

Abstract
Motivation
Intrinsically disordered regions of proteins play an essential role in the regulation of various biological processes. Key to their regulatory function is often the binding to globular protein domains via sequence elements known as molecular recognition features (MoRFs). Development of computational tools for the identification of candidate MoRF locations in amino acid sequences is an important task and an area of growing interest. Given the relative sparseness of MoRFs in protein sequences, the accuracy of the available MoRF predictors is often inadequate for practical usage, which leaves a significant need and room for improvement. In this work, we introduce MoRFCHiBi_Web, which predicts MoRF locations in protein sequences with higher accuracy compared to current MoRF predictors.
Methods
Three distinct and largely independent property scores are computed with component predictors and then combined to generate the final MoRF propensity scores. The first score reflects the likelihood of sequence windows to harbour MoRFs and is based on amino acid composition and sequence similarity information. It is generated by MoRFCHiBi using small windows of up to 40 residues in size. The second score identifies long stretches of protein disorder and is generated by ESpritz with the DisProt option. Lastly, the third score reflects residue conservation and is assembled from PSSM files generated by PSI-BLAST. These propensity scores are processed and then hierarchically combined using Bayes rule to generate the final MoRFCHiBi_Web predictions.
Results
MoRFCHiBi_Web was tested on three datasets. Results show that MoRFCHiBi_Web outperforms previously developed predictors by generating less than half the false positive rate for the same true positive rate at practical threshold values. This level of accuracy paired with its relatively high processing speed makes MoRFCHiBi_Web a practical tool for MoRF prediction.
Availability
Introduction
Intrinsically disordered regions (IDRs) are protein segments that do not adopt a unique 3D structure under physiological conditions [1–3]. Of particular interest to many researchers are relatively short segments within IDRs that can undergo disorder-to-order transitions during binding, which are known as molecular recognition features (MoRFs), a term that had only been coined a decade ago but had quickly gained recognition [4–8]. Interactions mediated by MoRFs are important and play key roles in regulatory processes and in signal transduction [3], as their structural flexibility grants MoRFs the ability to mold into a precise fit for a given binding surface and, thereby, achieve high interaction specificity, which is often desirable for protein interactions in signaling pathways [9]. Furthermore, long MoRFs form large interaction surfaces with their partners, which contain many specificity-enhancing electrostatic interactions [10]. Alternative splicing is another mechanism of regulating protein interactions that involves MoRFs. Exons that are alternatively spliced in a tissue specific manner are enriched with MoRFs [11], which allow protein interaction networks to be rewired by altering the availability of MoRF motifs in protein isoforms, and thus modulate the signal–integrating pathways.
Given the properties and functional importance of MoRFs, their identification has become an important computational challenge and several computational methods have been developed in recent years for that purpose, including the programs ANCHOR [12], MoRFpred [13], MFSPSSMpred [14], and DISOPRED3 [15]. ANCHOR and MoRFpred are among the most used MoRF predictors. ANCHOR, a downloadable and fast predictor, makes predictions based on the estimation of interaction energies between the residues in the protein sequence. ANCHOR searches for sequences in IDRs that have low stabilization energy on their own but have the propensity to interact with globular proteins. On the other hand, the web-based predictor MoRFpred predicts MoRFs using a linear kernel SVM-based approach to combine various properties extracted from the protein sequence, including physicochemical properties of amino acids, conservation of residues, predicted solvent accessibility of residues, and scores from five IDR predictors. MFSPSSMpred is a web-based predictor employing a SVM model with a radial basis function (RBF) kernel that utilizes sequence properties and conservation. The currently available prediction engine for MFSPSSMpred was trained on almost all known MoRF sequences, which limits the data available for external evaluation. However, an unreleased version of MFSPSSMpred that was trained and evaluated using the same training and test datasets used by MoRFPred showed a performance that was approximately equal to that of MoRFpred in terms of AUC, with a slight underperformance at low false positive rates (FPRs) [14]. DISOPRED3, just like MFSPSSMpred, is a web-based SVM-RBF model that was trained on all known MoRF sequences. In addition to sequence property and conservation, DISOPRED3 also relies on disorder predictions generated independently by a neural network model.
We recently introduced MoRFCHiBi, a downloadable predictor for identifying MoRF sequences of length between 5 and 25 residues using windows with up to 40 residues in size [16]. Conveniently, MoRFCHiBi’s exclusive reliance on local physicochemical properties of amino acids and the lack of dependence on any external component predictors makes it a good candidate for incorporating into a more complex predictor for high precision MoRF identification.
In this work, we introduce MoRFCHiBi_Web, the most accurate computational method to date for the prediction of MoRFs in protein sequences. MoRFCHiBi_Web utilizes Bayes rule to combine the output of MoRFCHiBi with the output of an intermediate predictor, MoRFDC, constructed from protein disorder and conservation information. The integration of disorder and conservation information resulted in significant improvements in the prediction quality of MoRFCHiBi_Web that is reflected in its ability to predict short MoRFs of up to 30 residues in length with an AUC of 0.87.
Methods
Three distinct and largely independent sources of information are used in MoRFCHiBi_Web (Fig 1).
Fig 1. MoRFCHiBi_Web structure.

The three components used by MoRFCHiBi_Web are MoRFCHiBi predictions, predictions of disorder by ESpritz with the DisProt option, and conservation predictions using PSI-BLAST. These three components are combined by using Bayes rules at multiple levels.
A) Sequence similarity and contrast in amino acid composition: In addition to sequence similarities between MoRFs, one can also exploit the fact that the amino acid composition of MoRFs is different from that of the general protein population [12, 13, 16] and contrasts most with the MoRFs’ flanking sequences (Flanks). MoRFCHiBi predicts MoRFs using two SVM models with two different noise tolerant kernels, SVMS and SVMT. SVMS, with a sigmoid kernel, extracts information related to the general contrast in amino acid composition between MoRFs and their Flanks. On the other hand, SVMT relies on a RBF kernel to identify sequence similarities between regions in a query sequence and MoRFs of the training set. Thus, in this work, MoRFCHiBi is used to generate a MoRF propensity score based on these two features.
B) Disorder: The residue composition of MoRFs is more similar to structured domains than to their surrounding disordered regions [7, 12, 13, 16]. As MoRFCHiBi relies solely on local sequence information, some of its identified MoRFs do not lie within disordered protein regions. Therefore, MoRFCHiBi_Web utilizes information on the level of protein disorder over longer regions encompassing putative MoRFs to improve its prediction accuracy. However, identifying such long stretches of disorder sequences requires disorder predictors that are designed for that purpose. Most disorder predictors are designed to identify short disorder segments and therefore generate low propensity scores at MoRF sites; in other words, their prediction scores dip in the vicinity of MoRFs [17]. Disorder predictors that target long disorder segments generate disorder propensity scores that do not dip at MoRF locations, so they tend to identify the entirety of a MoRF-harbouring IDR as disordered (more on this in Methods section: Selecting an appropriate disorder predictor).
C) Conservation: As functional elements, MoRF residues are more conserved on average than IDR residues [18, 19]. Furthermore, we assumed that the conservation is more pronounced for the subset of MoRF residues that are directly involved in binding and are part of the interaction interfaces (we documented the validation of this assumption in the Methods section “Binding residues and conservation“). Under this assumption, a MoRF consists of some highly conserved residues that are interspersed among less conserved residues. However, MoRFs would be computationally easier to identify if the high conservation scores extend throughout the entire MoRF sequence. Thus, we generated a MoRF conservation propensity score, mcs, such that the higher conservation scores of putative MoRF residues are extended to their neighboring residues (more on this in Methods section “Conservation propensity”).
These three sources of information–sequence similarity/contrast in amino acid composition, disorder, and conservation–are used in two steps to construct MoRFCHiBi_Web. First, an intermediate predictor, MoRFDC, utilizes Bayes rule to integrate disorder and conservation information into MoRF propensity scores. Then, Bayes rule is used again to combine the propensity scores of MoRFDC and MoRFCHiBi into the final MoRFCHiBi_Web scores.
In general, accumulating probabilities with Bayes rule should not be preceded by data processing. However, when Bayes rule is used to combine different propensities that do not reflect real probabilities, unjustified extreme values close to zero or one tend to dominate the prediction outcome. To reduce this effect, we transformed each set of input propensity scores from its unknown distribution into a Gaussian distribution with no extreme values (more on this in the Methods section: “Transforming data to normality”).
Datasets
Disfani et al. [13] collected a large set of structures containing protein-peptide interactions from the Protein Data Bank (PDB) deposited during or before March 2008 and filtered them on a number of principles to identify a set of 840 protein sequences. Each of these sequences includes a single peptide region with 5 to 25 residues presumed to be a MoRF. They divided these 840 sequences into a training set (TRAINING) with 421 sequences and the first test set with 419 sequences. Using similar criteria, Disfani et al. also collected a second test set from more recent PDB entries deposited between January 1st and March 11th, 2012. It consists of 45 sequences. All test sequences have less than 30% identity to those in TRAINING. We decided to use the same training set (TRAINING) used by Disfani et al. and Malhis and Gsponer [16] so that the performance of our predictor could be directly compared to theirs. TRAINING has a total of 245,984 residues including 5,396 MoRF residues. We evaluated our predictor using three test sets. The first test set, TEST464, includes all of the 464 sequences of the first and second test sets collected by Disfani et al. with a total of 296,362 residues including 5,779 MoRF residues. Although TEST464 is composed of sequence sets that have previously been used to test the performance of MoRF predictors [13, 14, 16], it may not be ideal for the task for two main reasons; Firstly, TEST464 is not free of redundancy as more than a third of its sequences share 90% or more identity with at least one other sequence in the set. Secondly, the selection procedure used by Disfani et al. does not verify that the identified peptides are disordered when alone in solution, i.e. that they are bona fide MoRFs. Therefore, we assembled an additional test set (EXP53) that includes only sequences that have been experimentally validated for their disordered properties in isolation. This second test set contains 53 non-redundant sequences that are sourced from four datasets consisting of MoRFs that have been experimentally validated to be disordered in isolation: First, a set of 8 sequences collected by Disfani et al. [13]. Second, a set of 40 sequences with short MoRFs of 30 residues or less which was used to train ANCHOR [12]. Third, a set of 26 sequences with long MoRFs of more than 30 residues that was also collected by Mészáros et al. [12]. And finally, a set of 21 sequences that was collected by Jones et al. [15]. We combined these four sets, and then removed sequences with more than 30% identity to those in TRAINING and redundant sequences with more than 30% identity with each other. We removed one additional sequence (Q51918|PDB:1YMH_E) because we could not find evidence for a disordered state in separation, its PDB structure resembles a globular domain, and the chain in the PDB structure, 1YMH_E, matched poorly to the protein sequence. Overall, EXP53 has 25,186 residues including 2,432 MoRF residues, which can be divided into 729 from sections with up to 30 residues identified as MoRF residues based one or more PDB structures and 1,703 from sections longer than 30 residues (please see “S2 Text”). The third test set, EXP9, consists of 9 sequences with experimentally validated MoRFs that were collected by Jones et al. These sequences are not homologous to any sequences used in the training of MFSPSSMpred, MoRFpred and ANCHOR. Hence, these 9 sequences do not overlap with our training data either, since we used the same training set as MoRFpred. EXP9 contains a total of 2,209 residues, including 12 MoRF regions with 163 MoRF residues. This third test set is used to compare prediction quality with MFSPSSMpred and DISOPRED3.
Selecting an appropriate disorder predictor
Several computational tools are available to predict the locations of disordered segments in proteins, including DISOPRED [20], ESpritz [21], IUPred [22, 23], and SPINE-D [24]. We evaluated the above disorder predictors on their performance in distinguishing MoRFs from non-MoRF residues by calculating the AUCMoRF (area under the ROCMoRF curve). As mentioned before, we require a disorder predictor that identifies MoRF residues as disordered. Our selection was made based on two measures: a high AUCMoRF value and a high true positive rate (TPR) at the lower left corner of ROCMoRF, which together represent high confidence predictions. Based on the ROCMoRF curves and AUCMoRF values generated by the above IDP predictors on TRAININIG (Fig 2A and 2B), ESpritz_D (ESpritz with the DisProt option) was selected because it provides the highest overall AUCMoRF (0.675) and the highest TPR at the lower left corner of the ROCMoRF curve.
Fig 2. ROCMoRF curves of multiple prediction tools.

Vertical axis is the true positive rate (TPR) and horizontal axis is the false positive rate (FPR). AUCMoRF values are in parentheses next to each label. (A) ROCMoRF curves of seven IDP predictors: DISOPRED (in black), ESpritz with the DisProt option (ESpritz_D in blue), ESpritz with the X-Ray option (ESpritz_X in yellow), ESpritz with the NMR option (ESpritz_N in green), IUPred with the long option (IUPred_l in brown), IUPred with the short option (IUPred_s in red), and SPINE-D (in light green). (B) The lower left corner of the ROCMoRF curves for the seven IDP predictors. (C) ROCMoRF curves of IPP (in blue), WOP (in gray), RWGRMP (in yellow) and its complement (Complement, in red). The Initial conservation propensity score, ics, (in green) was generated by joining IPP, Complement and WOP using Bayes rule. (D) ROCMoRF curves of ESpritz_D (in blue), ics (in green) and mcs (in red). mcs values are generated by processing ics.
Conservation propensity scores
First, three different conservation propensity scores from the PSI-BLAST [25] position specific scoring matrix (PSSM) files were calculated and tested for their ability to identify MoRFs: (1) the information per position (IPP), (2) the relative weight of gapless real matches to pseudocounts (RWGRMP), and (3) the weighted observed percentage of the query sequence residue rounded down (WOP). Alignment databases and PSI-BLAST parameters were selected to maximize AUCMoRF on TRAINING for each of the three scores independently (i.e. IPP, RWGRMP, and WOP). We tested the NCBI non-redundant, UniRef90, and UniProtKB/Swiss-Prot databases with and without filtering out coiled-coil and transmembrane regions using the program Pfilt [26]. We used three different amino acid substitution matrices (i.e. BLOSUM45, BLOSUM62, and BLOSUM80) in combination with a number of alignment iterations between 2 and 5. Note that when an AUCMoRF was less than 0.5, we used the propensity score’s complement. Thus, our selection criterion was to maximize the absolute value of |AUCMoRF—0.5|. For IPP and WOP, we selected UniRef90, BLOSUM62, and 2 alignment iterations, which resulted in AUCMoRF values of 0.586 and 0.604 respectively. For RWGRMP, the non-filtered UniProtKB/Swiss-Prot, BLOSUM62, and 2 alignment iterations resulted in an AUCMoRF of 0.422, which is equivalent to 0.578. These three scores were normalized and combined using Bayes rule to generate the initial conservation propensity score, ics (Fig 2 (C)). Although these three conservation values resulted from the alignment to two different databases, they are still highly related. Nonetheless, the ics generated from integrating these three scores provided a higher AUCMoRF than any of its subcomponents, and ics performs more consistently over various query sequences.
Next, the MoRF conservation propensity score, mcs, was determined. Its conception is based on the assumption that binding residues are the most conserved in MoRFs (see Methods: “Binding residues and conservation” for validation). The idea is to have a smooth conservation score such that disordered segments with an average disorder score > disorder 1 and more conserved residues than a certain minimum number (i.e. with ics > Conservation 1) are given high mcs scores (scenario 1). In addition, structured segments with an average disorder scores < disorder 2 and all their residues’ ics < Conservation 2 are given low mcs scores (scenario 2). The appropriate window sizes and thresholds for these scenarios were learned by testing many possible permutations and selecting those that generate the highest weighted AUCMoRF on TRAINING [16] (Fig 2 (D)). As a result, mcs is calculated in the two scenarios as follows: In scenario 1, if a residue r is at the center of a disordered window of 7 residues with an average normalized predicted disorder score > 0.45 and the window includes 3 or more conserved residues with an ics > 0.45, then this residue’s mcs is assigned to be equal to the average ics of the conserved residues. Furthermore, for every extra conserved residue above 3 in that window, a square root function is applied twice to further increase mcs. In scenario 2, if a residue r is at the center of a structured window of 15 residues with an average normalized predicted disorder score < 0.45 and no residue within the window has an ics > 0.60, then this residue’s mcs is assigned to be equal to its ics multiplied by the window’s average normalized predicted disorder score. For all other residues that do not fall under scenario 1 or 2, mcs is equal to ics. (For learning appropriate window sizes and other thresholds, please see: Learning the MoRF conservation propensity score thresholds values section in supplement).
Binding residues and conservation
To validate the assumption that residues involved in binding are more conserved than other residues in MoRFs, we measured the distances between MoRF residues and the surface of the binding partner using coordinates for the MoRF complex structures of the TEST464 dataset collected from the Protein Data Bank [27]. When we separated the MoRF residues into those that are in close proximity to the binding partner and those that are not, we found a significant difference between their median conservation (ics) values (distance >5Å ics = 0.536 ± 0.005; < = 5Å ics = 0.568 ± 0.004; p value < 2.2x10-16). The confidence intervals for the medians are calculated as , where IQR is the interquartile range. The IQR is calculated as the difference between the first and third quartile of a distribution of n values as defined in the boxplot.stats function of R [28] (see “S1 Excel” for data and example calculation). The p value is calculated using the Wilcoxon test in R. The distances measured correspond to those between the beta-carbon of the MoRF residues and the closest atom on the surface of the binding partner. The same trend was observable across a range of cut-off values (not shown). The lower conservation scores of the residues above the distance cut-off suggest that residues further away from the interaction surface are less conserved. We also measured the change in relative accessible surface area (rASA) upon binding using NACCESS [29]. The change in rASA of each residue was calculated by subtracting the rASA in the bound state from the rASA in the unbound state, where the unbound structure was obtained by removing all the atoms of the binding partner from the protein complex structure. Once again, we separated the MoRF residues into two groups for comparison. The group of residues that have a change in rASA greater than 20 percent upon binding has a higher median ics compared to the remaining residues (> 20% change in rASA ics = 0.570 ± 0.004; < = 20% change in rASA ics = 0.537 ± 0.005; p value < 2.2x10-16). This difference in ics values suggests that residues that contribute more to the interface surface tend to be more conserved.
Transforming data to normality (Normalization)
In this work, we utilized a number of component predictors to predict scores for features associated with MoRFs (eg. MoRFCHiBi, ESpritz_D, ics … etc). Then, we hierarchically integrated these predicted scores into a single propensity score that reflects the likelihood of each residue to be a MoRF residue. Because these component scores are positively correlated with the probability of MoRFs, we chose to integrate them using Bayes rule. Ideally, Bayes rule should be used to combine real probabilities. However, the scores we are integrating predict features that unequally reflect the likelihood of MoRFs. As a result, these component scores do not reflect real MoRF probabilities and include extreme values, which are close to zero or one. These extreme values dominate the outcome of Bayes rule and mask the effect of other features. Using TRAINING, a MapD function was created for each input feature D that transforms its propensity scores from its unknown distribution UD to approximately fit a Gaussian probability density function specified by the normal distribution N(μ = 0.5, σ2 = 0.01) while preserving their cumulative values. The cumulative value of the feature D score for a residue x, S(UD) x, is the probability of a residue in TRAINING to have a propensity score less than or equal to S(UD) x. Thus, the normalized score S(ND) x for S(UD) x is:
The MapD function for each feature is then used to preprocess the corresponding feature scores of query sequences. (For the implementation procedure, please see: “Defining MapD function and normalization section in S1 Text”). For this article, this process of transforming data to a normal distribution is what we call normalization.
Performance evaluation
We used AUC, the area under the receiver operator characteristics curve (ROC), to evaluate the performance of MoRFCHiBi_Web and compared it with that of MoRFCHiBi, MoRFpred and ANCHOR. Even though AUC is a common evaluation metric for prediction performance, it only provides an overall assessment about the separation of the two classes, which are MoRFs and non-MoRFs in this case. However, in most scenarios, we are only interested in high confidence predictions at some high threshold values represented by the lower left corner of the ROC curve. Thus, we also compared false positive rates, FPRs, at different true positive rate values, TPRs, for each predictor. TPR is defined as TPR = TP / NMoRF where true positive, TP, is the number of accurately predicted MoRF residues, and NMoRF is the total number of experimentally verified MoRF residues. False positive rate is defined as FPR = FP / Nnon-MoRF where FP is the number of inaccurately predicted MoRF residues, and Nnon-MoRF is the total number of non-MoRF residues. On occasion, to maintain consistency with evaluations presented by other groups, we also used sensitivity and specificity where sensitivity is equal to TPR and specificity is 1 –FPR.
Results
When comparing the performance of different machine learning predictors, it is crucial for the test data, or homologs of test data sequences, not to overlap with data used in the training of the predictors. In our case, MoRFCHiBi_Web was trained on the same TRAINING set used by MoRFpred and MoRFCHiBi, so we can make detailed performance comparisons with these predictors using the two datasets, TEST464 and EXP53, described in the Datasets section in Methods. Both TEST464 and EXP53 consist of sequences with less than 30% identity to those in TRAINING. These two test sets are also used for comparison with ANCHOR since it has minimal reliance on its training set. Note that all the predictors above are trained on short MoRFs with up to 30 residues, but EXP53 also includes MoRFs that are longer than 30 residues. Thus, when testing the predictors’ performance on EXP53, we also provided a separate performance evaluation for short and long MoRFs. Table 1 shows the AUC values for each of the four predictors and reveals that MoRFCHiBi_Web outperforms the other ones.
Table 1. AUC Results.
| Data | MoRFCHiBi_Web | MoRFCHiBi | MoRFpred | ANCHOR |
|---|---|---|---|---|
| TEST464 | 0.806 | 0.748 | 0.675 | 0.605 |
| EXP53_All | 0.789 | 0.714 | 0.620 | 0.615 |
| EXP53_Short | 0.877 | 0.790 | 0.673 | 0.683 |
| EXP53_Long | 0.751 | 0.681 | 0.598 | 0.586 |
AUC values of the four MoRF predictors using the TEST464 and EXP53 data sets. For EXP53_Short, only MoRF sections with up to 30 residues are considered as MoRFs, while longer MoRF sections were masked out. Similarly, for EXP53_Long, only MoRF sections longer than 30 residues are considered, while shorter MoRFs were masked out.
ROC curves generated for TEST464 (Fig C in S1 Text) and EXP53 (Fig 3) show that MoRFCHiBi_Web has a higher specificity (1- FPR) at any level of sensitivity (TPR) when compared to ANCHOR and MoRFpred. All four predictors performed better on EXP53_Short than on TEST464. This difference is expected because each sequence in TEST464 is labeled with only one MoRF, despite the fact that some sequences contain more MoRFs. For example, all MoRFs in the protein p53 are annotated on a single sequence in EXP53, whereas TEST464 contains eight almost identical sequences of p53 with only one annotated MoRF in each, which were all identified based on different PDB structures (Uniprot: P04637, Q2XSC7, Q2XN98, Q9TTA1 … etc.). Therefore, evaluations with the more sparsely annotated sequences of TEST464 results in some true MoRF residues with high prediction scores being erroneously treated as non-MoRF resides. Hence, we think that the overall performances are more accurately represented by test results on EXP53 than on TEST464.
Fig 3. Full ROC curves (A, C and E) and their lower left corners (B, D and F) for the EXP53 dataset.

ROC curves in A and B show the MoRF prediction performance for all MoRFs. C and D show the prediction performance of short MoRFs with up to 30 residues while longer MoRFs are masked out, and E and F show the performance of long MoRFs with more than 30 residues while shorter MoRFs are masked out. Vertical axis is the true positive rate (Sensitivity) and horizontal axis is the false positive rate (1-Specificity). MoRFCHiBi_Web (MCW) is in red, MoRFCHiBi (MC) in orange, MoRFpred (MP) in green, and ANCHOR (AN) in purple. The dashed line (Naïve) represents a random classifier. AUC values are in parentheses next to each label.
When compared to MoRFpred and ANCHOR, MoRFCHiBi_Web produces less than half the false positive rate for the same true positive rate at practical threshold values (Table 2).
Table 2. FPR as a Function of TPR.
| TPR (Sensitivity) | FPR (1—Specificity) | |||
|---|---|---|---|---|
| MoRFCHiBi_Web | MoRFCHiBi | MoRFpred | ANCHOR | |
| 0.2 | 0.016, 0.015, 0.018 | 0.030, 0.010, 0.038 | 0.081, 0.059, 0.099 | 0.103, 0.070, 0.128 |
| 0.3 | 0.032, 0.022, 0.041 | 0.064, 0.035, 0.091 | 0.140, 0.095, 0.168 | 0.173, 0.118, 0.217 |
| 0.4 | 0.060, 0.034, 0.083 | 0.124, 0.065, 0.165 | 0.218, 0.154, 0.252 | 0.263, 0.175, 0.310 |
| 0.5 | 0.113, 0.061, 0.144 | 0.201, 0.117, 0.264 | 0.306, 0.227, 0.353 | 0.345, 0.249, 0.398 |
FPR as a function of TPR computed on the EXP53 set (All MoRFs, short MoRFs, long MoRFs) for MoRF CHiBi_Web, compared to MoRF CHiBi, MoRFpred, and ANCHOR at different TPR cut off values.
As MFSPSSM and DISOPRED3 were trained on a large training set that includes all the sequences of TEST464 and many of the sequences in EXP53 (or homologs thereof), we compared the performance of MoRFCHiBi_Web with the results of DISOPRED3, MFSPSSMpred and MoRFpred that were reported by Jones and Cozzetto [15] based on the EXP9 set. This set contains experimentally validated MoRFs that have no sequence homology with MoRFs used in the training of all these predictors and thus MoRFCHiBi_Web. For each predictor, only one data point (i.e. specificity, sensitivity) was provided by Jones and Cozzetto [15]. On this small test set, MoRFCHiBi_Web also achieves higher sensitivity (TPR) for a given specificity (1-FPR) when compared to the other predictors (Fig 4).
Fig 4. Comparing the performance of MoRFCHiBi_Web (MCW in red), MoRFCHiBi (MC in orange), DISOPRED3, MFSPSSMpred and MoRFpred using EXP9.
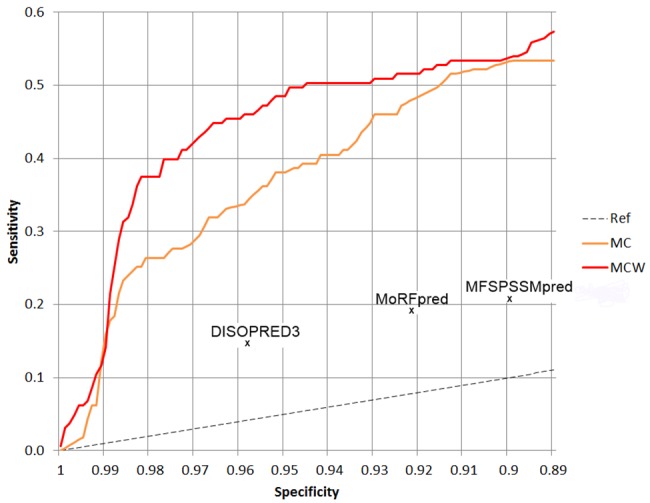
Curves of sensitivity as a function of specificity for MoRFCHiBi_Web and MoRFCHiBi are computed on a set of 9 protein sequences collected by Jones and Cozzetto [15]. The data points for each of the three predictors, DISOPRED3, MFSPSSMpred and MoRFpred, are from published results of Jones and Cozzetto [15].
As MoRF predictors are often used to screen large sets of proteins, we were also interested in their efficiencies. Prediction times of MoRFCHiBi and ANCHOR, both of which do not require PSI-BLAST alignments, were tested by scoring the entire TEST464 set on an Intel core i7, 3.44G desktop. MoRFCHiBi_Web, MoRFpred, DISOPRED3 and MFSPSSMpred were tested by repeatedly submitting a single sequence from the TEST464 set (Uniprot:Q38087) with 903 residues to their web sites and then selecting the fastest outcome. ANCHOR is the fastest with a processing speed of 4*106 r/m (residues/minute), MoRFCHiBi came in second with 6*103 r/m, and MoRFCHiBi_Web is third with 536 r/m. MFSPSSMpred processed 135 r/m, MoRFpred 41 r/m and DISOPRED3 13 r/m. This comparison might not be entirely fair as the underlying hardware of web-based predictors is unknown. Nevertheless, the comparison of computational costs of the tools that depend on PSI-BLAST alignment clearly shows that MFSPSSMpred, MoRFpred and DISOPRED3 are significantly slower than the predictor introduced here.
Discussion
We presented a new web-based approach, MoRFCHiBi_Web, for predicting MoRFs within protein sequences. We compared its performance to that of MoRFpred, MoRFCHiBi and ANCHOR using two different test sets: TEST464 with 464 sequences and EXP53 with 53 sequences that contain only experimentally validated MoRFs. The results demonstrate that MoRFCHiBi_Web outperforms all three predictors. Furthermore, MoRFCHiBi_Web generates less than half the false positive rate of other predictors at most cut-off values. Because MFSPSSMpred and DISOPRED3 use a different set of training data, we have to rely on a small set of only 9 sequences, EXP9, collected by the authors of the latter to compare the performance of these two predictors with MoRFCHiBi_Web. Although results from such a small dataset might not accurately reflect the actual performance of these predictors, MoRFCHiBi_Web‘s significantly higher sensitivity at selected specificities shows a consistent advantage over these predictors.
The high accuracy of MoRFCHiBi_Web can be attributed to the use of three mostly independent sources of information: sequence similarity and contrast in amino acid composition, disorder content of the MoRF-harbouring segment, and conservation of the MoRF sequence. The first source has been exploited in a stand-alone predictor (MoRFCHiBi) that we developed previously [16]. As MoRFCHiBi only considers local sequence information, it does not utilize information about the level of disorder of the protein segments in which the putative MoRFs are located. The location of MoRFs within IDRs is part of their definition and distinguishes them from a similar and overlapping class of protein sequences known as short linear motifs (SLiMs). SLiMs are sequences 3 to 10 residues in length that have been found to be enriched in IDRs and bind specific protein domains [30]. However, SLiMs are also found in structured domains, as exemplified by PY motifs buried in structured proteins that get exposed and recognized by E3 ubiquitin ligase upon heat shock [31]. MoRFCHiBi was trained and tested on MoRFs only and, therefore, the inclusion of disorder information was necessary and rewarding. Furthermore, residues in IDRs are known to be less conserved on average than those in MoRFs [18, 19]. Therefore, MoRFCHiBi_Web was also designed to exploit the difference in conservation to improve predictions.
The improvement derived from the addition of information on both intrinsic disorder and conservation to MoRFCHiBi can be exemplified in the case of the protein p53 (Fig 5). This protein contains two main regions where the existence of MoRFs has been verified: one in the N-terminal region and the other in the C-terminal region. The propensity scores generated by MoRFCHiBi correctly identified the N and C-terminal MoRFs but they also incorrectly identified the region around residue 161 as a MoRF segment, as shown by the grey curve in Fig 5. The region between residues 95 and 292 is known to be a globular domain (PDB: 1gzh; [32]). MoRFDC keeps the propensity scores low in this globular region, which suppresses the MoRFCHiBi_Web scores at the region around position 161 relative to the scores of the N and C-terminal MoRF regions. The addition of disorder and conservation information is thereby able to broaden the gap in propensity scores between MoRF and non-MoRF regions, which allows for a more stringent cutoff that generates a lower false positive rate. Interestingly, the remaining false positive region near the C-terminus is also known to make interactions, but it has not been verified to be intrinsically disordered [17].
Fig 5. MoRFCHiBi_Web Propensity scores for the protein p53.

Propensity scores for the protein p53 by MoRFCHiBi_Web (MCW) in orange, MoRFCHiBi (MC) in gray and MoRFDC (MDC) in green. The 2 verified MoRF sections in our EXP53 test set are marked in blue below the chart.
As some predictors have been trained on all available MoRFs, it is important to discuss issues regarding scoring bias towards the training data. In general, MoRFs in query sequences that are identical or very similar to those in the training set are scored favorably compared to other MoRFs. This scoring bias can often be misleading as we naturally expect the score quality to be consistent throughout the entire query sequence. Hence, the scoring bias can hamper the identification of novel MoRFs because their scores are overshadowed by those over-scored training MoRFs. Different machine learning tools are affected differently by training data. Support vector machines with RBF kernels are known to be prone to over-scoring their training data [33], whereas over-scoring is minimal with the use of linear and sigmoid kernels. One of MoRFCHiBi_Web‘s components is a SVM-RBF model, namely the SVMT model in MoRFCHiBi. However, the potential for bias brought on by SVMT is mitigated by contributions from the other models, MoRFCHiBi‘s SVMS and MoRFDC’s conservation and disorder components, which are all less susceptible to over-scoring. Specifically, MoRFCHiBi‘s SVMS employs a sigmoid kernel and was trained on synthetic data. Furthermore, MoRFDC’s conservation component has very limited reliance on training and its disorder component comes from ESpritz, which is based on a neural-network model that was trained on an independent dataset from DisProt. Using Bayes rule to integrate multiple propensity scores generated by different predictors targeting different features using different machine learning tools provides MoRFCHiBi_Web with less potential for biased scoring compared to predictors that are based on a single machine learning tool.
In summary, we introduced a new MoRF predictor, MoRFCHiBi_Web, which is based on hierarchically incorporating several independently computed propensity scores of features associated with MoRFs. MoRFCHiBi_Web is fast and its predictions include less than half the false positives compared to all available MoRF predictors. Regarding its usage, we like to stress that categorical prediction by assigning a static cut-off value can be a misleading oversimplification. Yet, if users need such a cut-off, we suggest a value of 0.66. At this cut-off, MoRFCHiBi_Web achieved sensitivities of 0.482, 0.615, 0.425 for all_MoRFs, short_MoRFs, and long_MoRFs from EXP53, respectively, at a specificity of 0.897.
Supporting Information
Excel file for data and example calculation.
(XLSX)
(DOCX)
(TXT)
Acknowledgments
We thank Duncan Ferguson for his helpful comments on this manuscript.
Data Availability
Four sets of sequences were used in this work: a training set and three test sets. TRAINING is the same training set used by Disfani et al. [13] and is publicly available from their web site: http://biomine-ws.ece.ualberta.ca/MoRFpred/index.html. TEST464 is the sum of two publicly available sets: Test and Test2012 from Disfani et al. [13]. EXP9 is publicly available from Jones et al. [15] supplementary material. EXP53: is collected from four publicly available experimentally validated sets: one from Disfani et al. [13], one from Jones et al. [15], supplementary material, two sets from Mészáros et al. [12]. EXP53 is in this paper's Supporting Information files.
Funding Statement
This work was supported by grants from the Canadian Institute of Health Research (CIHR) (http://www.cihr-irsc.gc.ca/e/193.html) (BOP-130835, to JG); Genome BC (http://www.genomebc.ca/) (Genome BC 175REG, to JG); and Michael Smith Foundation for Health Research (MSFHR) (http://www.msfhr.org/) (CI‐SCH‐03020(11‐1), to JG). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014. July 9; 114(13):6589–631. 10.1021/cr400525m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oldfield CJ, Dunker AK. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem. 2014; 83:553–84. 10.1146/annurev-biochem-072711-164947 [DOI] [PubMed] [Google Scholar]
- 3. Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015. January; 16(1):18–29. 10.1038/nrm3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cumberworth A, Lamour G, Babu MM and Gsponer J. Promiscuity as a functional trait: intrinsically disordered regions as central players of interactomes. Biochem. J., 2013. September 15; 454, 361–369. 10.1042/BJ20130545 [DOI] [PubMed] [Google Scholar]
- 5. Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, Uversky VN. Analysis of molecular recognition features (MoRFs). J Mol Biol. 2006. October 6; 362(5):1043–59. [DOI] [PubMed] [Google Scholar]
- 6. Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry, 44 (2005), pp. 12454–12470. [DOI] [PubMed] [Google Scholar]
- 7. Vacic V, Oldfield CJ, Mohan A, Radivojac P, Cortese MS, Uversky VN, Dunker AK. Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res. 2007. June; 6(6):2351–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng Y, Oldfield CJ, Meng J, Romero P, Uversky VN, Dunker AK. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry. 2007. November 27;46(47):13468–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Babu M, Lee RVD, DeGroot NS, Gsponer J. Intrinsically disordered proteins: regulation and disease. Current Opinion in Structural Biology, 2011. January 31; 21, 1–9. [DOI] [PubMed] [Google Scholar]
- 10. Wong ETC, NA D, Gsponer J. On the Importance of Polar Interactions for Complexes Containing Intrinsically Disordered Proteins. PLoS Comput Biol, 2013. August 22; 9(8), e1003192 10.1371/journal.pcbi.1003192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buljan M, Chalancon G, Eustermann S, Wagner GP, Fuxreiter M, Bateman A, et al. Tissue-Specific Splicing of Disordered Segments that Embed Binding Motifs Rewires Protein Interaction Networks. Mol Cell. 2012. June 29; 46(6):871–83. 10.1016/j.molcel.2012.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mészáros B, Simon I, Dosztányi Z. Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol., 2009. May 1; 5, e1000376 10.1371/journal.pcbi.1000376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Disfani FM, Hsu WL, Mizianty MJ, Oldfield CJ, Xue B, Dunker AK, et al. MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics, 2012. June 15; 28 (12): i75–i83. 10.1093/bioinformatics/bts209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fang C, Noguchi T, Tominaga D and Yamana H. MFSPSSMpred: identifying short disorder-to-order binding regions in disordered proteins based on contextual local evolutionary conservation. BMC Bioinformatics, 2013. October 4; 14:300 10.1186/1471-2105-14-300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones DT and Cozzetto D. DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics, 2015. March 15; 31 (6): 857–863. 10.1093/bioinformatics/btu744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Malhis N and Gsponer J. Computational Identification of MoRFs in Protein Sequences. Bioinformatics 2015. January 30; 31 (11): 1738–1744. 10.1093/bioinformatics/btv060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uversky VN, and Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010. June 1; 1804 (6):1231–64. 10.1016/j.bbapap.2010.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mészáros B, Tompa P, Simon I and Dosztányi Z. Molecular Principles of the Interactions of Disordered Proteins. JMB, 2007. September 14; Volume 372, Issue 2, Pages 549–561. [DOI] [PubMed] [Google Scholar]
- 19. Trudeau T, Nassar R, Cumberworth A, Wong ETC, Woollard G and Gsponer J. Structure and Intrinsic Disorder in Protein Autoinhibition. STRUCTURE 2013. March 5; 21, 332–341. 10.1016/j.str.2012.12.013 [DOI] [PubMed] [Google Scholar]
- 20. Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics, 2004; 20, 2138–2139. [DOI] [PubMed] [Google Scholar]
- 21. Walsh I, Martin AJ, Di Domenico T, Tosatto SC. ESpritz: accurate and fast prediction of protein disorder. Bioinformatics. 2012; 28(4):503–9. 10.1093/bioinformatics/btr682 [DOI] [PubMed] [Google Scholar]
- 22. Dosztányi Z, Csizmók V, Tompa P, Simon I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol 2005. April 8; 347: 827–839. [DOI] [PubMed] [Google Scholar]
- 23. Dosztányi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005. August 15; 21(16):3433–4. [DOI] [PubMed] [Google Scholar]
- 24. Zhang T, Faraggi E, Xue B, Dunker AK, Uversky VN, Zhou Y. SPINE-D: accurate prediction of short and long disordered regions by a single neural-network based method. J Biomol Struct Dyn. 2012; 29(4):799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl. Acids Res. 1997. September 1; 25(17), 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jones DT, Swindells MB. Getting the most from PSI-BLAST. Trends Biochem. Sci. 2002. March; 27, 161–164. [DOI] [PubMed] [Google Scholar]
- 27. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000. January 1 28(1):235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. R Core Team (2013). R: A language and environment for statistical computing R Foundation for Statistical Computing, Vienna, Austria: ISBN 3-900051-07-0. Available: http://www.R-project.org/. [Google Scholar]
- 29. Hubbard SJ, Thornton JM, 'NACCESS', Computer Program (Department of Biochemistry and Molecular Biology, University College London; ). 1993. Available: http://www.bioinf.manchester.ac.uk/naccess/. [Google Scholar]
- 30. Weatheritt R.J. and Gibson T.J. Linear motifs: lost in (pre)translation. Trends in Biochemical Sciences, August 2012, Vol. 37, No. 8: 333–41. 10.1016/j.tibs.2012.05.001 [DOI] [PubMed] [Google Scholar]
- 31. Fang NN, Chan GT, Zhu M, Comyn SA, Persaud A, Deshaies RJ, et al. Rsp5/Nedd4 is the main ubiquitin ligase that targets cytosolic misfolded proteins following heat stress. Nature Cell Biology, 2014. December; 16(12):1227–37. 10.1038/ncb3054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Derbyshire D.J., et al. (2002) Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. Embo J. 21:3863–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han H, Jiang X. Overcome support vector machine diagnosis overfitting. Cancer Inform. 2014. December 9 13: 145–58. 10.4137/CIN.S13875 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Excel file for data and example calculation.
(XLSX)
(DOCX)
(TXT)
Data Availability Statement
Four sets of sequences were used in this work: a training set and three test sets. TRAINING is the same training set used by Disfani et al. [13] and is publicly available from their web site: http://biomine-ws.ece.ualberta.ca/MoRFpred/index.html. TEST464 is the sum of two publicly available sets: Test and Test2012 from Disfani et al. [13]. EXP9 is publicly available from Jones et al. [15] supplementary material. EXP53: is collected from four publicly available experimentally validated sets: one from Disfani et al. [13], one from Jones et al. [15], supplementary material, two sets from Mészáros et al. [12]. EXP53 is in this paper's Supporting Information files.