

Abstract
Neurexins and neuroligins are cell-adhesion molecules that form trans-synaptic interactions. In this issue of Neuron, Choi and colleagues report that neurexin-neuroligin signaling plays a critical role in functional and structural synaptic plasticity underlying memory formation in Aplysia.
Synaptic plasticity is an essential cellular mechanism underlying learning and memory (Martin et al., 2000). During the course of memory formation, structural and functional modifications of both presynaptic and postsynaptic components of neurons have been widely reported. These changes can occur at both previously existing synapses as well as at synapses that are newly formed in response to learning-induced stimuli. Collectively these observations raise two basic questions. First, how are functional and structural alternations in both presynaptic and postsynaptic elements of pre-existing synapses dynamically coupled during the induction and maintenance of synaptic plasticity? Second, how do new synapses induced by learning mature and stabilize to maintain the storage of information?
The cell-adhesion molecules neurexin and neuroligin have emerged as a pair of interesting candidates to subserve both of these processes. Each contains an N-terminal extracellular region spanning the physical space of the synaptic cleft, a single trans-membrane region, and a C-terminal intracellular region with PDZ-binding domains (Dean and Dresbach, 2006; Südhof, 2008) (Figure 1). Neurexins are enriched at presynaptic terminals, with their extracellular region binding to neuroligins that project from postsynaptic membranes, and their intracellular regions interacting directly or indirectly, through scaffolding proteins such as CASK and Mint, with elements of neurotransmitter release machinery (Figure 1). On the postsynaptic side, neuroligins bind to scaffolding proteins, such as PSD-95 and Gephyrin, which in turn recruit glutamate receptors and GABA receptors, respectively.
Figure 1. Molecular interactions through neurexins and neuroligins at synapses.
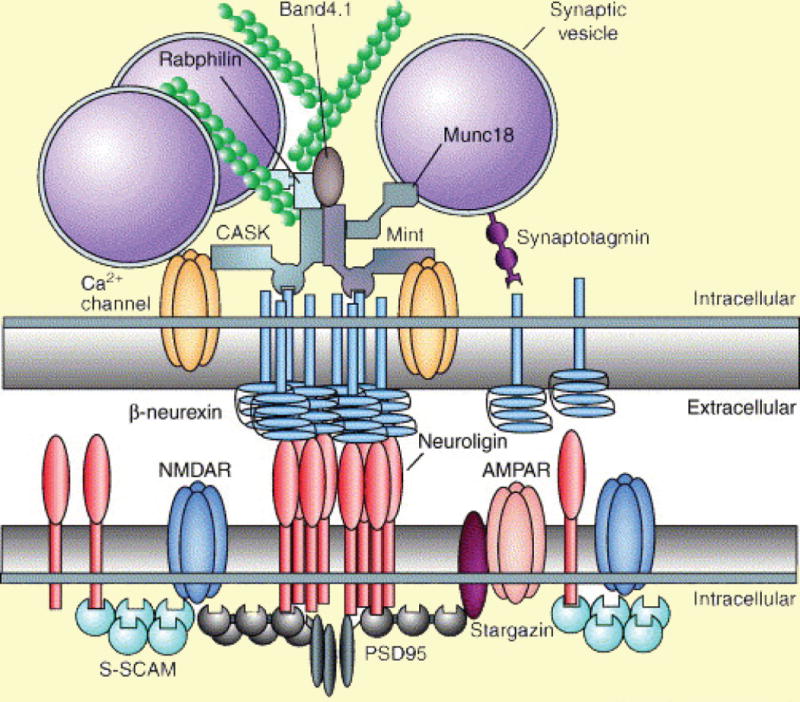
Neurexins and neuroligins are clustered in presynaptic and postsynaptic membranes, respectively, with their extracellular regions adhering to each other in the synaptic cleft (highlighted in white). Intracellularly (highlighted in yellow), neurexins and neuroligins interact with several fundamental components of the molecular machinery mediating synaptic transmission. Presynaptically, neurexins bind to the Ca2+ sensor synaptotagmin as well as to the scaffolding proteins CASK and Mint, which in turn interact with Ca2+ channels, synaptic vesicles and actin filaments. Postsynaptically, neuroligins interact with glutamate receptors via scaffolding proteins, such as PSD95 and S-SCAM. They can also interact with GABA receptors through the scaffolding protein Gephyrin at inhibitory synapses (not shown).
Reprinted by permission from Elsevier Ltd: Trends in Neurosciences (Dean and Dresbach, 2006)
Previous studies show that neurexins and neuroligins not only facilitate the assembly of functional units on their own side of the synapse, but they also regulate synaptic specialization on the opposite side of a nascent synapse through their trans-synaptic interactions (Dean and Dresbach, 2006). Furthermore, a series of recent studies suggest that during synaptogenesis in brain development, while these proteins are not important for initial stages of synapse differentiation, they do serve a fundamental role in subsequent synapse maturation and stabilization (Südhof, 2008).
A growing body of evidence suggests that development and learning are mechanistically related and, as described above, neurexins and neuroligins play critical roles in synapse formation during development. This raises a fascinating possibility: can trans-synaptic interactions between neurexins and neuroligins regulate functional and structural plasticity at synapses during learning and memory? In this issue of Neuron, Choi et al. (2011) explore this question in the marine mollusk Aplysia californica by taking advantage of the well characterized elementary circuit mediating the siphon-elicited gill withdrawal reflex, consisting of a monosynaptic connection between siphon sensory neurons (SNs) and gill motor neurons (MNs). Memory for sensitization in this reflex is supported in large measure by synaptic facilitation at the SN-MN synapse, where serotonin (5-HT) released in response to sensitizing stimuli enhances synaptic strength. A single pulse of 5-HT induces short-term facilitation (STF) lasting minutes, whereas repeated pulses of 5-HT induce intermediate-term and long-term facilitation (ITF and LTF) that last hours and days (Alberini et al., 1994; Sutton and Carew, 2000), and are thought to engage both presynaptic and postsynaptic modifications (Jin et al., 2011; Trudeau and Castellucci, 1995). Repeated 5-HT application also induces growth of new varicosities in SNs that contributes to the expression of LTF (Kim et al., 2003). By reconstituting the SN-MN connections in culture and restrictively manipulating the expression of neurexins and neuroligins in individual SNs and MNs, the authors examined the contribution of trans-synaptic neurexin-neuroligin signaling in different phases of 5-HT-induced synaptic facilitation and associated synaptic growth.
As a first step, the authors cloned a single homolog of neuroligin (ApNLG) and a single homolog of neurexin (ApNRX) in Aplysia, both of which contain all the critical internal structural domains and importantly, can bind to each other. ApNLG and ApNRX are clustered at synapses, especially on the initial segment and major neurites of MNs where most functional synapses are found. These two proteins also exhibit substantial colocalization in these regions. Moreover, the authors observed a pool of ApNRX clusters in MN neurites, consistent with a previous finding that neurexins can localize in postsynaptic compartments (Taniguchi et al., 2007).
To elucidate the role of trans-synaptic interactions between presynaptic ApNRX and postsynaptic ApNLG during synaptic facilitation, the authors injected antisense of ApNRX into SNs or ApNLG into MNs 3hrs before 5-HT application. They found that either of these manipulations resulted in a significant reduction in 24hr LTF induced by repeated 5-HT. In contrast, basal synaptic transmission and STF were not affected. Conversely, simultaneous overexpression of ApNRX in SNs and ApNLG in MNs led to an increase in synaptic strength, whereas overexpression of either one alone had no effect. Together, these loss-of-function and gain-of-function experiments highlight the importance of functional interaction between neurexins and neuroligins in the induction of synaptic plasticity. Although ApNRX and ApNLG are capable of recruiting synaptic elements within their own intracellular region, the trans-synaptic adhesion between the two proteins also appears to be critical for generating long-lasting changes at these synapses.
Previous studies have shown that repeated pulses of 5-HT induce the generation of new presynaptic varicosities, and recruitment of vesicles into pre-existing varicosities (Kim et al., 2003). In principle, both of these mechanisms could be mediated by neurexin-neuroligin signaling to generate LTF. Consistent with the first possibility, the authors found that injection of antisense of ApNRX into SNs or antisense of ApNLG into MNs indeed significantly reduced the increase in varicosities observed at 24hrs after repeated 5HT. They next examined the second possibility by expressing in SNs an ApNRX mutant that lacks the cytoplasmic tail. This mutant competes with endogenous ApNRX for ApNLG binding, but is not capable of binding to intracellular signaling partners for recruiting synaptic vesicles (Dean and Dresbach, 2006). Overexpression of the mutant significantly reduced 24hr LTF induced by 5-HT. In parallel with these experiments, the authors compared the distribution of ApNRX before and 24hrs after repeated 5-HT. They found an enrichment of ApNRX in newly formed varicosities as well as filling of pre-existing empty varicosities with ApNRX after 5-HT application. These data are consistent with the previous findings that enrichment of synaptic vesicles occurs in both newly formed and pre-existing varicosities after 5-HT (Kim et al., 2003). Taken together, these results suggest that ApNRX-ApNLG signaling contributes to LTF by activating pre-existing “silent” synapses, as well as by increasing the formation of newly functional synapses, thus coupling functional and structural synaptic plasticity.
Synaptic facilitation induced by repeated 5-HT can last at least 72hrs, and synaptic growth is thought to play a predominant role in this late (48–72hr) phase of LTF (Casadio et al., 1999). Since ApNRX and ApNLG are important for synaptic growth, the authors further examined their contribution to the persistence of LTF. For these experiments, antisense of ApNRX or ApNLG was injected into SNs or MNs, respectively, at 24hrs after repeated 5-HT application. Either of these treatments induced a significant decay of LTF at 48hrs after 5-HT, which further decayed to near baseline at 72hrs. Thus, trans-synaptic neurexin-neuroligin signaling is critical for the maintenance of persistent LTF.
Recent advances in a series of genetic analyses of neurological diseases have revealed a link between impaired neurexin-neuroligin signaling and autism (Pardo and Eberhart, 2007). For example, an Arginine to Cysteine (R451C) mutation in neuroligin-3, which reduces its surface expression and binding to neurexins, has been observed in autistic siblings (Jamain et al., 2003). Moreover, transgenic mice with the same mutation show increased inhibitory transmission, but no change in basal excitatory transmission (Südhof, 2008). In the present paper, the authors explore the physiological consequences of the homologous mutation in ApNLG. They report that expression of this mutant in MNs significantly reduced 1hr ITF and 24hr LTF after repeated 5-HT. Interestingly, autistic patients carrying this mutation also exhibit learning deficits (Jamain et al., 2003). Therefore the authors’ current findings can suggest at least a plausible (although certainly incomplete) explanation for these behavioral effects. Additionally, aberrant 5-HT transmission has also been implicated in autism (Pardo and Eberhart, 2007). Thus the results by Choi et al. can, at least in principle, provide critical traction in identifying functional interactions between two basic risk factors for autism.
Finally, the authors’ findings collectively suggest differential roles of ApNRX-ApNLG signaling in distinct phases of synaptic facilitation: ITF, LTF and the maintenance of LTF over days are critically dependent on ApNRX-ApNLG signaling, whereas STF and basal transmission are less affected. In Aplysia, ITF and LTF differ from STF by requiring de novo translation (Alberini et al., 1994; Sutton and Carew, 2000). Interestingly, the protein levels of ApNRX and ApNLG are increased following repeated 5-HT (Puthanveettil et al., 2008). Considering these data as a whole, the authors suggest a model in which repeated 5-HT upregulates ApNRX and ApNLG coordinately, which in turn leads to remodeling of pre-existing synapses and growth of new varicosities, resulting in long-lasting increases in synaptic strength. Since ITF and different stages of LTF also differ in their requirement of transcription and synaptic growth, it will now be of considerable interest to explore whether ApNRX-ApNLG signaling utilizes different mechanisms to regulate different phases of enduring plasticity.
Considering the paper by Choi et al. in a broader perspective, the authors have provided further compelling evidence that cell-adhesion molecules, once thought to function as the static backbones of synapses, can actually be dynamic regulators of synaptic plasticity that contribute to memory formation. Recently, an array of cell-adhesion molecules, such as Ephs and ephrins, cadherins, and immunoglobulin-containing cell adhesion molecules, have all been found to be engaged in a wide range of forms of synaptic plasticity (Dalva et al., 2007). These proteins all share two important features: first, they form homophilic or heterophilic protein-protein interactions spanning and maintaining the physical space of the synaptic cleft, and second, they interact with intracellular signaling partners on both sides of the synapses. Thus, these classes of adhesion molecules are well-equipped to couple the functional and structural dynamics of synapses. As a next step, it will now be important to explore how multiple cell adhesion molecules may collaborate to contribute to the induction and maintenance of synaptic plasticity, and ultimately, to examine how these molecules may contribute to the induction and expression of lasting memories. Furthermore, dysfunctional changes in synaptic strength is widely considered as a common contributing factor to a range of cognitive disorders, including Alzheimer’s disease, autism and Fragile X syndrome. Thus, elucidating the mechanisms by which cell adhesion molecules can structurally and functionally regulate synaptic strength has the additional potential of identifying possible therapeutic targets for these diverse neurological diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alberini CM, Ghirardi M, Metz R, Kandel ER. Cell. 1994;76:1099–1114. doi: 10.1016/0092-8674(94)90386-7. [DOI] [PubMed] [Google Scholar]
- Casadio A, Martin KC, Giustetto M, Zhu H, Chen M, Bartsch D, Bailey CH, Kandel ER. Cell. 1999;99:221–237. doi: 10.1016/s0092-8674(00)81653-0. [DOI] [PubMed] [Google Scholar]
- Choi YB, Li HL, Kassabov SR, Jin I, Puthanveettil SV, Karl KA, Lu Y, Kim JH, Bailey CH, Kandel ER. Neuron. 2011 doi: 10.1016/j.neuron.2011.03.020. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalva MB, McClelland AC, Kayser MS. Nat Rev Neurosci. 2007;8:206–220. doi: 10.1038/nrn2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C, Dresbach T. Trends Neurosci. 2006;29:21–29. doi: 10.1016/j.tins.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, et al. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin I, Kandel ER, Hawkins RD. Learn Mem. 2011;18:96–102. doi: 10.1101/lm.1949711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Udo H, Li HL, Youn TY, Chen M, Kandel ER, Bailey CH. Neuron. 2003;40:151–165. doi: 10.1016/s0896-6273(03)00595-6. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Pardo CA, Eberhart CG. Brain Pathol. 2007;17:434–447. doi: 10.1111/j.1750-3639.2007.00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthanveettil SV, Monje FJ, Miniaci MC, Choi YB, Karl KA, Khandros E, Gawinowicz MA, Sheetz MP, Kandel ER. Cell. 2008;135:960–973. doi: 10.1016/j.cell.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Carew TJ. Neuron. 2000;26:219–231. doi: 10.1016/s0896-6273(00)81152-6. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, Gollan L, Scholl FG, Mahadomrongkul V, Dobler E, Limthong N, Peck M, Aoki C, Scheiffele P. J Neurosci. 2007;27:2815–2824. doi: 10.1523/JNEUROSCI.0032-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau LE, Castellucci VF. J Neurosci. 1995;15:1275–1284. doi: 10.1523/JNEUROSCI.15-02-01275.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]