounted for the antidiabetic activity of CAR that we and others have reported (2, 3). The Cullin1 E3 ligase/SCF complex and SCF-like complexes belong to the largest family of E3 ligases whose substrates include a broad range of proteins involved in cell cycle progression, signal transduction, and gene transcription (36). Cdc4, an F-box component of the SCF complex, has been reported to target PGC1α for proteasomal degradation in a phosphorylation-dependent manner (37). In our study, we found no evidence that activation of CAR affected the phosphorylation of PGC1α. Instead, our data suggested that CAR interacted with the SCF complex in a ligand-independent manner, and served as an adaptor protein to bring PGC1α to the SCF complex for ubiquitination. Interestingly, the degradation of PGC1α after ubiquitination requires the presence of a CAR agonist, although the transcriptional targets of CAR are not required for the degradation because the DNA binding was dispensable. It is possible that the agonist-occupied conformation of CAR-PGC1α is required to recruit additional proteasome activators and trigger the degradation.
PGC1α is a transcriptional coactivator expressed in many tissues with high and fluctuating energy demands, such as the liver, skeletal muscle, heart, and brown adipose tissue. PGC1α has been established as a master regulator of mitochondrial biogenesis and energy expenditure. As a critical metabolic orchestrator, the activity of PGC1α is tightly regulated at both the transcriptional and posttranslational levels. To date, posttranslational modifications such as phosphorylation, methylation, acetylation, and GlcNAcylation on PGC1α have been reported to weave a multifaceted and flexible system to regulate its activity (25). PGC1α is a short-lived protein with a quick turnover rate (29), but little is known about the regulation of PGC1α protein stability. p38/MAPK-mediated phosphorylation of PGC1α has been reported to increase its stability (38), whereas glycogen synthase kinase-3β accelerates the proteasomal degradation of PGC1α in response to oxidative stress (39). Our results suggested that the regulation of PGC1α degradation by CAR was independent of phosphorylation. Instead, CAR modulates PGC1α activity by altering its subcellular localization and turnover rate. As the major gluconeogenic transcriptional factor, PGC1α has been strongly associated with diabetes. The hepatic PGC1α activity is robustly up-regulated in the diabetic animal models and human patients, which may have contributed to increased hepatic glucose production and hyperglycemia (11). Because the expression of CAR is highly enriched in the liver, suppressing PGC1α through CAR activation may provide a novel therapeutic strategy to specifically targeting the hepatic PGC1α in diabetic conditions, without interfering with the metabolic benefits of PGC1α in extrahepatic tissues, such as the skeletal muscle and heart.
Another interesting finding is the requirement of PML in the CAR-mediated PML-NBs-targeting and degradation of PGC1α. PML-NBs are macromolecular nuclear structures implicated in the regulation of diverse cellular functions. The current models envision PML-NBs as a glue to recruit and concentrate partners along with many protein-modifying enzymes, and subsequently enhance posttranslational modifications, leading to the activation, sequestration or degradation of proteins (40). Consistent with this model, our study showed PML-NBs were indispensable for CAR-induced degradation of PGC1α and suppression of gluconeogenesis. The Cullin1 E3 ligase/SCF complex, which was responsible for the ubiquitination of PGC1α, has been reported to reside within the PML-NBs (41). Our results are also consistent with a recent report that PML ablation in mice induced hepatic gluconeogenic genes, leading to glucose intolerance and insulin resistance (15).
Among the limitations, we are aware that the adenoviral overexpression system used in this study has its limitation in terms of the physiological relevance. In addition to the dynamics of the transfected proteins, we also included data showing that the proposed mechanism is applicable to the endogenous proteins (Figure 1, B–E, and Figure 4, C, D, and F–H). Nevertheless, future studies are necessary to further validate the molecular mechanism in more physiological conditions.
In summary, our study revealed a novel molecular mechanism, through which CAR posttranslationally antagonizes PGC1α. CAR and PGC1α are master regulators of drug metabolism and gluconeogenesis, respectively. Drug metabolism/detoxification is an essential cellular function that demands energy. Gluconeogenesis is also an energy-demanding process that requires a large amount of ATP to generate sufficient nicotinamide adenine dinucleotide phosphate, a reducing power also needed for the cytochrome P450 enzymes to eliminate noxious chemicals. The negative regulation of PGC1α by CAR may represent a cellular adaptive mechanism to cope with energy deficiency under energy-restricted conditions.
Acknowledgments
We thank Dr Marc R. Montminy (Salk Institute, La Jolla, CA) for the Ad-PGC1α RNAi and scrambled RNAi adenoviruses, Dr Richard M. O'Brien (Vanderbilt University, Nashville, TN) for the G6Pase-luciferase reporter plasmid, and Dr Jongsook Kim Kemper (University of Illinois at Urbana-Champaign, Champaign, IL) for the pcDNA-CAR (CC/AA) plasmid. We also thank Mengxi Jiang for her assistance in constructing some of the WT and mutant PGC1alpha and CAR plasmids.
This work was supported in part by National Institutes of Health Grants DK083952 and DK099232 (to W.X.). W.X. is also supported by the Joseph Koslow Endowed Professorship from the University of Pittsburgh School of Pharmacy.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Ad-Flag-CAR
- flag-tagged mouse CAR adenovirus
- Ad-HA-PGC1α
- HA-tagged PGC1α adenovirus
- CAR
- constitutive androstane receptor
- CBM
- coactivator-binding mutant
- ChIP
- chromatin immunoprecipitation
- coIP
- coimmunoprecipitation
- Cul1NT
- N terminus of Cullin1
- DMSO
- dimethyl sulfoxide
- DN-Cul1
- dominant-negative Cullin1
- FoxO1
- forkhead box protein O1
- FSK
- forskolin
- G6Pase
- glucose-6-phosphatase
- β-gal
- β-galactosidase
- HFD
- high-fat diet
- HNF4α
- hepatocyte nuclear factor 4α
- IPGTT
- ip glucose tolerance test
- PEPCK
- phosphoenolpyruvate carboxykinase
- PPAR
- peroxisome proliferator-activated receptor
- PGC1α
- PPARγ coactivator-1α
- PML-NB
- promyelocytic leukemia protein-nuclear body
- RNAi
- RNA interference
- SCF
- Skp1-Cullin1-F box protein
- TCPOBOP
- 1,4-Bis-[2-(3,5 dichloropyridyloxy)]benzene
- WT
- wild type.
References
- 1. Willson TM, Kliewer SA. PXR, CAR and drug metabolism. Nat Rev Drug Discov. 2002;1(4):259–266. [DOI] [PubMed] [Google Scholar]
- 2. Gao J, He J, Zhai Y, Wada T, Xie W. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J Biol Chem. 2009;284(38):25984–25992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dong B, Saha PK, Huang W, et al. Activation of nuclear receptor CAR ameliorates diabetes and fatty liver disease. Proc Natl Acad Sci USA. 2009;106(44):18831–18836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Masuyama H, Hiramatsu Y. Treatment with a constitutive androstane receptor ligand ameliorates the signs of preeclampsia in high-fat diet-induced obese pregnant mice. Mol Cell Endocrinol. 2012;348(1):120–127. [DOI] [PubMed] [Google Scholar]
- 5. Sotaniemi EA, Arranto AJ, Sutinen S, Stengård JH. Treatment of noninsulin-dependent diabetes mellitus with enzyme inducers. Clin Pharmacol Ther. 1983;33(6):826–835. [DOI] [PubMed] [Google Scholar]
- 6. Lahtela JT, Särkkä P, Sotaniemi EA. Phenobarbital treatment enhances insulin mediated glucose metabolism in man. Res Commun Chem Pathol Pharmacol. 1984;44(2):215–226. [PubMed] [Google Scholar]
- 7. Lahtela JT, Arranto AJ, Sotaniemi EA. Enzyme inducers improve insulin sensitivity in non-insulin-dependent diabetic subjects. Diabetes. 1985;34(9):911–916. [DOI] [PubMed] [Google Scholar]
- 8. Kodama S, Koike C, Negishi M, Yamamoto Y. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol Cell Biol. 2004;24(18):7931–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miao J, Fang S, Bae Y, Kemper JK. Functional inhibitory cross-talk between constitutive androstane receptor and hepatic nuclear factor-4 in hepatic lipid/glucose metabolism is mediated by competition for binding to the DR1 motif and to the common coactivators, GRIP-1 and PGC-1α. J Biol Chem. 2006;281(21):14537–14546. [DOI] [PubMed] [Google Scholar]
- 10. Herzig S, Long F, Jhala US, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413(6852):179–183. [DOI] [PubMed] [Google Scholar]
- 11. Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116(3):615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8(12):1006–1016. [DOI] [PubMed] [Google Scholar]
- 13. Ito K, Carracedo A, Weiss D, et al. A PML-PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18(9):1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carracedo A, Weiss D, Leliaert AK, et al. A metabolic prosurvival role for PML in breast cancer. J Clin Invest. 2012;122(9):3088–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheng X, Guo S, Liu Y, et al. Ablation of promyelocytic leukemia protein (PML) re-patterns energy balance and protects mice from obesity induced by a Western diet. J Biol Chem. 2013;288(41):29746–29759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang ZG, Delva L, Gaboli M, et al. Role of PML in cell growth and the retinoic acid pathway. Science. 1998;279(5356):1547–1551. [DOI] [PubMed] [Google Scholar]
- 17. Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature. 2000;407(6806):920–923. [DOI] [PubMed] [Google Scholar]
- 18. Monsalve M, Wu Z, Adelmant G, Puigserver P, Fan M, Spiegelman BM. Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Mol Cell. 2000;6(2):307–316. [DOI] [PubMed] [Google Scholar]
- 19. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92(6):829–839. [DOI] [PubMed] [Google Scholar]
- 20. Jin J, Ang XL, Shirogane T, Wade Harper J. Identification of substrates for F-box proteins. Methods Enzymol. 2005;399:287–309. [DOI] [PubMed] [Google Scholar]
- 21. Schilling MM, Oeser JK, Boustead JN, Flemming BP, O'Brien RM. Gluconeogenesis: re-evaluating the FOXO1-PGC-1α connection. Nature. 2006;443(7111):E10–E11. [DOI] [PubMed] [Google Scholar]
- 22. Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1α. Cell Metab. 2006;3(6):429–438. [DOI] [PubMed] [Google Scholar]
- 23. Koo SH, Flechner L, Qi L, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437(7062):1109–1111. [DOI] [PubMed] [Google Scholar]
- 24. Gao D, Wan L, Inuzuka H, et al. Rictor forms a complex with Cullin-1 to promote SGK1 ubiquitination and destruction. Mol Cell. 2010;39(5):797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-γ coactivator 1 α (PGC-1 α): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24(1):78–90. [DOI] [PubMed] [Google Scholar]
- 26. Shiraki T, Sakai N, Kanaya E, Jingami H. Activation of orphan nuclear constitutive androstane receptor requires subnuclear targeting by peroxisome proliferator-activated receptor γ coactivator-1 α. A possible link between xenobiotic response and nutritional state. J Biol Chem. 2003;278(13):11344–11350. [DOI] [PubMed] [Google Scholar]
- 27. Ding X, Lichti K, Kim I, Gonzalez FJ, Staudinger JL. Regulation of constitutive androstane receptor and its target genes by fasting, cAMP, hepatocyte nuclear factor α, and the coactivator peroxisome proliferator-activated receptor γ coactivator-1α. J Biol Chem. 2006;281(36):26540–26551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhong S, Hu P, Ye TZ, Stan R, Ellis NA, Pandolfi PP. A role for PML and the nuclear body in genomic stability. Oncogene. 1999;18(56):7941–7947. [DOI] [PubMed] [Google Scholar]
- 29. Sano M, Tokudome S, Shimizu N, et al. Intramolecular control of protein stability, subnuclear compartmentalization, and coactivator function of peroxisome proliferator-activated receptor γ coactivator 1α. J Biol Chem. 2007;282(35):25970–25980. [DOI] [PubMed] [Google Scholar]
- 30. Dussault I, Lin M, Hollister K, et al. A structural model of the constitutive androstane receptor defines novel interactions that mediate ligand-independent activity. Mol Cell Biol. 2002;22(15):5270–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang XW, Yan XJ, Zhou ZR, et al. Arsenic trioxide controls the fate of the PML-RARα oncoprotein by directly binding PML. Science. 2010;328(5975):240–243. [DOI] [PubMed] [Google Scholar]
- 32. Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol. 2008;9(9):679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zheng N, Schulman BA, Song L, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416(6882):703–709. [DOI] [PubMed] [Google Scholar]
- 34. Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. [DOI] [PubMed] [Google Scholar]
- 35. Suino K, Peng L, Reynolds R, et al. The nuclear xenobiotic receptor CAR: structural determinants of constitutive activation and heterodimerization. Mol Cell. 2004;16(6):893–905. [DOI] [PubMed] [Google Scholar]
- 36. Deshaies RJ. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;15:435–467. [DOI] [PubMed] [Google Scholar]
- 37. Olson BL, Hock MB, Ekholm-Reed S, et al. SCFCdc4 acts antagonistically to the PGC-1α transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev. 2008;22(2):252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Puigserver P, Rhee J, Lin J, et al. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Mol Cell. 2001;8(5):971–982. [DOI] [PubMed] [Google Scholar]
- 39. Anderson RM, Barger JL, Edwards MG, et al. Dynamic regulation of PGC-1α localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7(1):101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lallemand-Breitenbach V, de The H. PML nuclear bodies. Cold Spring Harb Perspect Biol. 2010;2(5):a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shima Y, Shima T, Chiba T, Irimura T, Pandolfi PP, Kitabayashi I. PML activates transcription by protecting HIPK2 and p300 from SCFFbx3-mediated degradation. Mol Cell Biol. 2008;28(23):7126–7138. [DOI] [PMC free article] [PubMed] [Google Scholar]