

Abstract
The melanocortin-4 receptor (MC4R) is a G protein-coupled receptor expressed in the brain, where it controls energy balance through pathways including α-melanocyte-stimulating hormone (α-MSH)-dependent signaling. We have reported that the MC4R can exist in an active conformation that signals constitutively by increasing cAMP levels in the absence of receptor desensitization. We asked whether synthetic MC4R agonists differ in their ability to increase intracellular cAMP over time in Neuro2A cells expressing endogenous MC4R and exogenous, epitope-tagged hemagglutinin-MC4R-green fluorescent protein. By analyzing intracellular cAMP in a temporally resolved Förster resonance energy transfer assay, we show that withdrawal of α-MSH leads to a quick reversal of cAMP induction. By contrast, the synthetic agonist melanotan II (MTII) induces a cAMP signal that persists for at least 1 hour after removal of MTII from the medium and cannot be antagonized by agouti related protein. Similarly, in mHypoE-42 immortalized hypothalamic neurons, MTII, but not α-MSH, induced persistent AMP kinase signal, which occurs downstream of increased cAMP. By using a fluorescence recovery after photobleaching assay, it appears that the receptor exposed to MTII continues to signal after being internalized. Similar to MTII, the synthetic MC4R agonists, THIQ and BIM-22511, but not LY2112688, induced prolonged cAMP signaling after agonist withdrawal. However, agonist-exposed MC4R desensitized to the same extent, regardless of the ligand used and regardless of differences in receptor intracellular retention kinetics. In conclusion, α-MSH and LY2112688, when compared with MTII, THIQ, and BIM-22511, vary in the duration of the acute cAMP response, showing distinct temporal signaling selectivity, possibly linked to specific cell compartments from which cAMP signals may originate.
The melanocortin-4 receptor (MC4R) is a G protein-coupled receptor (GPCR) expressed in the brain, where it controls food intake and energy expenditure. The MC4R is regulated by a natural agonist, α-melanocyte-stimulating hormone (α-MSH), and an antagonist/inverse agonist, agouti related peptide (AGRP). The binding of α-MSH to the MC4R leads to decreased food intake and increased energy expenditure, thus promoting weight loss (1–4). Exposure of MC4R to α-MSH induces a Gs-mediated increase of intracellular cAMP, which leads to decreased activity of AMP kinase (AMPK), a signaling pathway implicated in the regulation of food intake (5). In some cell types, α-MSH also increases intracellular calcium concentration (6–8). Several synthetic MC4R agonists have been generated in an effort to develop therapies to induce weight loss in obese humans. The relatively nonselective melanocortin receptor agonist melanotan II (MTII), and the more selective MC4R agonists THIQ, BIM-22511, and LY2112688 appear to affect energy balance in addition to other functions thought to be modulated to different extents in vivo by the MC4R. Those include glucose metabolism, cardiovascular tone, sexual function, gastric motility, and inflammation (9–14). The distinct physiological effects of MC4R agonists in vivo may derive from agonist-specific differences in receptor activation. A potential example of this is in the small molecule MC4R agonist THIQ, which activates cAMP similarly to α-MSH but is significantly impaired in calcium mobilization and receptor internalization (8). However, it is unknown whether functional selectivity of distinct MC4R agonists exists for the kinetics of cAMP signaling itself. It has been reported that, unlike α-MSH, the MC1R agonists Melanotan I, MTII, and 4-norleucine,7-D-phenylalanine-α-MSH have prolonged biological activity to darken frog skin (13–17). Frog skin darkening by α-MSH regulates melanin synthesis in melanocytes through the MC1R (15–19). Based on these observations we aimed to determine whether natural and synthetic MC4R agonists differ with respect to the temporal features by which the cAMP signal is generated upon acute agonist exposure. Chronic exposure to α-MSH leads to loss of MC4R activity, due to receptor desensitization (20, 21). Loss of MC4R function upon chronic exposure to MC4R agonists could limit the effects on food intake and body weight in vivo (22, 23). We have found that exposure of the MC4R to α-MSH in the endoplasmic reticulum leads to a sustained cAMP signal that is comparable with the level of cAMP activation obtained by acute exposure to α-MSH, without evidence of receptor desensitization (24). Interestingly, it has been proposed that the MC4R can exists in a complex with extracellular synthetic agonists possibly attaining multiple conformational states, that may each differ in their intracellular trafficking properties (8). Here, we have tested whether α-MSH and several MC4R synthetic agonists known to variably affect MC4R function in rodents, monkeys and humans (9–14) differ in the temporal characteristics by which the cAMP signal is generated in response to acute and chronic agonist exposure. To this end, we have used temporally resolved, fluorescence-based assays and biochemical assays to monitor the cAMP signal as well as the intracellular localization of tagged hemagglutinin (HA)-MC4R-green fluorescence protein (GFP) in neuronal cells that also express endogenous MC4R. The data indicate that distinct MC4R agonists can exert temporal selectivity to modulate the duration of the cAMP signal and potentially the cellular compartment from which the signal is generated in response to acute agonist stimulation.
Materials and Methods
Materials
Lipofectamine 2000 was purchased from Life technologies. Rat monoclonal anti-HA antibodies (3F10 clone), peroxidase-conjugated rat monoclonal anti-HA antibodies (3F10 clone), 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) tablets and buffer were purchased from Roche Applied Sciences. α-MSH, 3-isobutyl-1-methylxanthine (IBMX), and poly-L-lysine were from Sigma-Aldrich. Agouti-related protein (human, 86–132) was obtained from Peptides International. MTII and THIQ were obtained from Tocris (Bio-Techne). MTII [Gly]-rhodamine labeled was purchased from Phoenix Pharmaceuticals, Inc. Rabbit polyclonal anti-AMPK and antiphospho-AMPK antibodies (catalog numbers 2535 and 2532) were purchased from Cell Signaling Technology. The Calbiochem dynamin inhibitor V 34–2 (V 34–2) was purchased from EMD Millipore. Cy3-conjugated conjugated antirat IgG were from Jackson ImmunoResearch. Peroxidase Labeled Goat antirabbit IgG (H+L) was from Kirkegaard & Perry Laboratories Antibody. The bicinchoninic assay protein assay reagent, secondary peroxidase (POD)-conjugated antirat IgG, was from Pierce. Bicinchoninic assay protein assay reagent was purchased from PerkinElmer Life Sciences. Formaldehyde (16%) was from Ted Pella, Inc. The direct cAMP enzyme immunoassay kit was from Enzo Life Science, Inc (product ADI-900–066). G418 (30–234-CR) and Eagle's Medium (DMEM, 10–013-CV) were obtained from Corning Cellgro. Glass bottom dishes with a diameter of 35 mm coated with poly-D-lysine were obtained from MatTek Corp. The embryonic mouse hypothalamic cell line, mHypoE42, was purchased from Cedarlane Laboratories. V 34–2-Calbiochem was from EMD Millipore.
Cell culture and transfection
Cells that stably express HA-MC4R-GFP were derived from N2A cells (N2AHA-MC4R-GFP cells) (20). Cells were cultured in DMEM supplemented with L-glutamine and sodium pyruvate and 10% fetal bovine serum (Atlas Biologicals), penicillin and streptomycin, 4mM L-glutamine, and 100-μg/mL G418 to maintain expression of HA-MC4R-GFP. N2AHA-MC4R-GFP cells were transiently transfected with the indicated plasmids using Lipofectamine 2000, following the manufacturer's instructions.
Internalization HA-MC4R-GFP by ELISA
Internalization of HA-MC4R-GFP by immunoassay was measured as previously described (20). Briefly, N2AHA-MC4R-GFP cells were split into 24-well plates 24 hours before the assays were performed. Cells were washed 3 times with 250-μL DMEM per well and incubated in DMEM for 1 hour at 37°C. Cells were incubated in the presence or absence of agonist for 15 minutes at 4°C, then 25-mU/mL POD-conjugated anti-HA antibody was added to the incubation medium, and cells were further incubated for 1 hour at 4°C, to label the cell surface receptor. After the incubation with POD-conjugated anti-HA antibody cells were washed 3 times at 4°C, after which cells were incubated at 37°C for 0, 15, 30, or 60 minutes in the presence of the agonist, to allow endocytosis of the receptor. Cells were subsequently fixed in PBS containing 4% formaldehyde at 4°C for 15 minutes. Cells were washed with PBS 3 times, and POD activity was measured by incubating cells for 1 hour at 37°C with a membrane-impermeable POD substrate (2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid), 1 mg/mL) following the manufacturer's instructions. The oxidized product was detected by reading its absorbance at 405 nm using an mQuant universal microplate spectrophotometer (Bio-Tek Instruments, Inc).
Internalization HA-MC4R-GFP monitored by fluorescence recovery after photobleaching (FRAP)
N2AHA-MC4R-GFP cells were plated onto glass bottom dishes. Approximately 24 hours after plating, the cells were treated with 1μM α-MSH, 200nM MTII for 1 hour, or cells were maintained as untreated controls. Cells were transferred at 37°C using a temperature-control system (BioscienceTools) on the stage of the Olympus FluoView FV1000 confocal microscope. In each field of view, obtained by using the 100× apochromatic objective, a single cell was selected at random at a time, and region of interest (ROI) was drawn around the endosomal compartment. Images were collected at 30-second intervals for the 17 minutes of duration of the experiment using the 488-nm laser for each measurement. Photobleaching at the ROI took place at min 2.5 using the 405-nm diode laser and the Tornado scanning option. The average fluorescent intensity of ROI at each time point was monitored over time. Rate constants of FRAP experiments were derived by fitting the 1-phase exponential association model to the data by using GraphPad Software.
Changes in HA-MC4R-GFP distribution monitored by live-cell confocal fluorescence microscopy
Confocal fluorescence microscopy was used to monitor changes in distribution of intracellular HA-MC4R-GFP in the intracellular compartment. N2AHA-MC4R-GFP cells were plated onto glass bottom dishes at least 16 hours before the start of ligand treatment. Cells were washed 3 times with DMEM and incubated in DMEM for 1 hour at 37°C. Cells were further incubated in the presence or absence of agonist for 4 hours at 37°C. Live cells were kept at 37°C using a temperature-control system (BioscienceTools) on the stage of the Olympus FluoView FV1000 confocal microscope. Images were taken using the 488-nm laser. Fluorescence intensities of ROIs drawn immediately outside of the cell margin (total fluorescence) and around the intracellular GFP (intracellular fluorescence) were measured by using ImageJ software (Wayne Rasband, National Institutes of Health, http://rsbweb.nih.gov/ij/). Changes in intracellular fluorescence were monitored by measuring changes in the ratio intracellular GFP fluorescence intensity to total GFP fluorescence intensity.
Changes in HA-MC4R-GFP distribution by the V 34–2 monitored by fixed cell confocal fluorescence microscopy
N2AHA-MC4R-GFP cells were plated onto coverslips in 3-cm diameter dishes 24 hours before the experiments. Cells were washed 3 times with DMEM, incubated in DMEM for 1 hour at 37°C, and further incubated without and with 12.5μM V 34–2 at 37°C. Cells were transferred at 4°C, washed 3 times with ice-cold PBS, fixed with 4% formaldehyde (vol/vol) at 4°C for 30 minutes, washed 3 times with PBS, and incubated with rat anti-HA antibodies (1:25 dilution) in PBS containing 100-μg/mL ovalbumin. Secondary staining with Cy3-conjugated antirat antibodies was also performed without addition of detergents. Images were taken by using the Olympus FluoView FV1000 confocal microscope. The Cy3 fluorescence intensity monitors abundance of the HA epitope of HA-MC4R-GFP at the cell surface and GFP intensity monitors total abundance of HA-MC4R-GFP. The Fluorescence intensities of Cy3 and GFP in ROIs drawn immediately outside of the cell margin were measured by using ImageJ software.
cAMP determination by immunoassay
Assay to measure cAMP generation by N2AHA-MC4R-GFP cells in response to acute exposure to MC4R agonists
Cells were plated on 24-well plates 24 hours before the experiment was performed (25). Cells were washed 3 times with 250-μL DMEM and incubated in 250-μL DMEM for 1 hour at 37°C. Cells were treated with 250-μL DMEM containing 500μM IBMX for 10 minutes, and then with 250-μL DMEM containing the indicated MC4R agonists together with 500μM IBMX for additional 15 minutes. Cells were transferred on ice, lysed in 0.1M HCl containing 0.1% Triton X-100 and 500μM IBMX. Lysis volumes were 200 μL for N2A and N2AMC4R cells. Cell lysates were then cleared by centrifugation at 16 000g for 10 minutes. The supernatants were diluted 1:20, and cAMP was measured by using an enzyme immunoassay kit was from Enzo Life Science, Inc according to the manufacturers instruction. Data were analyzed by using GraphPad Prism software (nonlinear regression curve) to obtain the concentration of cAMP in the samples.
Desensitization assay
N2AHA-MC4R-GFP cells were plated onto 24-well plates approximately 24 hours before the experiment. Cells were washed 3 times with 250-μL DMEM and treated at 37°C for 4, 24, 48, 72, and 96 hours, as indicated in the figures, with no additions or with MC4R agonists at the indicated concentrations. In experiments using the chronic incubation with agonists (24, 48, 72, and 96 h), the cell medium was changed every 8 hours. Cells were then washed 3 times with 250-μL DMEM at room temperature and returned to 37°C for 10 minutes in DMEM containing 500μM IBMX. The cells pretreated with MC4R agonists were reexposed to the same agonist for 15 minutes in 250-μL DMEM containing 500μM IBMX. Cells that were not preexposed to agonists were used as controls to measure cAMP generation in response to acute exposure to the MC4R agonists in the same experiment.
Assay to measure cAMP signaling after agonist wash-off
N2AHA-MC4R-GFP cells were plated onto 24-well plates approximately 24 hours before the experiment. Cells were washed 3 times with 250-μL DMEM and treated for 1 hour with α-MSH and MTII at 37°C. Cells were then washed 3 times in 250-μL DMEM and returned to 37°C for 0, 30, 60, or 90 minutes. Samples were subsequently treated with 250-μL DMEM and with 500μM IBMX to measure the amount of cAMP generated in 15 at 37°C in the absence of any added agonists. Cells were lysed and the cell cAMP concentration was measured as described above.
Assay to measure α-MSH and MTII-induced AMPK signal in immortalized mouse hypothalamic mHypoE-42 neurons
The immortalized mouse hypothalamic neurons mHypoE-42 cells were plated onto 24-well plates approximately 24 hours before the start of the experiment. Cells were washed 3 times with 250-μL DMEM and incubated with DMEM at 37°C for 16 hours. Cells were then washed 1 time in 250-μL DMEM and returned to 37°C for 3 hours. Samples were subsequently treated as follows: 1) with and without 2nM MTII or 200nM α-MSH for 30 minutes (to monitor the acute AMPK signal); and 2) with 2nM MTII and 200nM α-MSH for 30 minutes followed by 3 washes with 250-μL DMEM and further incubated at 37°C for 1 hour in the absence of any added agonists (to monitor persistent AMPK signal). Cells were washed with 500 μL of ice-cold 1× PBS and incubated for 1 hour at 4°C in a lysis buffer (0.3 mL) containing 10mM Tris-HCL, 150mM NaCl, 1% Triton X-100, and phosphatase inhibitors and protease inhibitors. Cells were scraped and homogenized with 4 strokes in a 2-mL glass homogenizer equipped with a Teflon pestle. Samples were centrifuged at 16 000g for 20 minutes to obtain cleared cell lysates which were mixed with 2× sample buffer before loading onto SDS-PAGE gels for Western blot analysis as previously described (24). Quantification of the band intensity was carried out by using the Analyze →Gels →Plot Lanes commands of the ImageJ software.
Assay to measure MTII EC50
For determination of EC50, of MTII and THIQ data were analyzed by using GraphPad Prism software (sigmoid dose-response curve)
Real-time Förster resonance energy transfer (FRET)-based cAMP assay
The assay was carried out as previously described (24). Briefly, N2AHA-MC4R-GFP cells were transiently transfected with mTurquoise-Epac-YFP (TEPACVV) in 60-mm dishes. Cells were plated onto 35 mm glass bottom dishes approximately 24 hours after transfection. Approximately 24 hours after plating, cells were washed 3 times in 250-μL DMEM incubated in 250-μL DMEM for 1 hour at 37°C. The experiments were performed at 37°C using the Olympus FluoView FV1000 confocal microscope equipped with a heating stage under a temperature-control system (BioscienceTools). Cells with above-background YFP and mTurquoise fluorescence were treated with MC4R agonists and with the MC4R antagonist AgRP as indicated in the figures. Cells were imaged at least twice per minute. mTurquoise was excited at 458 nm, and the intensities of fluorescence emitted at 480–495 nm (mTurquoise) and at 535–565 nm (YFP) were recorded over time. The ratio intensity of mTurquoise fluorescence to intensity of YFP fluorescence was calculated using the FluoViewTM software from Olympus and graphed using GraphPad Prism version 5.0. Bleed-through of mTurquoise (14.8 ± 3.08%, number of cells = 55) and cross talk of YFP (3.08 ± 0.62%, number of cells = 15) were calculated by using cells transfected with either mTurquoise or YFP, respectively, to obtain the corrected FRET ratio as described by others (26). Where indicated, the ratio intensity of mTurquoise fluorescence to intensity of YFP fluorescence was then set as 0% of the signal and the ratio intensity of mTurquoise fluorescence to intensity of YFP fluorescence in response to addition of either forskolin/IBMX or to initial exposure to the agonist was set as 100% of the signal. Averages and SD of at least 5 cells derived from at least 2 independent experiments are shown, unless indicated otherwise.
Statistical analysis
Data are expressed as mean ± SD. The experiments were performed in triplicate and performed at least 3 times. Data were analyzed by using GraphPad Prism version 5.0 software and were compared using the Student's t test and one-way ANOVA as indicated.
Results
Time course of HA-MC4R-GFP desensitization in N2AHA-MC4R-GFP cells in response to chronic exposure to MTII
We generated N2A cells (N2AHA-MC4R-GFP cells), which in addition to endogenous MC4R, express a relatively low level (<10-fold endogenous receptor) of an exogenous human MC4R modified with a HA tag at the N terminus of the protein, exposed to the extracellular medium, and with a GFP tag at the intracellular C terminus, facing the cytoplasm (20, 27, 28). Unlike neurons, which express only endogenous MC4R, HA-MC4R-GFP can be monitored in the N2AHA-MC4R-GFP cells by a variety of assays. Importantly, the activity and desensitization properties of the HA-MC4R-GFP in these cells appear to mirror those of hypothalamic cells expressing only endogenous MC4R (24, 27). Treatment of lean and obese mice with the synthetic peptide agonist MTII, a nonselective and potent MC4R agonist, initially leads to decreased food intake and weight loss. However, at day 2 of the treatment, these effects appear blunted with significantly diminished outcomes by day 3 and day 4 (22, 23). Here, we tested whether the time course of HA-MC4R-GFP desensitization in N2AHA-MC4R-GFP cells reflects the time frame by which MTII's effects on food intake cease in vivo. Acute exposure of cells to 2nM MTII increased intracellular cAMP levels by more than 20-fold. However, when cells were chronically exposed to MTII for 24 and 48 hours and rechallenged with the same agonist, the cAMP response was decreased to less than 30% (Figure 1, A and B). The response to chronic MTII exposure was further decreased, to less than 10% of the acute effect observed, when cells were preexposed to MTII for an increased time interval (d 3 and d 4 of treatment). Therefore, the time course of HA-MC4R-GFP desensitization upon chronic incubation of N2AHA-MC4R-GFP cells with MTII appears to parallel the effects of MTII on food intake in vivo. We subsequently used N2AHA-MC4R-GFP cells as a model to study effects on cAMP and receptor internalization after acute and chronic exposure to diverse MC4R agonists.
Figure 1.
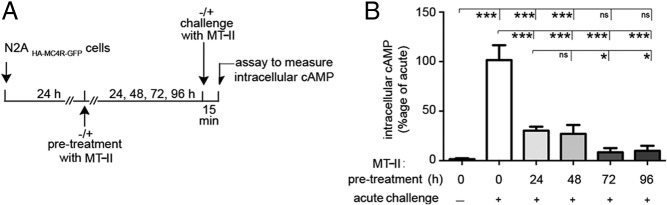
Chronic exposure to MTII induces profound loss of MC4R signaling in N2AHA-MC4R-GFP cells. A, Schematic diagram of the time course of the experiment. N2AHA-MC4R-GFP cells were prechallenged for the indicated time with 2nM MTII. Cells were washed and challenged acutely with 2nM MTII in the presence of IBMX. B, Amount of cAMP generated in response to chronic exposure to MTII (2nM) is expressed as percentage of that obtained in by acute MTII challenge in cells not pretreated with agonist.
Temporal selectivity of the natural agonist α-MSH and the synthetic agonist MTII in the cAMP signaling pathway
It has been reported that MTII and another nonselective MC4R agonists, 4-norleucine,7-D-phenylalanine-α-MSH, have more prolonged biological activity than α-MSH to darken frog skin, an effect mediated by the MC1R (15–19). We determined whether MTII could induce prolonged MC4R-mediated cAMP signaling after removal of the agonist from the medium. We found previously that in the N2AHA-MC4R-GFP cells the EC50 of α-MSH to induce the cAMP signal is approximately 14nM (25). In these cells, MTII appeared to be a more potent MC4R agonist with lower EC50 (∼0.5nM) (Figure 2A), consistent with earlier findings (29). Next, N2AHA-MC4R-GFP cells were exposed to 200nM α-MSH or 2nM MTII for 1 hour. Thereafter, the cells were washed and incubated in medium free of agonist at 37°C for varying time points, up to 90 minutes. Subsequently the cells were incubated for 15 minutes in the presence of the phosphodiesterase inhibitor IBMX, harvested, and the amount cAMP generated in the presence of IBMX was measured (Figure 2, B and C). In cells previously exposed to α-MSH, the level of cAMP generated 90 minutes after withdrawal of the agonist was less than 20% of that measured at the end of the agonist incubation period. By contrast, when cells were previously exposed to MTII, the cAMP signal remained constant at the same amplitude for at least 90 minutes after washout of MTII by medium exchange. These experiments suggest that MTII may stabilize the MC4R in an active conformation for a more prolonged time interval when compared with α-MSH, as concluded from the difference in the persistent cAMP signal obtained with MTII, after medium exchange.
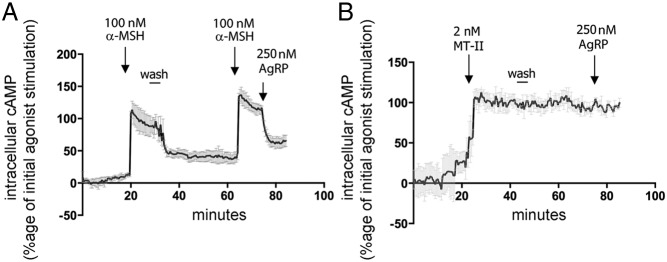
Figure 2.

MTII, unlike α-MSH, induces persistent cAMP signaling after agonist withdrawal. A, Dose-response curve to MTII. The amount of cAMP generated is measured by immunoassay as described in Materials and Methods. B, Schematic diagram of the experiments shown in C. C, N2AHA-MC4R-GFP cells were treated with 200nM α-MSH or 2nM MTII for 1 hour. Cells were then washed with the medium being replaced for a total of 3 times and incubated without any agonist. At the indicated time, cells previously treated with 500μM IBMX for 15 minutes were lysed, and intracellular cAMP was measured by ELISA. D, Live N2AHA-MC4R-GFP cells were incubated with 2nM MTII-Rh for 30 minutes at 37°C, transferred to a heated stage at 37°C to visualize the distribution of MTII-Rh and of HA-MC4R-GFP by confocal microscopy. E, Live N2AHA-MC4R-GFP cells were treated with 2nM MTII-Rh as in D (before wash) and then washed, with the medium being replaced for a total of 6 times. The distribution of MTII-Rh before the cell wash and at 5, 10, and 25 minutes after medium replacement is monitored by confocal microscopy. F and G, N2AHA-MC4R-GFP cells were transfected with TEpacVV and transferred to a heated stage at 37°C for confocal microscopy. Cells were treated with agonists as indicated followed by a wash step where the medium was replaced for a total of 6 times. The cAMP response was measured by a real-time FRET-based assay as described in Materials and Methods. The fluorescence of mTurquoise was excited at 458 nm, and the intensities of fluorescence emitted at 480–495 nm (mTurquoise) and at 535–565 nm (YFP) were recorded over time. In the graphs, the corrected ratio of the intensity of mTurquoise fluorescence to the intensity of YFP fluorescence is shown for a representative cell. The experiments in F and G were run in parallel using the same population of transfected cells.
Prolonged effects of MTII after medium replacement, even at the relatively low concentrations used here to elicit a maximal cAMP signaling response, might be due to its ability to cross cell membranes, rather than to specific binding to MC4R and internalization of the agonist/receptor complex. To test this possibility, live N2AHA-MC4R-GFP cells were exposed to 2nM rhodamine-conjugated MTII (MTII-Rh) for 30 minutes at 37°C, and the distribution of MTII-Rh and of HA-MC4R-GFP was visualized by confocal microscopy. We previously found that the MC4R at the cell surface can internalize rapidly, whether as ligand-free receptor or in a complex with α-MSH (20, 25). MTII-Rh colocalized with HA-MC4R-GFP at the cell surface (Figure 2D, arrowhead) and at the same intracellular location where the receptor internalized. After MTII-Rh was removed from the cell medium, although there was a drop in total cell fluorescence, the ligand remained distributed to the same cell localizations for at least 25 minutes (Figure 2E). These observations indicate that MTII, rather than partitioning nonspecifically to cell membranes, exists in a complex with HA-MC4R-GFP at the cell surface and in endosomes and that such association may persist, at least in part, long after the removal of the agonist from the cell medium.
We have recently used a FRET-based cAMP assay to measure intracellular changes of cAMP in N2AHA-MC4R-GFP cells with high degree of temporal resolution (24). The FRET-based sensor, mTurquoise-Epac-YFP (TEpacVV), is composed of the cAMP-responsive protein Epac sandwiched between 2 fluorescent, FRET compatible tags, mTurquoise (donor) and YFP (acceptor) (30). When intracellular cAMP concentrations increase, Epac binds cAMP and undergoes a conformational change, with an increased ratio of donor fluorescence/acceptor fluorescence (31). In N2AHA-MC4R-GFP cells transiently transfected with TEpacVV and treated with α-MSH, the FRET ratio mTurqouise to YFP increased, thus indicating an increase of intracellular cAMP (Figure 2F). After α-MSH was washed off, the cAMP signal decreased to baseline, within less than 15 minutes, thus indicating that generation of cAMP ceases rapidly upon agonist withdrawal. Reexposing N2AHA-MC4R-GFP cells to α-MSH about 60 minutes later, again increased the FRET ratio of the TEpacVV sensor, indicating that cells remain responsive to the hormone. Conversely, when cells were exposed to MTII after which the medium containing agonist was removed, the FRET ratio of TEpacVV remained elevated for at least 60 minutes (Figure 2G). The data in this experiment are consistent with those obtained in Figure 2C by an independent, ELISA-based, assay to measure intracellular cAMP. Together, these data indicate that initial binding of MTII, but not of α-MSH, to the MC4R induces the receptor to continue to generate cAMP even when the agonist has been removed from the medium. Given that these effects persisted, at even higher MTII agonist concentrations (200nM MTII, see Figure 5 below), it is unlikely that varying levels of receptor occupancy provide an explanation for the variable persistence of cAMP signaling.
Figure 5.

Persistent cAMP signal by MTII is maintained after rapid redistribution of HA-MC4R-GFP to intracellular compartments by a dynamin inhibitor. A and B, N2AHA-MC4R-GFP cells were incubated at 37°C without and with the dynamin inhibitor, V 34–2 at 12.5μM, for the indicated time. Cells were fixed and stained with anti-HA (Cy3, red fluorescence). A, Merged image with both GFP and Cy3 fluorescence. Arrowhead indicates HA-MC4R-GFP at cytoplasmic vesicles clustered near the nucleus; arrows indicate HA-MC4R-GFP in scattered cytoplasmic vesicles. Scale bar, 10 μm. B, Cy3 fluorescence showing cell surface HA-MC4R-GFP in the same cells as in A. C, The graph shows the quantification of the experiments in A where the fraction of total HA-MC4R-GFP residing at the cell surface is monitored by measuring the ratio Cy3 pixel intensity to GFP pixel intensity for each individual cell (n cells = 60). D, N2AHA-MC4R-GFP cells were transfected with TEpacVV, transferred to a heated stage at 37°C for confocal microscopy to monitor changes in intracellular cAMP by real-time FRET assay. The time course of the experiment is shown above the graph. After addition of MTII (2nM), cells were incubated for approximately 5 minutes, washed, and incubated either with vehicle (black trace) or with V 34–2 (12.5μM, red trace), followed by forskolin (1μM) and IBMX (100μM). In the graph, the amount of cAMP being generated was derived from the corrected ratio of the intensity of mTurquoise fluorescence to the intensity of YFP fluorescence normalized to that induced by the addition of forskolin/IBMX as described in Materials and Methods. Data are shown in the graphs as averages and SEMs (n cells = 20 per condition from 2 independent experiments where the 2 conditions, vehicle and V 34–2, are run in parallel). E, N2AHA-MC4R-GFP cells were transfected as in D. MTII was added to the cells at t = 0 and kept in the incubation medium for approximately 3 minutes. The graph shows a representative experiment where the averages and SEMs of the amount of cAMP being generated, expressed as percentage of that induced by forskolin, are derived as in D (n cells = 3). The t1/2 of the cAMP response was derived by fitting the 1-phase association model to the data by using GraphPad Software.
AgRP does not antagonize the cAMP signal by MTII
AgRP acts as a competitive antagonist of α-MSH (32–34). Because the cAMP signal by MC4R is temporally different when induced by α-MSH and by MTII, it is also possible that properties of AgRP to abolish such signal differ. To test this possibility, we used the temporally resolved FRET assay to measure changes in intracellular cAMP. When AgRP was added to a cell medium already containing α-MSH, the previously elevated level of intracellular cAMP declined rapidly, similar to that of cells where the agonist was removed from the cell medium (Figure 3A). This effect by AgRP is similar to our previous finding (24). Differently, AgRP added to N2AHA-MC4R-GFP cells previously exposed to MTII, even when the agonist was washed off, did not decrease the already elevated amount of cAMP being generated (Figure 3B). These experiments indicate that, when MC4R has been previously exposed to MTII, AgRP cannot act as an antagonist of the signal that mediates continued cAMP production, possibly due to the signal being derived from an intracellular compartment, not immediately accessible to AgRP, or to an inability of AgRP to displace MTII.
Figure 3.
Persistent cAMP signaling by MTII is not antagonized by AgRP. A and B, N2AHA-MC4R-GFP cells were transfected with TEpacVV. Cells were treated with α-MSH and MTII followed by a wash step where the medium was replaced for a total of 6 times before the addition of AgRP. Changes in corrected FRET ratio were monitored as in Figure 2C, normalized to that observed in response to the initial exposure to the agonist (Materials and Methods), and shown in the graphs as averages and SDs (n cells per condition = 5). Experiments in A and B were run in parallel using the same population of transfected cells.
Persistent cAMP signaling induced by MTII takes place after the pool of HA-MC4R-GFP exposed to the agonist has been endocytosed
It is possible that the prolonged cAMP signal generated by MTII after the exchange to medium without ligand, originates, at least in part, from MC4R after it has been internalized from the plasma membranee. To address this option we used a FRAP-based experimental set-up to visualize the dynamic properties of HA-MC4R-GFP in the absence and in the presence of agonist. To discriminate potential cellular HA-MC4R-GFP compartments, cells kept in basal conditions were photobleached at the intracellular HA-MC4R-GFP compartment and the rate by which the GFP fluorescence reappeared at this location was measured over time (Figure 4, A and B). The t1/2 by which HA-MC4R-GFP reappeared in the intracellular compartment after photobleaching was approximately 3 minutes whether cells were incubated in the absence of any agonist or whether cells were exposed to 200nM α-MSH or 200nM MTII (Figure 4, C–E). These experiments indicate that MC4R is internalized rapidly from the plasma membrane, whether or not the cells were exposed to ligand. The experiments presented in Figure 2 indicate that 1) in N2AHA-MC4R-GFP cells exposed to MTII, elevated cAMP levels persists for more than 60 minutes after agonist removal from the medium, whereas 2) the cAMP signal by α-MSH disappeared quickly; and 3) that MTII likely remains bound to MC4R once the receptor is internalized. These observations, together with the data of Figure 4 suggest that the rate of MC4R internalization appears independent of ligand exposure and that a pool of MC4R, likely in a complex with MTII, may continue to signal for a prolonged time after being internalized.
Figure 4.

HA-MC4R-GFP in a complex with MTII internalizes rapidly and at the same rate as the receptor in a complex with α-MSH or the ligand-free receptor. A, N2AHA-MC4R-GFP cells were transferred to a heated stage at 37°C for photobleaching/confocal microscopy. A, ROI is drawn around the HA-MC4R-GFP intracellular compartment of N2AHA-MC4R-GFP cells kept in basal conditions (t = 0, white circle). Photobleaching is carried out at the ROI (t = 2.5 min, white circle highlighted by the arrow). Fluorescence recovery is visualized at the ROI (t = 15 min, white circle). Scale bar, 10 μm. B–D, Integrated fluorescence intensity at the intracellular ROI is monitored over time in a cell either kept in basal conditions (A) or treated with α-MSH (C) and with MTII (D). The agonists, at a concentration of 200nM, were added to the cell medium 45 minutes before the start of the FRAP experiment. E, The graph shows the average half-life of FRAP in the selected ROIs (data are derived from ∼6 cells per condition).
We have previously found that MC4R cycles constitutively between the plasma membrane and the endosomal compartment in the absence and in the presence of agonists (20, 25). Further, experiments of Figure 4 indicate that the cycling is rapid, in the order of minutes and experiments of Figure 2 indicate that association of MTII-rhodamine with MC4R may persist for at least 25 minutes after the removal of the agonist from the cell medium. These data suggest that MTII may stay bound to MC4R through multiple cycles of MC4R endocytosis/exocytosis. Thus, it is possible that the MTII-dependent MC4R signal is prolonged due to either of the next 2 explanations: 1) MTII-bound MC4R signals at the plasma membrane, ceases to signal intracellulary, and then returns to the plasma membrane to signal again; or 2) MTII-bound MC4R signals at the plasma membrane and from the intracellular compartment. To test these hypotheses, we treated N2A HA-MC4R-GFP cells with V 34–2 (35) which inhibits dynamin I, a protein functioning in the endocytosis of clathrin-coated vesicles, and dynamin II, which functions also to inhibit vesicle fission at the trans-Golgi network (35). We reasoned that inhibition of MC4R traffic by the dynamin inhibitor should lead to changes in the cellular distribution of the receptor. To test whether this is the case, we monitored the total abundance of the receptor (Figure 5A, GFP, green fluorescence) and the abundance of the HA-epitope of HA-MC4R-GFP, expressed extracellularly (Figure 5, A, Cy3, red fluorescence, and B), in populations of fixed and unpermeabilized N2AHA-MC4R-GFP cells pretreated with vehicle or with V 34–2. After 5–10 minutes of cell incubation at 37°C with V 34–2, the fraction of cell receptors exposed at the cell surface was reduced by 60% as compared with that of control cells treated without the dynamin inhibitor (Figure 5C). This rapid redistribution of HA-MC4R-GFP upon cell exposure to V 34–2 is consistent with HA-MC4R-GFP cycling rapidly (Figure 4), and likely results from V 34–2 inhibiting receptor recycling more efficiently than endocytosis. To monitor by the temporally resolved FRET assay the prolonged cAMP generation by MTII in cells treated with and without the dynamin inhibitor, cells transiently transfected with TEpacVV were exposed to the agonist for approximately 5 minutes, washed, and then incubated in agonist-free medium containing either vehicle or V34–2 (Figure 5D, upper panel). In cells that were treated for 10 minutes with the dynamin inhibitor, the cAMP signal was decreased by only approximately 17% as compared with cells that were incubated under the same conditions but in a dynamin inhibitor-free medium (value at end of the 10-min V 34–2 treatment) (P < .05; Figure 5D, lower panel). Together, the data in Figure 5, C and D, indicate that although shortly after the addition of V 34–2 to the cell medium, there is a robust redistribution of MC4R away from the cell surface, the loss of the cAMP signal by MTII-exposed MC4R is modest in comparison. These data suggest that the persistent cAMP signal induced by MTII originates not only from the plasma membrane, but also from already internalized receptors (Figure 5C, 10-min time point). MTII-exposed cells incubated with V 34–2 for 30 minutes had pronounced loss of intracellular cAMP abundance as compared with MTII-exposed cells incubated in dynamin inhibitor-free medium (by 47%, P < .0001, value at end of the 30-min V 34–2 treatment). At this time point, the intracellular receptor appeared in scattered cytoplasmic vesicles (Figure 5A, arrows) rather than at the vesicles clustered near the nucleus of cells not treated with V 34–2 (Figure 5A, arrowhead). These observations suggest that, in cells treated with V 34–2, intracellular MC4R accumulates over time in a compartment from where it does not signal. In conclusion, the data suggest that MTII-bound MC4R signals both at the plasma membrane and from an intracellular localization, and that receptor cycling is critical for continued MC4R signaling.
It has been reported that agonist binding to GPCRs can induce fast changes in the conformation of the receptor that can take place within 1 second and are then followed by a slower cAMP response that can be measured by the FRET-based EPAC sensors (36, 37). Here, we measured the kinetics by which intracellular cAMP signal appeared in response to the MC4R agonist by FRET ratios being recorded approximately every 3 seconds. The cAMP response to 200nM MTII appeared to take place with a t1/2 of approximately 30 seconds (mean = 31.5 s ± SEM = 5.0 s; n cells = 6), similar to that of A(2A)-adenosine receptor (37), but slower than that of another GPCR, the parathyroid hormone receptor (36). It is possible that, in the case of MC4R, the slowness to generate the cAMP response to agonist might be due to the contribution of multiple populations of receptors to the final signal observed. We postulate that these populations of receptors include MC4Rs at the cell surface and MC4Rs that have been internalized.
MTII, but not α-MSH, induces persistent AMPK signaling in mHypoE-42 immortalized hypothalamic neurons
MC4R agonists inhibit food intake in the hypothalamus by signaling though a pathway that includes a decrease in the phosphorylated form of AMPK (AMPKp) and reduced activity of the enzyme. Such signal is thought to occur downstream of increased cAMP and activation of Protein Kinase A (5, 38). Here, we asked whether the persistent signal induced by MTII but not by α-MSH takes place in immortalized hypothalamic mHypoE-42 neurons, which express endogenous MC4R (27). Addition of 200nM α-MSH to the medium of mHypoE-42 neurons induced dephosphorylation of AMPK at Thr172 (Figure 6, B and C), consistent with a similar result in other immortalized hypothalamic neurons (38). However, when α-MSH was removed from the medium and cells were further incubated at 37°C for 1 hour in the absence of agonists, the level of AMPKp increased significantly (by ∼70%) when compared with the signal intensity observed immediately after acute α-MSH exposure. Also exposure to 2nM MTII induced a decrease in AMPKp abundance to the same level as that induced by α-MSH. However, 1 hour after MTII removal from the medium, the level of AMPKp remained reduced and at the same level as that observed after the acute exposure to the MTII. The data of Figure 6 indicate that α-MSH and MTII have temporal specificity in respect to the AMPK signal in immortalized hypothalamic neurons expressing only endogenous MC4R.
Figure 6.
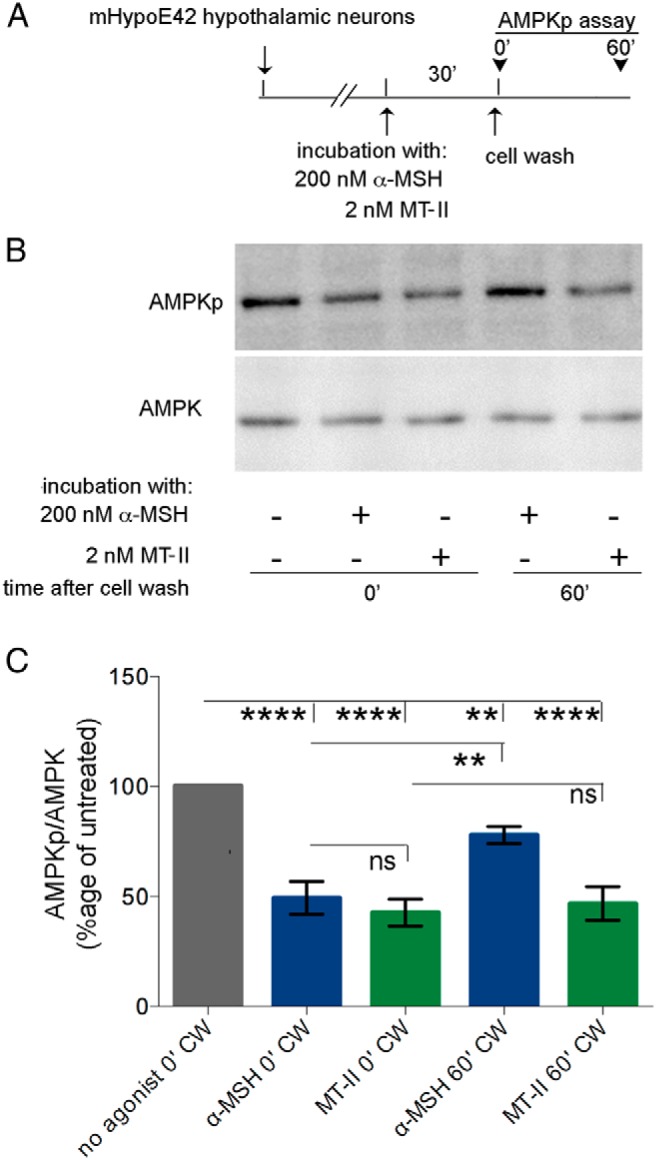
MTII induces persistent AMPK signal in mHypoE-42 hypothalamic neurons. A, mHypoE-42 hypothalamic cells were treated as indicated by the schematic diagram. B, Western blot analysis of cleared mHypoE-42 hypothalamic neurons lysates with the indicated antibodies. C, The intensity of the AMPKp and AMPK bands were measured as described in Materials and Methods, and data were expressed as the percentage of the untreated control. Averages and SDs were derived from 3 independent experiments.
Induction of persistent cAMP signaling is a specific property of some synthetic MC4R agonists
The experiments shown so far indicate that α-MSH and MTII have specific effects on temporal features of the cAMP signal. We tested whether other MC4R agonists shared MTII's property to prolong cAMP signaling even after MTII has been eliminated from the medium. THIQ, BIM-22511, and LY2112688 are selective MC4R agonists that reduce food intake and body weight in animal models (9, 11, 12, 39–42). BIM-22511 and LY2112688 at a concentration of 100nM (>10-fold EC50) induced a maximal cAMP response similar to that induced by 200nM α-MSH (>10-fold EC50) or 200nM MTII (>10-fold EC50). THIQ, which was reported to be a high affinity MC4R ligand (39), was instead found to have an EC50 approximately 95nM when tested in N2AHA-MC4R-GFP cells (data not shown) and was therefore used at the more elevated concentration of 1μM (>10-fold EC50), consistent with another report (43) to obtain a maximal cAMP signal (Figure 7A). In N2AHA-MC4R-GFP cells expressing TEpacVV, exposure to THIQ and BIM-22511 generated a cAMP signal that continued to persist for at least 30 minutes after the agonist has been removed from the medium, similar to what has been observed with MTII (Figure 7, C–E). When N2AHA-MC4R-GFP cells exposed to MTII, THIQ, and BIM-22511 were washed and reexposed to a second challenge with α-MSH, the increase of cAMP over the already elevated signal was 40% or less of that observed in response to the initial stimulation (Figure 7, C–E). By contrast cAMP levels induced by LY2112688, like those induced by α-MSH, extinguished rapidly after the agonist was removed from the medium and a rechallenge with α-MSH increased cAMP levels to the same extent as that induced by the initial exposure to the agonist (Figure 7, B and F). Thus, cells previously exposed to LY2112688, like those exposed to α-MSH, although being unable to signal upon agonist withdrawal, nevertheless remain responsive to MC4R agonists, at least within the time interval of the experiment. These data indicate that MC4R agonists, on the basis of their temporal selectivity, can be divided in 2 groups, of which one, which includes α-MSH and LY2112688, induces a cAMP signal ceasing rapidly upon removal of the agonists, and the second group, which includes MTII, THIQ, and BIM-22511, induces continued production of cAMP even after agonist withdrawal from the medium.
Figure 7.

Persistent cAMP signaling is a property of some synthetic MC4R agonists. A, Intracellular cAMP in N2AHA-MC4R-GFP cells was measured by ELISA in response to no additions, or addition of α-MSH and MTII (200nM), THIQ (1μM), BIM-22511 and LY2112688 (100nM). B–F, N2AHA-MC4R-GFP cells were transfected with TEpacVV, transferred to a heated stage at 37°C for confocal microscopy. Changes in intracellular cAMP were monitored by real-time FRET assay as in Figure 2. In all experiments, N2AHA-MC4R-GFP cells were treated with different agonists, as in A. Cells were washed approximately 10 minutes after addition of the agonists and then rechallenged by using 500nM α-MSH. The experiments in C–F were carried out after an initial experiment where the same population of transfected cells was treated with α-MSH followed by agonist wash-off, as in A. Changes in corrected FRET ratio were monitored as in Figure 2C, normalized to that observed in response to the initial exposure to the agonist (Materials and Methods), and shown in the graphs as averages and SDs (n cells per condition >/= 5).
The different extent by which HA-MC4R-GFP is retained intracellularly in response to chronic exposure to MC4R agonists suggests that the receptor can exist in multiple active conformations
Functional selectivity of MC4R agonists has been proposed to be linked to distinct conformational states of the receptor that can be discriminated based on their unique intracellular trafficking properties (8). We determined whether the MC4R agonists used in this study induced specific receptor trafficking and associated intracellular localization properties. We found that MC4R cycles constitutively and that, upon exposure to α-MSH, the receptor is retained intracellularly due to an inhibition of recycling rather than a change in the rate of internalization (20, 24). The internalization rate of HA-MC4R-GFP in N2AHA-MC4R-GFP cells populations exposed either to α-MSH, MTII, THIQ, BIM-22511, or LY2112688 were measured in parallel experiments and compared with controls. We used an immunoassay that monitors cointernalization of a POD-conjugated antibody raised against the extracellular HA-tag of the receptor (20). Firstly, we noted that the internalization rate of HA-MC4R-GFP by the immunoassay was found to be similar for all agonists (Figure 6A), albeit slower than that detected by FRAP. We next reasoned that if an inhibition of MC4R recycling upon exposure to agonist occurs over time, an intracellular accumulation of undegraded receptors should be visible in N2AHA-MC4R-GFP cells by confocal fluorescence microscopy. For chronic (4 h) incubations of N2AHA-MC4R-GFP cells, agonists were used at elevated concentrations, because their stability over time in the cell medium has been reported to vary, with MTII being the more stable peptide (15). Prolonged exposure to 1μM α-MSH for 4 hours indeed changed the distribution of HA-MC4R-GFP with reduced receptor abundance at the cell surface and increased receptor abundance at the intracellular localization (Figure 8, B and C). These changes in the distribution of HA-MC4R-GFP were quantified by calculating changes in the ratio intensity of GFP fluorescence in the intracellular compartment to total cell intensity of GFP fluorescence in ROIs drawn over a single confocal slice for each cell. This ratio was significantly increased in samples treated with α-MSH as compared with those not exposed to the agonist, thus indicating intracellular retention of the receptor (Figure 8D). Exposure of N2AHA-MC4R-GFP cells to BIM-22511 or LY2112688, for 4 hours (also at elevated concentrations of 500nM), induced intracellular accumulation of HA-MC4R-GFP to the same level as observed for α-MSH (Figure 8D). Exposure of N2AHA-MC4R-GFP cells to 500nM MTII for 4 hours also changed the cell distribution of HA-MC4R-GFP. However, such change in distribution of HA-MC4R-GFP by MTII occurred to a lower extent than that induced by α-MSH (Figure 8, C and D). Similarly, exposure to 1μM THIQ was less effective in inducing intracellular accumulation of the receptor than induced by α-MSH. Thus, based on the MC4R propensity to be retained in the intracellular location upon prolonged agonist exposure at high receptor occupancy, the receptors internalized after MTII or THIQ exposure appear to be retained to a different extent than the receptors internalized after exposure to α-MSH, BIM-22511, or LY2112688. Of the agonists leading to most efficient intracellular retention of the MC4R, BIM-22511 stands out, because it differs from α-MSH and LY2112688 by evoking a sustained cAMP signal after being withdrawn from the medium (Figure 7). These experiments suggest that the properties of the MC4R to induce sustained cAMP generation after agonist withdrawal are not directly related to the extent by which the receptor is retained in the intracellular compartment. Possibly, upon binding to specific agonists the receptor can exist in multiple active conformations, of which one, when MC4R is likely in a complex with BIM-22511, leads both to receptor retention in the intracellular compartment upon chronic agonist exposure and to sustained cAMP signaling upon acute agonist exposure and withdrawal.
Figure 8.

The extent by which HA-MC4R-GFP redistributes to the intracellular localization in response to prolonged treatment with synthetic agonist is specific to the type of agonist being used. A, Live N2AHA-MC4R-GFP cells were treated with no additions or exposed to α-MSH and MTII (200nM), THIQ (1μM), BIM-22511 and LY2112688 (100nM), and to POD-conjugated anti-HA antibodies at 4°C. Cells were washed and transferred at 37°C in the presence of agonist to measure internalization of the pool of HA-MC4R-GFP by monitoring the abundance of agonist-free and agonist-bound receptor at the cell surface as outlined in Materials and Methods. B, N2AHA-MC4R-GFP cells are treated at 37°C for 4 hours with no additions or with α-MSH (1μM) and MTII (500nM) as shown in the schematic. C, Confocal images of live N2AHA-MC4R-GFP cells treated as in B and transferred to a heated stage at 37°C for confocal microscopy. Scale bar, 10 μm; blue tracing, ROI of intracellular GFP fluorescence; red tracing, ROI of GFP fluorescence within the cell perimeter. D, Experiments as outlined in B were carried out exposing cells to α-MSH (1μM), MTII (500nM), THIQ (1μM), and BIM-22511 and LY2112688 (500nM) for 4 hours. Changes in the cellular distribution of HA-MC4R-GFP upon prolonged agonist exposure were monitored by measuring the ratio intracellular GFP fluorescence intensity to total GFP fluorescence intensity as outlined in Materials and Methods. Each symbol corresponds to a single cell (∼60 cells per condition). E, Schematic of experiments where N2AHA-MC4R-GFP cells were pretreated at 37°C with no additions or with α-MSH, MTII, THIQ, BIM-22511, and LY2112688 for 4 hours as in Figure 6F. F, HA-MC4R-GFP desensitization is monitored by the cAMP levels in N2AHA-MC4R-GFP cells pretreated with the indicated agonists expressed as a percentage of that of control cells pretreated in the absence of MC4R ligands.
It is possible that the temporal effects of MC4R agonists noted above or the changes in receptor distribution under chronic exposure conditions lead to variable desensitization of the MC4R. To test this possibility, N2AHA-MC4R-GFP cells were pretreated for 4 hours with α-MSH, MTII, THIQ, BIM-22511, or LY2112688, and rechallenged with each of the same agonists (Figure 8E). In all cases, upon chronic agonist pretreatment, followed by rechallenge with agonist intracellular cAMP was decreased by approximately 40% when compared with that induced by acute exposure to the same agonist (Figure 8F). These experiments indicate that desensitization of MC4R takes place regardless of the extent to which MC4R is retained in the intracellular compartment upon chronic agonist exposure.
Discussion
We show that distinct MC4R agonists exhibit temporal selectivity, namely that the agonists MTII, THIQ, and BIM-22511, in contrast to α-MSH and LY2112688, show prolonged cAMP signaling after the agonist is eliminated from the medium. To reach this conclusion, we have used a temporally resolved FRET-based cAMP assay where the intracellular level of the cyclic nucleotide can be monitored constantly without confounding alterations resulting from the addition of phoshodiesterase inhibitors. For example, elimination of MTII from the cell medium, did not lead to a rapid loss of intracellular cAMP, which instead remained at similar elevated level (∼80% of max) for at least 60 minutes after withdrawal of the agonist. It has been reported that the EC50 of MTII to induce MC4R cAMP signal is lower than that of α-MSH (44, 45), an observation reproduced here (Figure 2A) and in a previous report (25) using the N2AHA-MC4R-GFP cells to test function of the receptor (EC50 of MTII = 0.5nM and EC50 of α-MSH = 14nM). We have carried out the FRET-based experiments to measure MC4R signaling using MTII both at concentration approximately 5-fold and approximately 500-fold its EC50 (2nM and 200nM, respectively) and α-MSH at concentration approximately 10-fold its EC50 (200nM). In the N2AHA-MC4R-GFP cells MTII, at both concentrations, induced the same prolonged cAMP signal for at least 1 hour after removal of the agonist from the medium, negating the possibility that such ligand specific effects reflect variability in receptor occupancy. We also observed that the distribution of MTII-Rh, with the ligand being delivered to live N2AHA-MC4R-GFP cells at 2nM concentration, was virtually identical to that of HA-MC4R-GFP, which localizes to the cell surface and to endosomes. These observations suggest that intracellular MTII-Rh is endocytosed from the cell surface in a complex with MC4R, and that effects of MTII on the duration of the cAMP signal are dependent on specific interactions with the receptor, rather than on unspecific association of the agonist from cell membranes. Importantly, in immortalized mHypoE-42 immortalized hypothalamic neurons 2nM MTII, but not α-MSH, induced persistent signaling by endogenous MC4R by reducing phosphorylation of Thr172 of AMPK for at least 1 hour after the agonist was eliminated from the medium. This data suggests that the MTII-induced persistence of MC4R signal is physiologically relevant because MC4R-induced reduction of AMPK activity by decreased phosphorylation of the kinase is thought to take place downstream of increased cAMP and, in the hypothalamus, appears to control appetite (5, 38). These data also converge with our earlier observations that tagged exogenous HA-MC4R-GFP expressed in N2A cells recapitulates effects observed with endogenous receptor in immortalize hypothalamic neurons (25–26). On the basis of the ability to induce a persistent cAMP signal, synthetic MC4R agonists can be divided into 3 groups where, in one group, α-MSH and LY2112688 do not have such property, and, in the second group, THIQ and MTII facilitate signaling even after removal of the ligand from the medium. BIM-22511 appears to stand apart from each of these ligands, because it shares the continued cAMP signaling capability of MTII and THIQ while mimicking α-MSH and LY2112688 in its receptor intracellular retention behavior. It therefore appears that MC4R agonists have temporal selectivity within the cAMP signaling pathway, which may contribute to unique agonist-specific pharmacology observed in vivo. Our observation that AgRP cannot antagonize the persistent MTII initiated cAMP signaling strengthens our conclusion that MTII-activated receptors are at an intracellular compartment, not accessible to AgRP and/or that MTII remains bound to MC4R at the plasma membrane and endosomes after agonist wash-off. Interestingly, the half-life of HA-MC4R-GFP internalization, measured by FRAP experiments, indicates that MC4R, whether as ligand-free receptor or in a complex with α-MSH, undergoes endocytosis with a t1/2 of approximately 3 minutes. Thus, the data presented here are consistent with a model where the intracellular cAMP signal disappears within minutes after internalization of the MC4R when the receptor is stabilized in the active conformation by α-MSH. These observations are in agreement with the general concept that agonist-bound GPCRs couple to Gs at the plasma membrane (46) and can internalize after ligand-mediated GPCR activation. Conversely, the prolonged generation of cAMP by MTII, which lasts for at least 1 hour, occurs in the face of HA-MC4R-GFP being endocytosed together with MTII by a process which appears to take place within minutes, at a rate similar to that of the receptor bound to α-MSH, and under conditions where receptor occupancy by the ligand is expected to be virtually complete. The persistent cAMP response in the face of rapid endocytosis of MTII-exposed MC4R suggests that the receptor continues to signal from the intracellular compartment. Another possibility is that MTII-exposed MC4R continues to signal, rather than at the intracellular localization, when it recycles back to the cell surface. In this respect, we find that shortly after the addition of a dynamin inhibitor, the cell distribution of MC4R is shifted to the intracellular localization, with 60% less receptor being exposed at the cell surface, and yet a modest decrease (<20%) in the cAMP signal being generated. These data offer evidence that MTII-exposed MC4R can continue to signal from an intracellular localization after being internalized. The observation that the cAMP signal can originate not only when the receptors are localized at the plasma membrane, but also after receptor internalization has been described for some GPCRs (36, 37). For instance, the TSH receptor has been reported to signal by increasing intracellular cAMP after being internalized to endosomes, and the PTH receptor has been found to induce a cAMP signal from endosomes in response to specific agonists (47, 48). It has been proposed that melanotan 1 and MTII have more prolonged biological activity than α-MSH resulting in more profound frog skin darkening due to their slower off-rate from the MC1R when compared with α-MSH (15–19). Here, we find that at least some fluorescently labeled MTII remains in the same plasma membrane and endosome localization of HA-MC4R-GFP for at least 25 minutes after the agonist is eliminated from the medium. The findings reported by others (15–19) together with the work described here opens up the possibility that slow dissociation of MTII from MC4Rs after the receptor is internalized contributes to the receptor's ability to maintain the cAMP signal once the MC4R/MTII complex has been internalized. In conclusion, it is possible that the ability of MTII, but not α-MSH, to induce prolonged cAMP signaling might modulate specific downstream responses, which may contribute to unique physiological effects of agonists, examples of which have been described by others (14).
Drugs targeting GPCRs appear to alter the active conformation of these receptors, so that desensitization is, in some cases, blunted (49–52). In the case of MC4R, it has been proposed that extracellular agonists can induce multiple ligand-specific conformational states of the receptor associated with specific trafficking properties (8). We find here that MC4R, when exposed chronically (∼4 h) to α-MSH, LY2112688, or BIM-22511, has an increased propensity to be retained intracellularly as compared with that of the receptor exposed to MTII and THIQ. Thus, these MC4R agonists may stabilize MC4R in at least 2 distinct conformations, distinguishable by their propensity to be retained intracellularly. It is unclear whether the temporal selectivity of MTII, THIQ, and BIM-22511, found here to induce a persistent cAMP signal for at least 30–60 minutes after being eliminated from the medium, extends to longer time frames. However, all agonists studied here, appear to desensitize the MC4R to the same extent as α-MSH after 4 hours of exposure. The data presented in this paper also suggests that the cellular localization of MC4R in response to chronic agonist exposure cannot be used as unique parameter to predict the extent by which the receptor is desensitized. Interestingly, a separate study has highlighted that MC4R at the plasma membrane can also exist in an active or inactive conformation of the receptor (25). It has been reported that endosome acidification is a potential mechanism by which intracellular signal by GPCRs is turned off (31, 53). These findings, together with the observation that MC4R in a complex with MTII might signal from the endosomal compartments, suggest the possibility, to be explored in the future, that persistence of agonist/receptor complexes that are less sensitive to the reduced pH of endosomes may contribute to the persistent signal induced by MTII, THIQ, and BIM-22511.
It has been suggested that the tachyphylaxis observed in rodents upon chronic treatment with MTII is due to desensitization of MC4R and/or to an increase of AgRP expression taking place in the hypothalamus (22, 23). In this study we find that, although MC4R exposed to MTII can elicit signaling that is insensitive to antagonism by AgRP, the time course of MC4R desensitization by MTII in Neuro2A cells parallels the tachyphylaxis that takes place in mice treated with MTII. Together, these observations suggest that MTII-mediated MC4R desensitization may significantly contribute to the loss of a response to MTII in rodents. We conclude that unique ligand-mediated MC4R receptor interactions contribute by diverse mechanisms to the kinetics by which the receptor remains stabilized in the active conformation.
Acknowledgments
We thank Dr Kees Jalink (The Netherlands Cancer Institute) for the kind gift of TEpacVV.
This work was supported by National Institutes of Health Grants R01-DK080424 and R01-DK102206 (to G.B.) and F30 DK095569 (to B.M.M.) and by Intramural Funding Support from the University of Arkansas for Medical Sciences College of Medicine Research Council.
Disclosure Summary: B.M.M., K.A.C., K.W., and G.B. have nothing to disclose. L.H.T.V.D.P. is an employee of Rhythm Pharmaceuticals, Inc, a privately held biotechnology company, which is developing MC4R agonists for the treatment of obesity.
Footnotes
- AgRP
- agouti related peptide
- AMPK
- AMP kinase
- AMPKp
- phosphorylated form of AMPK
- FRAP
- fluorescence recovery after photobleaching
- FRET
- Förster resonance energy transfer
- green fluorescent protein
- (GFP)
- GPCR
- G protein-coupled receptor
- HA
- hemagglutinin
- IBMX
- 3-isobutyl-1-methylxanthine
- MC4R
- melanocortin-4 receptor
- α-MSH
- α-melanocyte-stimulating hormone
- MTII
- melanotan II
- MTII-Rh
- rhodamine-conjugated MTII
- V 34–2
- dynamin Inhibitor V 34–2
- POD
- peroxidase
- ROI
- region of interest.
References
- 1. Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8(5):571–578. [DOI] [PubMed] [Google Scholar]
- 2. Tao YX. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr Rev. 2010;31(4):506–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Williams KW, Elmquist JK. From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat Neurosci. 2012;15(10):1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen KY, Muniyappa R, Abel BS, et al. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab. 2015:100(4):1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Minokoshi Y, Alquier T, Furukawa N, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428(6982):569–574. [DOI] [PubMed] [Google Scholar]
- 6. Mountjoy KG, Kong PL, Taylor JA, Willard DH, Wilkison WO. Melanocortin receptor-mediated mobilization of intracellular free calcium in HEK293 cells. Physiol Genomics. 2001;5(1):11–19. [DOI] [PubMed] [Google Scholar]
- 7. Newman EA, Chai BX, Zhang W, Li JY, Ammori JB, Mulholland MW. Activation of the melanocortin-4 receptor mobilizes intracellular free calcium in immortalized hypothalamic neurons. J Surg Res. 2006;132(2):201–207. [DOI] [PubMed] [Google Scholar]
- 8. Nickolls SA, Fleck B, Hoare SR, Maki RA. Functional selectivity of melanocortin 4 receptor peptide and nonpeptide agonists: evidence for ligand-specific conformational states. J Pharmacol Exp Ther. 2005;313(3):1281–1288. [DOI] [PubMed] [Google Scholar]
- 9. Greenfield JR, Miller JW, Keogh JM, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360(1):44–52. [DOI] [PubMed] [Google Scholar]
- 10. Van der Ploeg LH, Martin WJ, Howard AD, et al. A role for the melanocortin 4 receptor in sexual function. Proc Natl Acad Sci USA. 2002;99(17):11381–11386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kumar KG, Sutton GM, Dong JZ, et al. Analysis of the therapeutic functions of novel melanocortin receptor agonists in MC3R- and MC4R-deficient C57BL/6J mice. Peptides. 2009;30(10):1892–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kievit P, Halem H, Marks DL, et al. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes. 2013;62(2):490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuo JJ, Silva AA, Hall JE. Hypothalamic melanocortin receptors and chronic regulation of arterial pressure and renal function. Hypertension. 2003;41(3 pt 2):768–774. [DOI] [PubMed] [Google Scholar]
- 14. Richardson J, Cruz MT, Majumdar U, et al. Melanocortin signaling in the brainstem influences vagal outflow to the stomach. J Neurosci. 2013;33(33):13286–13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sawyer TK, Sanfilippo PJ, Hruby VJ, et al. 4-Norleucine, 7-D-phenylalanine-α-melanocyte-stimulating hormone: a highly potent α-melanotropin with ultralong biological activity. Proc Natl Acad Sci USA. 1980;77(10):5754–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fitzgerald LM, Fryer JL, Dwyer T, Humphrey SM. Effect of MELANOTAN, [Nle(4), D-Phe(7)]-α-MSH, on melanin synthesis in humans with MC1R variant alleles. Peptides. 2006;27(2):388–394. [DOI] [PubMed] [Google Scholar]
- 17. Haskell-Luevano C, Miwa H, Dickinson C, et al. Characterizations of the unusual dissociation properties of melanotropin peptides from the melanocortin receptor, hMC1R. J Med Chem. 1996;39(2):432–435. [DOI] [PubMed] [Google Scholar]
- 18. Al-Obeidi F, Castrucci AM, Hadley ME, Hruby VJ. Potent and prolonged acting cyclic lactam analogues of α-melanotropin: design based on molecular dynamics. J Med Chem. 1989;32(12):2555–2561. [DOI] [PubMed] [Google Scholar]
- 19. Al-Obeidi F, Hruby VJ, Castrucci AM, Hadley ME. Design of potent linear α-melanotropin 4–10 analogues modified in positions 5 and 10. J Med Chem. 1989;32(1):174–179. [DOI] [PubMed] [Google Scholar]
- 20. Mohammad S, Baldini G, Granell S, Narducci P, Martelli AM, Baldini G. Constitutive traffic of melanocortin-4 receptor in Neuro2A cells and immortalized hypothalamic neurons. J Biol Chem. 2007;282(7):4963–4974. [DOI] [PubMed] [Google Scholar]
- 21. Shinyama H, Masuzaki H, Fang H, Flier JS. Regulation of melanocortin-4 receptor signaling: agonist-mediated desensitization and internalization. Endocrinology. 2003;144(4):1301–1314. [DOI] [PubMed] [Google Scholar]
- 22. Pierroz DD, Ziotopoulou M, Ungsunan L, Moschos S, Flier JS, Mantzoros CS. Effects of acute and chronic administration of the melanocortin agonist MTII in mice with diet-induced obesity. Diabetes. 2002;51(5):1337–1345. [DOI] [PubMed] [Google Scholar]
- 23. Blüher S, Ziotopoulou M, Bullen JW, Jr, et al. Responsiveness to peripherally administered melanocortins in lean and obese mice. Diabetes. 2004;53(1):82–90. [DOI] [PubMed] [Google Scholar]
- 24. Granell S, Molden BM, Baldini G. Exposure of MC4R to agonist in the endoplasmic reticulum stabilizes an active conformation of the receptor that does not desensitize. Proc Natl Acad Sci USA. 2013;110(49):E4733–E4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McDaniel FK, Molden BM, Mohammad S, et al. Constitutive cholesterol-dependent endocytosis of melanocortin-4 receptor (MC4R) is essential to maintain receptor responsiveness to α-melanocyte-stimulating hormone (α-MSH). J Biol Chem. 2012;287(26):21873–21890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vilardaga JP, Romero G, Feinstein TN, Wehbi VL. Kinetics and dynamics in the G protein-coupled receptor signaling cascade. Methods Enzymol. 2013;522:337–363. [DOI] [PubMed] [Google Scholar]
- 27. Cragle FK, Baldini G. Mild lipid stress induces profound loss of MC4R protein abundance and function. Mol Endocrinol. 2014;28(3):357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Granell S, Serra-Juhe C, Martos-Moreno GA, et al. A novel melanocortin-4 receptor mutation MC4R-P272L associated with severe obesity has increased propensity to be ubiquitinated in the ER in the face of correct folding. PLoS One. 2012;7(12):e50894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xiang Z, Proneth B, Dirain ML, Litherland SA, Haskell-Luevano C. Pharmacological characterization of 30 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists, synthetic agonists, and the endogenous agouti-related protein antagonist. Biochemistry. 49(22):4583–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klarenbeek JB, Goedhart J, Hink MA, Gadella TWJ, Jalink K. A mTurquoise-based cAMP sensor for both FLIM and ratiometric read-out has improved dynamic range. PLoS One. 2011;6(4):e19170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gidon A, Al-Bataineh MM, Jean-Alphonse FG, et al. Endosomal GPCR signaling turned off by negative feedback actions of PKA and v-ATPase. Nat Chem Biol. 2014;10(9):707–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ollmann MM, Wilson BD, Yang YK, et al. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278(5335):135–138. [DOI] [PubMed] [Google Scholar]
- 33. Lu X, Williams JA, Deadman JJ, et al. Preferential antagonism of the interactions of the integrin α IIb β 3 with immobilized glycoprotein ligands by snake-venom RGD (Arg-Gly-Asp) proteins. Evidence supporting a functional role for the amino acid residues flanking the tripeptide RGD in determining the inhibitory properties of snake-venom RGD proteins. Biochem J. 1994;304(pt 3):929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang YK, Ollmann MM, Wilson BD, et al. Effects of recombinant agouti-signaling protein on melanocortin action. Mol Endocrinol. 1997;11(3):274–280. [DOI] [PubMed] [Google Scholar]
- 35. Jones SM, Howell KE, Henley JR, Cao H, McNiven MA. Role of dynamin in the formation of transport vesicles from the trans-Golgi network. Science. 1998;279(5350):573–577. [DOI] [PubMed] [Google Scholar]
- 36. Castro M, Nikolaev VO, Palm D, Lohse MJ, Vilardaga JP. Turn-on switch in parathyroid hormone receptor by a two-step parathyroid hormone binding mechanism. Proc Natl Acad Sci USA. 2005;102(44):16084–16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hein P, Rochais F, Hoffmann C, et al. Gs activation is time-limiting in initiating receptor-mediated signaling. J Biol Chem. 2006;281(44):33345–33351. [DOI] [PubMed] [Google Scholar]
- 38. Damm E, Buech TR, Gudermann T, Breit A. Melanocortin-induced PKA activation inhibits AMPK activity via ERK-1/2 and LKB-1 in hypothalamic GT1–7 cells. Mol Endocrinol. 2012;26(4):643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sebhat IK, Martin WJ, Ye Z, et al. Design and pharmacology of N-[(3R)-1,2,3,4-tetrahydroisoquinolinium- 3-ylcarbonyl]-(1R)-1-(4-chlorobenzyl)- 2-[4-cyclohexyl-4-(1H-1,2,4-triazol- 1-ylmethyl)piperidin-1-yl]-2-oxoethylamine (1), a potent, selective, melanocortin subtype-4 receptor agonist. J Med Chem. 2002;45(21):4589–4593. [DOI] [PubMed] [Google Scholar]
- 40. Xiang Z, Pogozheva ID, Sorenson NB, et al. Peptide and small molecules rescue the functional activity and agonist potency of dysfunctional human melanocortin-4 receptor polymorphisms. Biochemistry. 2007;46(28):8273–8287. [DOI] [PubMed] [Google Scholar]
- 41. Muceniece R, Zvejniece L, Vilskersts R, et al. Functional evaluation of THIQ, a melanocortin 4 receptor agonist, in models of food intake and inflammation. Basic Clin Pharmacol Toxicol. 2007;101(6):416–420. [DOI] [PubMed] [Google Scholar]
- 42. Maier T, Hoyer J. Modulation of blood pressure by central melanocortinergic pathways. Nephrol Dial Transplant. 2010;25(3):674–677. [DOI] [PubMed] [Google Scholar]
- 43. Huang H, Tao YX. A small molecule agonist THIQ as a novel pharmacoperone for intracellularly retained melanocortin-4 receptor mutants. Int J Biol Sci. 2014;10(8):817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qu H, Cai M, Mayorov AV, et al. Substitution of arginine with proline and proline derivatives in melanocyte-stimulating hormones leads to selectivity for human melanocortin 4 receptor. J Med Chem. 2009;52(12):3627–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xiang Z, Litherland SA, Sorensen NB, et al. Pharmacological characterization of 40 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists and the agouti-related protein (AGRP) antagonist. Biochemistry. 2006;45(23):7277–7288. [DOI] [PubMed] [Google Scholar]
- 46. Daaka Y, Luttrell LM, Ahn S, et al. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem. 1998;273(2):685–688. [DOI] [PubMed] [Google Scholar]
- 47. Calebiro D, Nikolaev VO, Gagliani MC, et al. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 2009;7(8):e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ferrandon S, Feinstein TN, Castro M, et al. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol. 2009;5(10):734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Keith DE, Murray SR, Zaki PA, et al. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996;271(32):19021–19024. [DOI] [PubMed] [Google Scholar]
- 50. Yu Y, Zhang L, Yin X, Sun H, Uhl GR, Wang JB. Mu opioid receptor phosphorylation, desensitization, and ligand efficacy. J Biol Chem. 1997;272(46):28869–28874. [DOI] [PubMed] [Google Scholar]
- 51. Raehal KM, Schmid CL, Groer CE, Bohn LM. Functional selectivity at the μ-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol Rev. 2011;63(4):1001–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hoffmann C, Ziegler N, Reiner S, Krasel C, Lohse MJ. Agonist-selective, receptor-specific interaction of human P2Y receptors with β-arrestin-1 and -2. J Biol Chem. 2008;283(45):30933–30941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tsvetanova NG, Irannejad R, von Zastrow M. G protein-coupled receptor (GPCR) signaling via heterotrimeric G proteins from endosomes. J Biol Chem. 2015;290(11):6689–6696. [DOI] [PMC free article] [PubMed] [Google Scholar]