

Abstract
Synthesis of the p53 tumor suppressor and its subsequent activation following DNA damage are critical for its protection against tumorigenesis. We previously discovered an internal ribosome entry site (IRES) at the 5′ untranslated region of the p53 mRNA. However, the connection between IRES-mediated p53 translation and p53's tumor suppressive function is unknown. In this study, we identified two p53 IRES trans-acting factors, translational control protein 80 (TCP80), and RNA helicase A (RHA), which positively regulate p53 IRES activity. Overexpression of TCP80 and RHA also leads to increased expression and synthesis of p53. Furthermore, we discovered two breast cancer cell lines that retain wild-type p53 but exhibit defective p53 induction and synthesis following DNA damage. The levels of TCP80 and RHA are extremely low in both cell lines, and expression of both proteins is required to significantly increase the p53 IRES activity in these cells. Moreover, we found cancer cells transfected with a shRNA against TCP80 not only exhibit decreased expression of TCP80 and RHA but also display defective p53 induction and diminished ability to induce senescence following DNA damage. Therefore, our findings reveal a novel mechanism of p53 inactivation that links deregulation of IRES-mediated p53 translation with tumorigenesis.
INTRODUCTION
One of the most important tumor suppressors identified thus far is p53. Under normal conditions, p53 is inactive, and its cellular levels are low. In response to various genotoxic or cytotoxic stress, including DNA damage, p53 becomes activated (1). The activated p53 causes cell growth arrest or apoptosis, allowing damaged cells to self-repair or be eliminated. In this manner, p53 protects against malignant transformation of normal cells (2, 3). Activation of p53 involves both accumulation and posttranslational modification. It is thought that the control of p53 induction or accumulation occurs mainly at the translational and posttranslational levels (1). Although levels of p53 are known to be regulated by the ubiquitin ligase mouse double minute 2 (MDM2), now there is clear evidence showing that enhanced p53 translation is essential for its induction following DNA damage (4). More specifically, the 5′ untranslated region (5′ UTR) of p53 mRNA was found to be a major site for regulation of p53 translation (5, 6). The mechanism underlying translational regulation of p53 induction via its 5′ UTR has started to emerge.
Cap-dependent initiation of protein translation is used by the majority of mRNAs, since almost all eukaryotic mRNAs have an N7-methylguanosine cap structure at their 5′ ends. Eukaryotic translation initiation factor 4E (eIF-4E), a translation initiation protein that binds to the cap structure, initiates the process (7). In situations where cap-dependent translation is impaired, including apoptosis or DNA damage, cap-independent protein translation, mediated by an internal ribosomal entry site (IRES) that does not require the participation of eIF-4E, is needed in eukaryotes for the synthesis of key regulatory proteins (8, 9).
We found that exposure to etoposide, a DNA damaging agent that causes DNA double-strand breaks and suppresses cap-dependent translation, results in an increase in the association of p53 mRNA with polyribosomes (10). Using a bicistronic reporter vector, we discovered an IRES sequence in the 5′ UTR of the p53 mRNA (4, 10). We also found that the IRES activity increases in response to DNA damage in MCF-7 cells (4, 10). MCF-7 is a breast cancer cell line that expresses wild-type p53 protein and exhibits increased p53 synthesis in response to DNA damage (10). Therefore, these results suggest that IRES-mediated p53 translation or synthesis is important for the accumulation of p53 protein following DNA damage.
The presence of an IRES sequence in an isoform of p53, p47 (also known as p53/p47, Δ40p53, and ΔNp53), and a p53 homologue, p73, has also been discovered (11–13). It was recently found that mouse p53 mRNA also contains an IRES sequence (14), which is consistent with a previous finding that mouse p53 synthesis is upregulated by DNA damage (5). The increase in p53 IRES activity following different cellular stress has been observed by a number of recent reports (15–20). For instance, it was found that during DNA damage or oncogene-induced senescence (OIS), the p53 IRES exhibits enhanced activity to facilitate p53 translation (17), which provides further evidence that the p53 IRES plays a key role in regulation of p53 synthesis following geno- or cytotoxic stress. However, little is still known regarding how the p53 IRES regulates p53 synthesis in response to DNA damage and other cellular stress and whether there is a functional link between defective IRES-mediated p53 translation and tumorigenesis.
Control of translational initiation at cellular IRESs requires the presence of auxiliary factors that are known as IRES trans-acting factors (ITAFs) (21, 22). ITAFs are proteins that can positively or negatively affect IRES activity (8). A number of proteins have been identified as binding to the p53 5′ UTR in vitro (6). Many of them are also known to be involved in protein translation and ribosomal biogenesis. Therefore, some of these proteins could be p53 ITAFs that regulate p53 IRES activity and p53 synthesis.
While p53 mutations are common (mutation rate ≥ 50%) in cancer, certain types of cancer display a much lower rate of p53 mutation. For instance, only ca. 20% of breast cancers harbor p53 mutations (23). It is unclear why 80% of breast tumors develop into cancer despite containing the wild-type p53 coding region. We made the initial hypothesis that inefficient or defective p53 translation in response to DNA damage could result in tumorigenic transformation (4). It is conceivable that in many tumors retaining wild-type p53, p53 may lose the ability to respond to DNA damage due to defective IRES-mediated p53 synthesis.
In the present study, we identified two ITAFs of p53 that positively regulate p53 IRES activity and p53 synthesis. Moreover, we discovered two breast cancer cell lines that express wild-type p53 but do not exhibit normal p53 induction and p53 IRES activation following DNA damage. The expression of both ITAFs in these breast cancer cell lines and in MCF-7 cells was further analyzed to determine the potential role of these ITAFs in p53 induction and malignant transformation. Our results suggest a link between reduced expression of positive p53 ITAFs and the defective response of p53 to DNA damage in cancer cells.
MATERIALS AND METHODS
Materials.
Etoposide and cycloheximide were from Calbiochem. The antibodies include anti-DRBP76 (TCP80) antibody (BD Transduction Laboratories), the anti-DHX9 (RHA) antibody (Bethyl Laboratories), the anti-p53 primary antibody (Calbiochem), and the horseradish peroxidase (HRP)-conjugated p53 antibody (Santa Cruz Biotechnology). MCF-7/pCDNA3.1 and MCF-7/shTCP80 cells were obtained by stably transfecting MCF-7 cells with either a pCDNA3.1 plasmid or a pCDNA3.1 plasmid containing a shRNA against TCP80.
Cell culture and transfection.
Cells were grown in Dulbecco modified Eagle medium or RPMI medium supplemented with antibiotics and 10% fetal bovine serum. All plasmid transfections were performed using Fugene 6 transfection reagent (Roche). Cells were allowed to grow to subconfluence and were then transfected with 1.5 μg of plasmid and lysed 24 or 48 h after transfection.
RNA pulldown assay.
The p53 IRES sequence was amplified from the pR5UTRF vector by PCR. The amplified fragment was then transcribed in vitro using an AmpliScribe T7 Flash transcription kit (Epicenter Biotechnologies) in the presence of biotin-14-CTP according to the manufacturer's instructions. Next, 1 μg of biotinylated RNA was used to coat streptavidin M-280 DynaBeads (Invitrogen). MCF-7 cells were lysed in a cytoplasmic extraction buffer containing 10 mM HEPES, 3 mM MgCl2, 40 mM KCl, 5% glycerol, 0.2% NP-40, 1 mM dithiothreitol, and protease inhibitors. The cell lysate was incubated with the RNA-coated beads. The beads were then washed extensively with cytoplasmic extraction buffer before the addition of sodium dodecyl sulfate (SDS) sample loading buffer.
Dual-luciferase assays.
Cells were lysed with 1× passive lysis buffer. The Dual-Luciferase reporter assay system (Promega) was then used in conjunction with a Berthold luminometer to determine firefly and Renilla luciferase activities according to the manufacturer's instructions.
Cell extract preparation, SDS-PAGE, and Western blotting.
Cells were washed twice with phosphate-buffered saline (PBS) and lysed with TGN lysis buffer (10) containing 1% NP-40 and a protease inhibitor cocktail tablet (Roche). Protein concentration was measured using the Lowry assay method. Equal amounts of protein were loaded onto an SDS-PAGE gel and later transferred onto nitrocellulose or polyvinylidene difluoride (PVDF) membranes for immunoblotting. Densitometry analysis for proteins bands was done using an UN-SCAN-IT gel analysis software.
[35S]Met labeling of newly synthesized p53 protein.
Cells were incubated with cysteine and methionine-free medium supplemented with dialyzed fetal bovine serum for 2 h. After incubation, 90 μCi of [35S]methionine ([35S]Met) was added to the medium. The cells were then lysed, and p53 protein was immunoprecipitated by an anti-p53 antibody. The beads were washed three to four times, and SDS loading buffer was added to the beads, followed by SDS-PAGE and Western blotting. Newly synthesized p53 protein was detected using a Typhoon phosphorimager.
Determination of p53 half-life.
Relative p53 density is determined as the ratio of p53 to β-actin levels following densitometric analysis. The relative p53 density was then normalized so that the highest value corresponds to 100. The log10 of normalized p53 density was then plotted against time of CHX treatment, and the best-fit linear trend line was generated. The equation of the trend line (y = ax + b) was used to determine the half-life of the protein, where y = log1050 and x is the p53 half-life.
Reverse transcription-PCR (RT-PCR) analysis.
Cells were grown to subconfluence. Total cellular RNA was purified from cell lysates by an Aurum total RNA minikit (Bio-Rad) according to the manufacturer's instructions and treated with RNase-Free DNase (Qiagen). RNA was then reverse transcribed using the iScript RT Supermix kit (Bio-Rad). PCR was then carried out for detecting p53 and β-actin or GAPDH mRNA using the GoTaq green master mix from Promega. The forward and reverse primers of the p53 sequence were 5′-TGG AAA CTA CTT CCT GAA AAC AAC G-3′ and 5′-GGG AGT ACG TGC AAG TCA CAG A-3′, respectively. Reactions were done using a MasterCycler gradient (Eppendorf) PCR machine, and products were run on a 1% agarose gel stained with ethidium bromide.
SA-β-galactosidase staining for cellular senescence.
Cells were stained using an SA-β-galactosidase staining kit (Cell Signaling). Briefly, cells were washed with PBS and fixed for 15 min with the fixative solution. Cells were then stained overnight in a dry 37°C incubator using the staining solution containing X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside). Pictures were taken using a Leica DM IRB microscope at ×40 magnification.
Flow cytometry assay.
Cells were seeded onto six well plates and grown to subconfluence. After treatment with 10 μM etoposide for 24 h, the cells were gently removed from the plates, washed once with cold PBS, and fixed with cold 70% ethanol. The cells were then stained with propidium iodide and subjected to flow cytometry (Becton Dickinson FACSCalibur). The cell population at each phase was analyzed by using Modfit 2 software.
Data analysis.
One-way analysis of variance with the Newman-Keuls post test was used to compare means of multiple groups (see Fig. 6B and C). Comparisons between two individual groups were evaluated by using a Student t test and Microsoft Office Excel software. P values of <0.05 were considered statistically significant.
FIG 6.
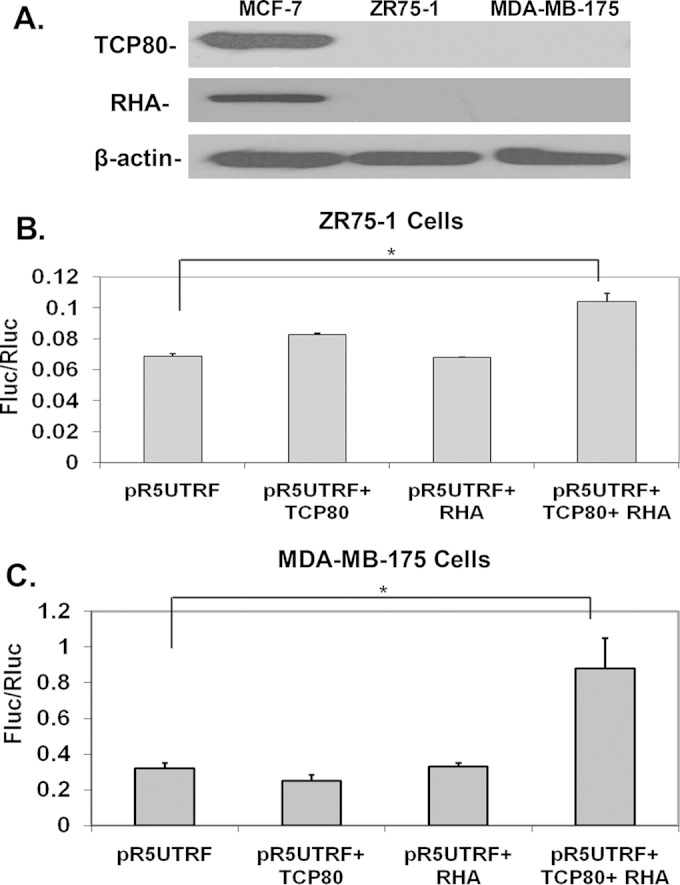
Both TCP80 and RHA are required to induce increased p53 IRES activity in ZR75-1 and MDA-MB-175 cells. (A) Expression levels of TCP80 and RHA in various cell lines. MCF-7, ZR75-1, and MDA-MB-175 cells were grown to subconfluence. Cells were then lysed and immunoblotting was performed to determine TCP80, RHA, and β-actin levels in each cell line. The results presented are representative of three separate experiments. (B and C) Effects of TCP80 and RHA overexpression on p53 IRES activity in ZR75-1 and MDA-MB-175 cells. ZR75-1 (B) and MDA-MB-175 (C) cells were transfected with pR5UTRF alone or pR5UTRF, along with the TCP80 expression vector, the RHA expression vector, or both. At 48 h after transfection, the cells were lysed, and a dual-luciferase assay was performed as previously described. The results presented in panels B and C are averages ± the SEM from three individual experiments (*, P < 0.05).
RESULTS
Translational control protein 80 (TCP80), also known as nuclear factor 90 (NF90) or double-stranded RNA binding protein 76 (DRBP76), is one of the proteins that was found to bind to the p53 5′ UTR in vitro (6). It is a double-stranded-RNA binding protein (24) that has documented roles in the regulation of protein translation (25). TCP80 is also involved in IRES-mediated protein translation by acting as an ITAF of the rhinovirus type 2 IRES (26). In addition, RNA helicase A (RHA) or nuclear DNA helicase II (NDH II), was also identified to bind to the p53 5′ UTR in vitro (6). Interestingly, RHA is known to associate with TCP80 in vitro (27) and has a known role in the regulation of protein translation as well (28). Since these findings suggest that both TCP80 and RHA may modulate the p53 IRES activity by binding to p53 IRES, we tested whether these two proteins can act as p53 ITAFs.
TCP80 and RHA bind to the p53 IRES sequence in vitro.
A previous study determined that the major transcriptional start site of human p53 is 145 nucleotides upstream of the start codon (29). This indicates that the majority of human p53 mRNAs have a 145-nucleotide 5′-UTR sequence. This fragment has been demonstrated to contain the IRES sequence of the p53 5′ UTR using dual-luciferase reporter assays (10, 11), and the IRES sequence has been further confirmed by DNA regional deletions and structural analysis (10, 30). Since the sequence (145 nucleotides) harboring the p53 IRES is shorter than the p53 5′-UTR sequence (192 nucleotides) used in Takagi et al.'s assay to pull down TCP80 and RHA (6), we first tested whether or not TCP80 and RHA can bind specifically to the p53 IRES in vitro (10).
We used an RNA pulldown assay to test the ability of TCP80 and RHA to bind to the p53 IRES sequence in MCF-7 cells. Dynabeads coated with biotinylated p53 IRES RNA were incubated with cytoplasmic extracts of MCF-7 to allow for proteins to bind to the RNA. Our results showed that both TCP80 and RHA were bound to the Dynabeads containing p53 IRES RNA (Fig. 1A). However, beads not coated with biotinylated RNA of the p53 IRES (Fig. 1A) or coated with biotinylated RNA of a firefly luciferase coding region (data not shown) were incapable of binding to either protein. These results indicate that both TCP80 and RHA can specifically bind to the p53 IRES sequence.
FIG 1.

TCP80 and RHA positively regulate the p53 IRES activity. (A) TCP80 and RHA bind to the p53 IRES sequence in vitro. Lanes 1 to 3 show levels of TCP80 and RHA protein in MCF-7 cytoplasmic extracts (in triplicate). Lanes 4 to 6 show the amount of TCP80 and RHA protein pulled down from MCF-7 cytoplasmic extracts by streptavidin-beads coated with biotinylated p53 IRES RNA (in triplicate) as described in Materials and Methods. TCP80 and RHA were detected by Western blotting with their respective antibodies. The last lane represents a sample pulled down with streptavidin-beads that were not coated with biotinylated p53 IRES RNA. Beads coated with biotinylated RNA of a firefly luciferase coding region were tested separately and were incapable of binding to either protein (data not shown). (B) Effect of TCP80 overexpression on p53 and EMCV IRES activity. MCF-7 cells were cotransfected with pRF, pR5UTRF, or pREMCVF, along with either pCDNA3.1 or pCDNA3.1/HisB/TCP80. At 24 h after the transfection, the cells were lysed and a dual-luciferase assay was performed to detect firefly (Fluc) and Renilla (Rluc) luciferase activities as described in Materials and Methods. (C) RHA positively affects the p53 IRES activity. MCF-7 cells were transfected with pRF, pR5UTRF, or pREMCVF, along with either pCDNA3.1 or pCDNA3.1/RHA. At 24 h after transfection, cells were lysed and a dual-luciferase assay was performed as described above. The results presented in panels B and C are averages ± the standard errors of the mean (SEM) from three individual experiments (*, P < 0.05).
TCP80 and RHA upregulate p53 IRES activity in MCF-7 cells.
We then tested whether TCP80 can modulate p53 IRES activity in vivo in cultured MCF-7 cells. The bicistronic dual-luciferase reporter vector pR5UTRF, which contains the p53 IRES sequence, was used to determine p53 IRES activity (10). MCF-7 cells were cotransfected with pR5UTRF and a plasmid expressing TCP80. p53 IRES activity was measured as the ratio of firefly (controlled by the p53 IRES) to Renilla (controlled by cap-dependent translation machinery, used as an internal control) luciferase activity (10). The pREMCVF vector, which contains the IRES sequence of the EMCV virus, and the empty vector (pRF) were used as controls for the pR5UTRF vector. A 2-fold increase in p53 IRES activity was observed in MCF-7 cells overexpressing TCP80 compared to the control cells (Fig. 1B). In contrast, no change was seen in the EMCV IRES activity of MCF-7 cells overexpressing TCP80 (Fig. 1B). This result suggests that TCP80 is a specific positive regulator of p53 IRES activity. Furthermore, we found that overexpression of RHA in MCF-7 cells led to a 1.8-fold increase in p53 IRES activity, whereas it did not affect EMCV IRES activity (Fig. 1C), suggesting that RHA is also a specific positive regulator of the p53 IRES activity. Positive regulation of the p53 IRES activity by RHA has been further demonstrated in MCF-7 cells transfected with a small interfering RNA (siRNA) against RHA. Our results show that these cells not only display reduced expression of RHA but also exhibit a nearly 60% decrease in p53 IRES activity compared to those transfected with a control siRNA (data not shown).
To further confirm the role of TCP80 and RHA in regulation of the p53 IRES activity, we also cotransfected plasmids encoding TCP80 or RHA, along with the pRDNF vector into MCF-7 cells. The pRDNF vector was generated by deleting the first 70 nucleotides of the p53 IRES sequence from the pR5UTRF vector that contains the IRES sequence (∼140 nucleotides) of the p53 mRNA. This regional deletion results in over 98% decrease of the p53 IRES activity (10). Similar to the results in MCF-7 cells cotransfected with plasmids encoding TCP80 or RHA plus the empty vector pRF (Fig. 1B and C), we observed that TCP80 or RHA has no significant effect on Fluc/Rluc activity of the pRDNF vector (data not shown). These results have further confirmed the specificity of both TCP80 and RHA in positively regulating the p53 IRES activity.
Overexpression of TCP80 and RHA leads to increased expression and synthesis of p53.
Next, we examined the effect of overexpression of TCP80 and RHA on p53 expression in H1299 (p53-null) lung carcinoma cells. Transfection of the pC53-SN3 vector, which contains the p53 IRES sequence (∼140 bp) and p53 open reading frame, in H1299 cells resulted in expression of the p53 protein (Fig. 2A). When H1299 cells were cotransfected with the pC53-SN3 vector and a plasmid encoding either TCP80 or RHA, a substantial increase in p53 levels was observed compared to cells cotransfected with pC53-SN3 and the empty vector pCDNA3.1 (Fig. 2A). It is worth noting that the levels of increase in p53 expression in cells transfected with either TCP80 or RHA plasmid were similar to that observed in cells transfected with only pC53-SN3 and treated with etoposide (data not shown). To confirm the role of RHA in the regulation of p53 expression, H1299 cells were transfected with the pC53-SN3 vector along with either siRNA against RHA or control siRNA. We observed a dramatic decrease in p53 expression in H1299 cells transfected with RHA siRNA compared to those transfected with control siRNA (Fig. 2B).
FIG 2.
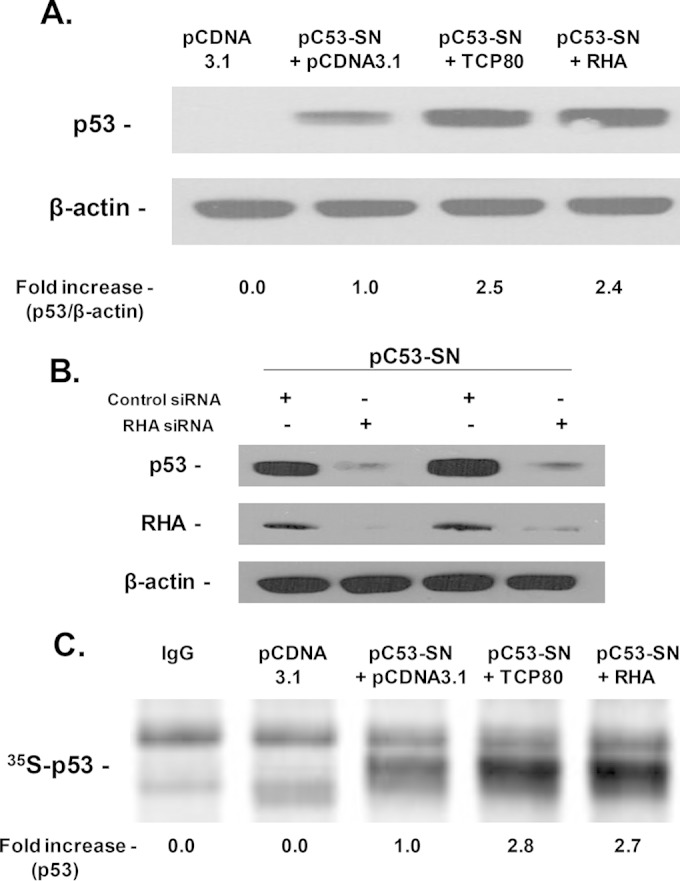
Overexpression of TCP80 and RHA leads to increased p53 expression and synthesis. (A) Overexpression of TCP80 and RHA leads to increased p53 expression in H1299 cells transfected with the pC53-SN3 vector. H1299 lung carcinoma cells (p53-null) were cotransfected with the p53 expression vector pC53-SN3 along with the empty pCDNA 3.1 vector, the TCP80 expression vector, or the RHA expression vector. At 24 h after transfection, the cells were lysed, and equal amounts of protein were subjected to SDS-PAGE and transferred to a nitrocellulose membrane. The p53 protein and β-actin were then detected by their respective antibodies. (B) Effect of RHA knockdown on p53 expression in H1299 cells transfected with the pC53-SN3 vector. H1299 cells were cotransfected with pC53-SN3, along with RHA siRNA or control siRNA. At 72 h after transfection, the cells were lysed, and equal amounts of protein were subjected to SDS-PAGE and later transferred to a nitrocellulose membrane. The p53 protein was detected by immunoblotting with an HRP-conjugated p53 antibody. RHA and β-actin were detected with their respective antibodies as well. (C) Effect of TCP80 and RHA on p53 synthesis in H1299 cells transfected with the pC53-SN3 vector. H1299 cells were transfected with pCDNA 3.1, pC53-SN3, pC53-SN3 plus the TCP80 expression vector, or the RHA expression vector. After transfection, the cells were incubated for 2 h in Cys- and Met-free medium and then metabolically labeled for 1 h with 90 μCi of [35S]Met. Cell lysates were immunoprecipitated with an antibody against p53 as described in Materials and Methods. Newly synthesized 35S-p53 was detected using a Typhoon phosphorimager. The results presented in panels A to C are representative of three individual experiments.
Next, we metabolically labeled H1299 cells that were transfected with pCDNA3.1, pC53-SN3, pC53-SN3 plus the TCP80 expression vector, or pC53-SN3 plus the RHA expression vector. As expected, cells transfected with pcDNA 3.1 did not contain newly synthesized p53 protein. In cells transfected with pC53-SN3, we detected a small amount of newly synthesized p53 following metabolic labeling. However, when H1299 cells were cotransfected with pC53-SN3 along with either TCP80 or RHA, a substantial increase in the amount of newly synthesized p53 protein was observed (Fig. 2C). These results further demonstrate that overexpression of TCP80 and RHA in H1299 cells leads to increased synthesis of de novo p53 protein.
Identification of breast cancer cell lines expressing wild-type p53 but not exhibiting p53 induction following DNA damage.
Although the MCF-7 cell line still maintains its ability to induce p53 following DNA damage, we suspect that many breast cancer cells with wild-type p53 may lose their ability to induce p53 due to defective p53 synthesis (4). To determine whether or not some breast cancer cell lines known to harbor wild-type p53 have defective p53 responses to DNA damage, we tested several such cell lines that have been shown to harbor the wild-type p53 coding region (31, 32), using MCF-7 as their positive control. Interestingly, two of these breast cancer cell lines, ZR75-1 and MDA-MB-175, were identified as having an abrogated p53 induction following treatment with etoposide (Fig. 3A) or short-wavelength UV (Fig. 3B). We also metabolically labeled newly synthesized p53 in these cell lines with [35S]Met after etoposide treatment. A dramatic increase in the levels of de novo-synthesized p53 protein was observed in MCF-7 cells treated with etoposide (Fig. 3C). However, in ZR75-1 cells, there was no significant change in the amount of newly synthesized p53 following etoposide treatment. Similarly, newly synthesized p53 protein was undetectable in MDA-MB-175 cells even after treatment with etoposide (Fig. 3C). In addition, we measured p53 mRNA levels in these cell lines, and the results show that there are no significant changes in the levels of p53 mRNA in cells treated with or without etoposide (Fig. 3D), which further suggests that the lack of p53 induction in ZR75-1 and MDA-MB-175 cells is caused by defective p53 synthesis.
FIG 3.
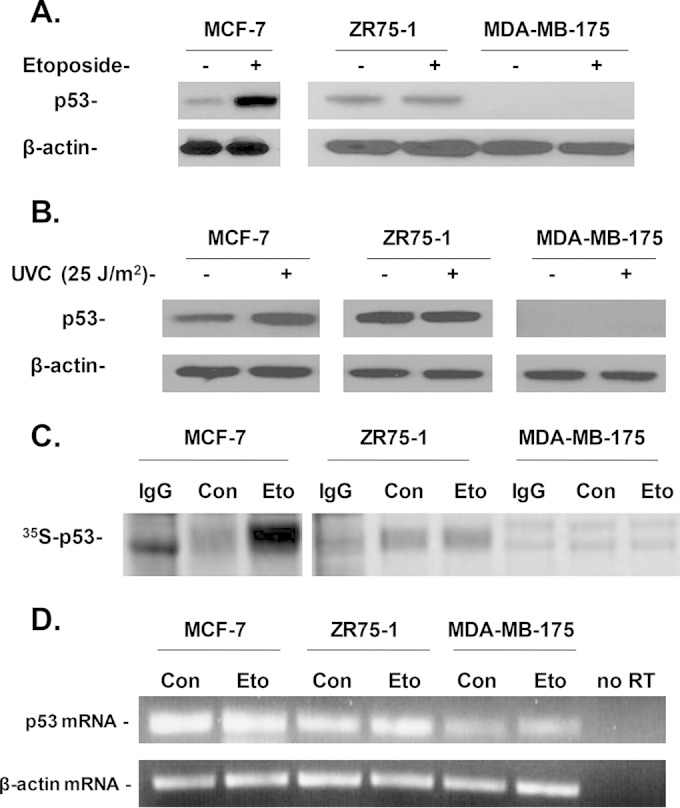
Effect of DNA damage on p53 induction and synthesis in breast cancer cell lines harboring wild-type p53. (A) Effect of etoposide treatment on p53 induction in MCF-7, ZR75-1, and MDA-MB-175 cells. Cells were grown to confluence. Each cell line was then treated with or without 10 μM etoposide for 2 h and lysed with TGN lysis buffer. The p53 protein was then detected by immunoblotting with an HRP-conjugated p53 antibody. The levels of β-actin were also detected by immunoblotting. (B) Effect of UVC irradiation on p53 levels in MCF-7, ZR75-1, and MDA-MB-175 cells. Cells were grown on 60-mm plates. The medium was aspirated, and the cells were washed twice with PBS. The cells were then exposed to 25 J/m2 of short UV light (UVC) irradiation using a UV cross-linker. The cells were then supplemented with medium containing 10% fetal bovine serum and incubated at 37°C for 1 h. After incubation, the cells were lysed. p53 and β-actin levels were detected as described above. (C) Etoposide induces de novo p53 synthesis in MCF-7 cells but not in ZR75-1 and MDA-MB-175 cells. Cells were grown to confluence and then incubated for 2 h in Met- and Cys-free medium. Next, cells were treated with 10 μM etoposide for 30 min. After treatment, 90 μCi of S35-Met was added to the cells for 30 min. The cells were then lysed, and the p53 protein was immunoprecipitated from the cell lysates as described in Materials and Methods. The samples were subjected to SDS-PAGE and transferred onto a nitrocellulose membrane. The newly synthesized 35S-p53 protein was detected using a Typhoon phosphorimager. (D) Effect of etoposide treatment on p53 mRNA levels in MCF-7, ZR75-1, and MDA-MB-175 cells. Subconfluent cells were either left untreated or were treated with 10 μM etoposide for 2 h. Total RNA was then extracted, and an RT-PCR was performed as described in Materials and Methods to detect p53 mRNA levels. The midpoint for linearity of the exponential phase of amplification of the p53 mRNA was determined to be 25 cycles (denaturation at 95°C for 60 s, annealing at 55°C for 30 s, and elongation at 72°C for 60 s). The results presented in panels A to D are representative of three individual experiments.
Lack of p53 induction in ZR75-1 and MDA-MB-175 cells is not caused by increased p53 degradation or decreased p53 half-life.
DNA damage is also known to lead to an increase in p53 half-life through regulation of the MDM2 activity (33), which dissociates from p53 following DNA damage to prevent the degradation of p53. Therefore, we next tested whether the lack of p53 response following DNA damage is caused by a decrease in p53 half-life in ZR75-1 and MDA-MB-175 cell lines. Treatment of MCF-7 cells with cycloheximide, an inhibitor of protein elongation, led to a gradual decrease in p53 levels over the course of 3 h (Fig. 4A). However, a similar treatment did not affect p53 levels in ZR75-1 cells, suggesting a stabilization of the p53 protein in these cells (Fig. 4B). Moreover, p53 half-life was similar in MCF-7 and MDA-MB-175 cells (Fig. 4C). These results indicate that the lack of p53 induction in these two cell lines is not caused by increased p53 degradation or decreased p53 half-life compared to MCF-7 cells.
FIG 4.

Comparison of the p53 degradation rates in MCF-7, ZR75-1, and MDA-MB-175 cells. Confluent MCF-7 (A), ZR75-1(B), and MDA-MB-175 (C) cells were either left untreated or treated with 50 μg of cycloheximide/ml for 30 min, 1 h, 2 h, or 3 h. The cells were collected and lysed at each time point. Cell lysates were then subjected to SDS-PAGE and transferred onto a PVDF membrane. Immunoblotting to detect p53 was performed using a primary antibody against p53 and an HRP-conjugated secondary antibody for enhanced p53 signal. The levels of β-actin were also detected by Western blotting. The results shown (left panels) are representative of three experiments. Calculation of the p53 degradation rates from three individual experiments performed in different cell lines (right panels) was as described in Materials and Methods. The results indicate that the p53 half-lives were 56, 1,003, and 74 min for MCF-7, ZR75-1, and MDA-MB-175 cells, respectively.
ZR75-1 and MDA-MB-175 cells display an abrogated p53 IRES response to DNA damage.
Next, we measured the p53 IRES activity following etoposide-induced DNA damage in ZR75-1 and MDA-MB-175 cells. Both cell lines were transfected with pRF or pR5UTRF vectors and subsequently treated with etoposide. We observed a slight decrease in p53 IRES activity in ZR75-1 cells treated with etoposide compared to control cells (Fig. 5A). In MDA-MB-175 cells, etoposide treatment did not affect p53 IRES activity (Fig. 5B). These results are in contrast to those obtained in MCF-7 cells which show that p53 IRES activity increases in response to DNA damage (10), suggesting that the lack of p53 induction and synthesis in ZR75-1 and MDA-MB-175 cells is the result of the inability of p53 IRES to respond to DNA damage.
FIG 5.

Effect of etoposide treatment on p53 IRES activity in ZR75-1 and MDA-MB-175 cells. ZR75-1 (A) and MDA-MB-175 (B) cells were transfected with either pRF or pR5UTRF as described in Materials and Methods. At 48 h after transfection, the cells were treated with 10 μM etoposide for 2 h or were left untreated. The cells were then lysed with passive lysis buffer, and a dual-luciferase assay was performed as described in Materials and Methods. The results presented in panels A and B are averages ± the SEM from three individual experiments.
TCP80 and RHA levels are extremely low in ZR75-1 and MDA-MB-175 cells and overexpression of both proteins is needed to stimulate p53 IRES activity in these cell lines.
TCP80 and RHA are known to associate with each other in vitro (27). In addition, the expression levels of TCP80 and RHA are also correlated in various cell lines (34). Therefore, we tested whether the defective p53 IRES response to DNA damage is linked to altered expression of TCP80 and RHA in ZR75-1 and MDA-MB-175 cells. Interestingly, immunoblotting results indicate that levels of TCP80 and RHA are greatly reduced in both cell lines compared to MCF-7 cells (Fig. 6A).
In ZR75-1 cells, overexpression of TCP80 alone only led to a slight increase in p53 IRES activity, while overexpression of RHA did not affect p53 IRES activity. However, overexpression of both proteins led to a significant increase (1.7-fold) in p53 IRES activity (Fig. 6B). Similar results were obtained with MDA-MB-175 cells in which overexpression of either TCP80 or RHA alone did not result in any increase of p53 IRES activity, whereas the overexpression of both proteins together led to a significant increase (2.8-fold) in p53 IRES activity (Fig. 6C). These results suggest a cooperative or synergistic effect between these two proteins that causes enhanced p53 IRES activity.
Decreased expression of TCP80 and RHA in MCF-7 cells leads to reduced p53 IRES activity and decreased induction of p53 following DNA damage.
To further determine the role of positive p53 ITAFs, such as TCP80 and RHA, in the regulation of p53 IRES activity and p53 induction, TCP80 expression in MCF-7 cells was knocked down using a plasmid harboring an shRNA against TCP80 (Fig. 7A). Since the expression levels of TCP80 and RHA are known to be correlated (34), we tested whether a decrease in TCP80 expression would also result in reduced levels of RHA. Our results showed that this is indeed the case, as the MCF-7/shTCP80 cell line has decreased expression of both TCP80 and RHA (Fig. 7A). Moreover, in these cells (MCF-7/shTCP80), we observed a marked decrease in p53 IRES activity compared to the controls cells (MCF-7/pCDNA3.1) stably transfected with an empty plasmid (Fig. 7B). Subsequently, we found that the p53 expression is also reduced in MCF-7/shTCP80 cells. More importantly, our results indicate that following treatment with etoposide, p53 induction is dramatically decreased in MCF-7/shTCP80 cells compared to MCF-7/pCDNA3.1 cells (Fig. 7C). Furthermore, p53 mRNA levels were measured in both cell lines. The results show that there are no significant changes in the levels of p53 mRNA between these two cell lines (Fig. 7D), suggesting that the decreased expression and induction of p53 in MCF-7/shTCP80 cells are not caused by reduced transcription of p53 mRNA. The combined effect of TCP80 and RHA on p53 expression has been further confirmed by our experiments showing that in H1299 cells cotransfected with pC53-SN3 along with plasmids encoding TCP80, RHA, or both, the overexpression of both TCP80 and RHA leads to a markedly higher expression of p53 in H1299 cells than those transfected with only TCP80 or RHA (data not shown).
FIG 7.

Knockdown of TCP80 in MCF-7 cells leads to reduced expression of RHA and defective p53 induction following DNA damage. (A) MCF-7/shTCP80 cells have reduced expression of both TCP80 and RHA. Cells were grown to subconfluence. The cells were then lysed, and equal amounts of protein were subjected to SDS-PAGE and Western blotting. TCP80, RHA, and β-actin were detected by immunoblotting. (B) Effect of reduced expression of TCP80 and RHA on p53 IRES activity in MCF-7 cells. MCF-7/pCDNA3.1 and MCF-7/shTCP80 cells were plated on six-well plates and grown to subconfluence. The cells were then transfected with pR5UTRF vector. At 24 h after transfection, the cells were lysed with the passive lysis buffer, and the firefly and Renilla luciferase activities were measured by performing a dual-luciferase assay. The results presented are averages ± the SEM from three individual experiments (*, P < 0.05). (C) MCF-7/shTCP80 cells exhibit diminished ability to induce p53 following DNA damage. MCF-7/pCDNA3.1 and MCF-7/shTCP80 cells were grown to subconfluence. The cells were then treated with 10 μM etoposide for 2 or 3 h. After the treatment, the cells were lysed, and equal amounts of protein were subjected to SDS-PAGE and transferred to PVDF membranes. p53 and β-actin proteins were detected with their respective antibodies. (D) Comparison of p53 mRNA levels in MCF-7/pCDNA3.1 and MCF-7/shTCP80 cells. MCF-7/pCDNA3.1 and MCF-7/shTCP80 cells were grown to subconfluence. Total RNA was then extracted, and an RT-PCR was performed as described in Materials and Methods to detect p53 mRNA levels. The midpoint for linearity of the exponential phase of amplification of the p53 mRNA was determined to be 25 cycles (denaturation at 95°C for 60 s, annealing at 55°C for 30 s, and elongation at 72°C for 60 s). The results presented in panels C and D are representative of three individual experiments.
MCF-7/shTCP80 cells exhibit defective induction of p21Cip1 and diminished ability to induce senescence following DNA damage.
As a key regulator of the cellular senescence process, p53 can initiate cellular senescence in response to DNA damage by upregulating its downstream target p21Cip1 (35). After treatment with etoposide, we found that MCF-7/shTCP80 cells exhibit much lower levels of p21Cip1 induction than those in MCF-7/pCDNA3.1 cells (Fig. 8A). Consistent with decreased p21Cip1 induction, MCF-7/shTCP80 cells exhibit diminished induction of cellular senescence following etoposide treatment compared to MCF-7/pCDNA3.1 cells (Fig. 8B and C). Since G1 cell cycle arrest usually precedes DNA damage-induced senescence (36), we performed a cell cycle distribution analysis after treating MCF-7/shTCP80 and MCF-7/pCDNA3.1 cells with etoposide. Although both cell lines display no significant differences in cell cycle distribution under normal growth conditions (data not shown), incubation of MCF-7/pCDNA3.1 cells with etoposide results in a substantial increase in cell numbers at the G1 phase and a dramatic decrease at the S phase (Fig. 8D). Compared to MCF-7/shTCP80 cells, a nearly 2-fold increase in the G1/S ratio, an indicator of G1 cell cycle arrest (37), was observed in MCF-7/pCDNA3.1 cells treated with etoposide (Fig. 8E).
FIG 8.

MCF-7/shTCP80 cells exhibit diminished ability to induce G1 cell cycle arrest and senescence following DNA damage compared to MCF-7/pCDNA3.1 cells. (A) p21Cip1 induction following DNA damage is decreased in MCF-7/shTCP80 cells. Subconfluent MCF-7/pCDNA3.1 and MCF-7/shTCP80 cells were treated with 10 μM etoposide for 4 or 6 h. After treatment, the cells were lysed, and equal amounts of protein were subjected to SDS-PAGE and Western blotting. p21Cip1 and β-actin proteins were detected with their respective antibodies. The results presented are representative of three separate experiments. (B and C) MCF-7/shTCP80 cells exhibit diminished ability to induce senescence following etoposide treatment. Subconfluent MCF-7/pCDNA3.1 and MCF-7/shTCP80 cells were treated with 5 or 10 μM etoposide for 4 days. After treatment, the medium was removed, and the cells were washed with PBS. The cells were then stained for SA-β-galactosidase using a kit from Cell Signaling as described in Materials and Methods, and pictures were taken with a microscope (Leica DM IRB) at ×40 magnification. The number of SA-β-Gal-positive cells versus total cells per field in three independent fields of view was counted. The results in panel C represent the means ± the SEM of three independent measurements (**, P < 0.01). (D and E) Etoposide causes stronger G1 cell cycle arrest in MCF-7/pCDNA3.1 cells. Subconfluent MCF-7/pCDNA3.1 and MCF-7/shTCP80 cells were treated with 10 μM etoposide for 24 h. After treatment, cells were collected and subjected to flow cytometry analysis after being fixed with ethanol and stained with propidium iodide. The cell population at each phase was analyzed by the Modfit 2 software. The G1/S ratio presented in panel E represents the average ± the SEM from three individual experiments (*, P < 0.05).
DISCUSSION
In this study, we identified two novel ITAFs of p53, TCP80 and RHA, which positively regulate p53 IRES activity and p53 synthesis. We have also identified two breast cancer cell lines, MDA-MB-175 and ZR-75-1, that express wild-type p53 protein but exhibit diminished p53 induction and synthesis following DNA damage. Our results have further shown that the defective p53 induction and synthesis is likely caused by reduced expression of TCP80 and RHA in these cancer cells. Moreover, we found that MCF-7 breast cancer cells with decreased expression of TCP80 and RHA show defective induction of p53 and diminished ability to induce senescence following DNA damage. Our findings thus not only have provided a better understanding of the mechanism that regulates IRES-mediated p53 translation in response to genotoxic stress but also have revealed a novel mechanism by which defective IRES-mediated p53 translation in response to DNA damage leads to tumorigenesis.
TCP80 contains multiple RNA binding domains (24). It is known to regulate the translation of the acid beta-glucosidase mRNA by binding to its coding sequence (25). The involvement of TCP80 in cellular IRES-mediated protein translation is further supported by a previous report indicating that TCP80 is an ITAF of the rhinovirus type 2 IRES (26). RHA plays a crucial role in the translation of some viral and cellular mRNAs that contain a posttranscriptional control element (PCE) within their 5′ UTR by disrupting the secondary structure of PCE, thus allowing for a more efficient scanning of the 40S ribosomal subunit and translation initiation (28). Since the region containing the p53 IRES is known to have a strong secondary structure (6, 30), TCP80 and RHA could both exert a positive effect on the p53 IRES by aiding in the unwinding of its secondary structure.
In addition to the individual effect of TCP80 and RHA on the p53 IRES activity, the interaction between TCP80 and RHA may also be important for the stimulation of p53 IRES activity. Consistent with previous results, we found that these two proteins associate with each other in MCF-7 cells (data not shown). Our results also indicate that knockdown of TCP80 in MCF-7 cells results in decreased expression of RHA. These observations corroborate our findings that levels of both TCP80 and RHA are low in ZR75-1 and MDA-MB-175 cells. Although ZR75-1 and MDA-MB-175 show diminished p53 induction and defective activation of p53 IRES after DNA damage, expression of both TCP80 and RHA are required to significantly increase the p53 IRES activity in these cells. It is conceivable that TCP80 and RHA could interact to facilitate each other's ability to remodel or unwind the secondary structure of p53 IRES, thereby allowing more efficient translation of the p53 mRNA. The detailed mechanism regarding how TCP80 and RHA interact to enhance p53 IRES activity requires further investigation.
We have identified two breast cancer cell lines, ZR75-1 and MDA-MB-175, that exhibit defective induction of p53 in response to DNA damage, even though they harbor wild-type p53. These results imply that tumorigenic transformation of these cells could be caused by the inability of their p53 IRES to respond to DNA damage. Our results are the first to show that p53 synthesis in response to etoposide is abrogated in these two cancer cell lines. We further discovered that these cells display an abrogated p53 IRES response to DNA damage, suggesting that the IRES-mediated p53 translation is defective in these two cell lines. These findings thus provide strong support for our hypothesis (4) that inefficient or defective IRES-mediated p53 translation in response to DNA damage is linked to tumorigenesis.
Previous studies have shown that IRES/ITAF interactions could be important regulators of tumorigenesis. For example, a single C-U transition in the proto-oncogene c-myc IRES sequence leads to enhanced binding of its positive ITAF hnRNPK and increased c-myc translation, which explains increased c-myc levels in patients with multiple myelomas (8). Another study found that increased binding of a positive ITAF to the insulin-like growth factor 1 receptor (IGF-IR) IRES sequence in breast cancer cells is postulated to lead to IGF-IR overexpression and stimulate tumorigenic transformation (38). Our results indicate that p53 IRES activity cannot be further enhanced by DNA damage in MDA-MB-175 or ZR75-1 cells. To determine whether the defect in p53 induction was caused by mutations in the p53 IRES, we sequenced the p53 mRNA in these two cell lines. Sequencing results revealed that these two cell lines harbor wild-type p53 5′ UTR, as well as the wild-type p53 coding sequence (data not shown). Since mutations were not found within the p53 IRES, defects in p53 IRES response following DNA damage are most likely caused by alterations of p53 ITAFs in expression, function, or localization in these two cancer cell lines.
Indeed, we found that expression of TCP80 and RHA, two positive ITAFs of p53, is very low in ZR75-1 and MDA-MB-175 cells compared to MCF-7 cells that exhibit normal p53 response to DNA damage. It was found that RHA maps to chromosome band 1q25, which is the site of a major prostate cancer susceptibility locus (39). Prostate cancer, similar to breast cancer, also has a low mutation rate (7 to 18%) of p53 (40). In line with RHA's role in stimulating p53 synthesis, RHA also upregulates activity of several other tumor suppressors, such as Werner syndrome helicase (WRN), that are involved in the DNA repair process through interaction with these proteins in the DNA damage foci (41, 42). Therefore, it is possible that alteration or deletion of this locus results in abrogated RHA function or expression and prevent induction of the p53 IRES and other tumor suppressors, thereby increasing the risk of tumorigenic transformation. Similarly, the expression of TCP80 is known to be greatly reduced in malignant brain tumors of glial origin, and the subcellular localization of TCP80 is altered in these malignant tumors as well (43). These results suggest abnormal expression or subcellular localization of TCP80 is linked to malignant transformation of cells in addition to breast tumor cells.
Our results show that knockdown of TCP80 in MCF-7 cells results in decreased expression of RHA, which is consistent with previous findings showing that expression levels of TCP80 and RHA are correlated in various cell lines (34). We found that when expression of TCP80 and RHA is reduced, MCF-7 cells exhibit a marked decrease in the induction of p53 and its downstream target p21Cip1 following DNA damage, which results in its diminished ability to induce cellular senescence. Furthermore, we found that MCF-7/shTCP80 cells show decreased induction of the p53 upregulated modulator of apoptosis (PUMA) (44, 45), a proapoptotic protein whose expression is stimulated by p53 (data not shown). Since both cellular senescence and apoptosis are important events for preventing the development of malignant tumors in vivo (46), these findings have not only linked reduced expression of positive ITAFs of the p53 IRES with tumorigenesis but also provided an explanation as to why both ZR75-1 and MDA-MB-175 develop into malignant cancer cells despite retaining wild-type p53.
To date, very limited studies have been done to investigate the role of p53 translation in the prevention of tumorigenesis. An earlier report shows that the lack of p53 synthesis is responsible for the abrogation of p53 accumulation following DNA damage and the development of malignant peripheral nerve sheath tumors in zebrafish (47). A recent study has observed defective IRES-mediated p53 synthesis in X-linked dyskeratosis congenita, a tumor susceptible syndrome, in response to DNA damage and OIS (17). Our present study has further linked reduced expression of positive ITAFs of the p53 IRES with tumorigenesis in tumor cells still expressing wild-type p53, thus uncovering a novel mechanism of p53 inactivation that makes normal cells susceptible to cancer development.
Recently, multiple new ITAFs of the p53 IRES have been identified and have been shown to affect the translation of p53 and its isoform p47 under different stressful conditions (15, 18–20). However, it is not known how altered expression/localization and/or function of these ITAFs link to defective p53 function and cancer development. In contrast, our results, for the first time, provide a mechanistic link between altered expression of positive p53 ITAFs and defective p53 induction, as well as diminished tumor suppressive function of p53 in cancer cells.
In addition to the discovery of many proteins that regulate p53 translation through binding to the p53 IRES in the 5′ UTR, it was reported that the 3′ UTR of p53 mRNA also plays an important role in regulating p53 translation in response to DNA damage (48). Recently, an interaction between the 5′ UTR (harboring the p53 IRES sequence) and 3′ UTR of the p53 mRNA has been uncovered (49), and it was shown that proteins bound to the p53 3′ UTR also modulate p53 synthesis following DNA damage (50, 51). Therefore, the novel insights as to how dysfunctional IRES-driven translation of p53 mRNA leads to tumorigenesis presented in the present study may open the door for further investigation of the roles of various regulatory proteins of p53 synthesis in the pathogenesis of cancer.
ACKNOWLEDGMENTS
We thank Vitaly Polunovsky and Peter Bitterman (University of Minnesota) for helpful comments on the results presented in the manuscript. We also thank Suisheng Zhang (Institute of Molecular Biotechnology, Germany) and Michael B. Matthews (University of Medicine and Dentistry of New Jersey) for providing the pcDNA3.1/RHA and pcDNA3.1/HisB/TCP80 expression vectors, respectively.
Funding for this research was provided by an Idea Award (BC051719) from the Department of Defense, three research grants (1RO1 CA084325, 1RO3 ES017869, and 1RO3 CA177954) from the National Institutes of Health, an Institutional Research Grant (118198-IRG-58-001-52-IRG99) from American Cancer Society, and the Hormel Foundation.
REFERENCES
- 1.Giaccia AJ, Kastan MB. 1998. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev 12:2973–2983. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- 2.Vogelstein B, Lane D, Levine AJ. 2000. Surfing the p53 network. Nature 408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 3.Horn HF, Vousden KH. 2007. Coping with stress: multiple ways to activate p53. Oncogene 26:1306–1316. doi: 10.1038/sj.onc.1210263. [DOI] [PubMed] [Google Scholar]
- 4.Halaby MJ, Yang DQ. 2007. p53 translational control: a new facet of p53 regulation and its implication for tumorigenesis and cancer therapeutics. Gene 395:1–7. doi: 10.1016/j.gene.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 5.Mosner J, Mummenbrauer T, Bauer C, Sczakiel G, Grosse F, Deppert W. 1995. Negative feedback regulation of wild-type p53 biosynthesis. EMBO J 14:4442–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takagi M, Absalon MJ, McLure KG, Kastan MB. 2005. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 123:49–63. doi: 10.1016/j.cell.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 7.Gingras AC, Raught B, Sonenberg N. 1999. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem 68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 8.Stoneley M, Willis AE. 2004. Cellular internal ribosome entry segments: structures, trans-acting factors and regulation of gene expression. Oncogene 23:3200–3207. doi: 10.1038/sj.onc.1207551. [DOI] [PubMed] [Google Scholar]
- 9.Holcik M, Sonenberg N. 2005. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- 10.Yang DQ, Halaby MJ, Zhang Y. 2006. The identification of an internal ribosomal entry site in the 5′-untranslated region of p53 mRNA provides a novel mechanism for the regulation of its translation following DNA damage. Oncogene 25:4613–4619. doi: 10.1038/sj.onc.1209483. [DOI] [PubMed] [Google Scholar]
- 11.Ray PS, Grover R, Das S. 2006. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep 7:404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Candeias MM, Powell DJ, Roubalova E, Apcher S, Bourougaa K, Vojtesek B, Bruzzoni-Giovanelli H, Fahraeus R. 2006. Expression of p53 and p53/47 are controlled by alternative mechanisms of messenger RNA translation initiation. Oncogene 25:6936–6947. doi: 10.1038/sj.onc.1209996. [DOI] [PubMed] [Google Scholar]
- 13.Sayan AE, Roperch JP, Sayan BS, Rossi M, Pinkoski MJ, Knight RA, Willis AE, Melino G. 2007. Generation of DeltaTAp73 proteins by translation from a putative internal ribosome entry site. Ann N Y Acad Sci 1095:315–324. doi: 10.1196/annals.1397.035. [DOI] [PubMed] [Google Scholar]
- 14.Kim DY, Kim W, Lee KH, Kim SH, Lee HR, Kim HJ, Jung Y, Choi JH, Kim KT. 2013. hnRNP Q regulates translation of p53 in normal and stress conditions. Cell Death Differ 20:226–234. doi: 10.1038/cdd.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grover R, Ray PS, Das S. 2008. Polypyrimidine tract binding protein regulates IRES-mediated translation of p53 isoforms. Cell Cycle 7:2189–2198. doi: 10.4161/cc.7.14.6271. [DOI] [PubMed] [Google Scholar]
- 16.Montanaro L, Calienni M, Bertoni S, Rocchi L, Sansone P, Storci G, Santini D, Ceccarelli C, Taffurelli M, Carnicelli D, Brigotti M, Bonafe M, Trere D, Derenzini M. 2010. Novel dyskerin-mediated mechanism of p53 inactivation through defective mRNA translation. Cancer Res 70:4767–4777. doi: 10.1158/1538-7445.AM10-4767. [DOI] [PubMed] [Google Scholar]
- 17.Bellodi C, Kopmar N, Ruggero D. 2010. Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. EMBO J 29:1865–1876. doi: 10.1038/emboj.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharathchandra A, Lal R, Khan D, Das S. 2012. Annexin A2 and PSF proteins interact with p53 IRES and regulate translation of p53 mRNA. RNA Biol 9:1429–1439. doi: 10.4161/rna.22707. [DOI] [PubMed] [Google Scholar]
- 19.Weingarten-Gabbay S, Khan D, Liberman N, Yoffe Y, Bialik S, Das S, Oren M, Kimchi A. 2013. The translation initiation factor DAP5 promotes IRES-driven translation of p53 mRNA. Oncogene 33:611–618. [DOI] [PubMed] [Google Scholar]
- 20.Khan D, Katoch A, Das A, Sharathchandra A, Lal R, Roy P, Das S, Chattopadhyay S, Das S. 2015. Reversible induction of translational isoforms of p53 in glucose deprivation. Cell Death Differ 33:611–618. doi: 10.1038/onc.2012.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hellen CU, Sarnow P. 2001. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev 15:1593–1612. doi: 10.1101/gad.891101. [DOI] [PubMed] [Google Scholar]
- 22.Jackson RJ. 2005. Alternative mechanisms of initiating translation of mammalian mRNAs. Biochem Soc Trans 33:1231–1241. doi: 10.1042/BST20051231. [DOI] [PubMed] [Google Scholar]
- 23.Gasco M, Shami S, Crook T. 2002. The p53 pathway in breast cancer. Breast Cancer Res 4:70–76. doi: 10.1186/bcr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fierro-Monti I, Mathews MB. 2000. Proteins binding to duplexed RNA: one motif, multiple functions. Trends Biochem Sci 25:241–246. doi: 10.1016/S0968-0004(00)01580-2. [DOI] [PubMed] [Google Scholar]
- 25.Xu YH, Grabowski GA. 1999. Molecular cloning and characterization of a translational inhibitory protein that binds to coding sequences of the human acid beta-glucosidase and other mRNAs. Mol Genet Metab 68:441–454. doi: 10.1006/mgme.1999.2934. [DOI] [PubMed] [Google Scholar]
- 26.Merrill MK, Gromeier M. 2006. The double-stranded RNA binding protein 76:NF45 heterodimer inhibits translation initiation at the rhinovirus type 2 internal ribosome entry site. J Virol 80:6936–6942. doi: 10.1128/JVI.00243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reichman TW, Parrott AM, Fierro-Monti I, Caron DJ, Kao PN, Lee CG, Li H, Mathews MB. 2003. Selective regulation of gene expression by nuclear factor 110, a member of the NF90 family of double-stranded RNA-binding proteins. J Mol Biol 332:85–98. doi: 10.1016/S0022-2836(03)00885-4. [DOI] [PubMed] [Google Scholar]
- 28.Hartman TR, Qian S, Bolinger C, Fernandez S, Schoenberg DR, Boris-Lawrie K. 2006. RNA helicase A is necessary for translation of selected messenger RNAs. Nat Struct Mol Biol 13:509–516. doi: 10.1038/nsmb1092. [DOI] [PubMed] [Google Scholar]
- 29.Tuck SP, Crawford L. 1989. Characterization of the human p53 gene promoter. Mol Cell Biol 9:2163–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grover R, Sharathchandra A, Ponnuswamy A, Khan D, Das S. 2011. Effect of mutations on the p53 IRES RNA structure: implications for deregulation of the synthesis of p53 isoforms. RNA Biol 8:132–142. doi: 10.4161/rna.8.1.14260. [DOI] [PubMed] [Google Scholar]
- 31.Concin N, Zeillinger C, Tong D, Stimpfl M, Konig M, Printz D, Stonek F, Schneeberger C, Hefler L, Kainz C, Leodolter S, Haas OA, Zeillinger R. 2003. Comparison of p53 mutational status with mRNA and protein expression in a panel of 24 human breast carcinoma cell lines. Breast Cancer Res Treat 79:37–46. doi: 10.1023/A:1023351717408. [DOI] [PubMed] [Google Scholar]
- 32.Wasielewski M, Elstrodt F, Klijn JG, Berns EM, Schutte M. 2006. Thirteen new p53 gene mutants identified among 41 human breast cancer cell lines. Breast Cancer Res Treat 99:97–101. doi: 10.1007/s10549-006-9186-z. [DOI] [PubMed] [Google Scholar]
- 33.Vousden KH, Prives C. 2005. P53 and prognosis: new insights and further complexity. Cell 120:7–10. doi: 10.1016/S0092-8674(04)01252-8. [DOI] [PubMed] [Google Scholar]
- 34.Parker LM, Fierro-Monti I, Mathews MB. 2001. Nuclear factor 90 is a substrate and regulator of the eukaryotic initiation factor 2 kinase double-stranded RNA-activated protein kinase. J Biol Chem 276:32522–32530. doi: 10.1074/jbc.M104408200. [DOI] [PubMed] [Google Scholar]
- 35.Itahana K, Dimri G, Campisi J. 2001. Regulation of cellular senescence by p53. Eur J Biochem 268:2784–2791. doi: 10.1046/j.1432-1327.2001.02228.x. [DOI] [PubMed] [Google Scholar]
- 36.Schmitt E, Paquet C, Beauchemin M, Bertrand R. 2007. DNA-damage response network at the crossroads of cell-cycle checkpoints, cellular senescence and apoptosis. J Zhejiang Univ Sci B 8:377–397. doi: 10.1631/jzus.2007.B0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Yang DQ. 2010. The ATM inhibitor KU-55933 suppresses cell proliferation and induces apoptosis by blocking Akt in cancer cells with overactivated Akt. Mol Cancer Ther 9:113–125. doi: 10.1158/1535-7163.MCT-08-1189. [DOI] [PubMed] [Google Scholar]
- 38.Meng Z, Jackson NL, Choi H, King PH, Emanuel PD, Blume SW. 2008. Alterations in RNA-binding activities of IRES-regulatory proteins as a mechanism for physiological variability and pathological dysregulation of IGF-IR translational control in human breast tumor cells. J Cell Physiol 217:172–183. doi: 10.1002/jcp.21486. [DOI] [PubMed] [Google Scholar]
- 39.Lee CG, Eki T, Okumura K, Nogami M, Soares Vda C, Murakami Y, Hanaoka F, Hurwitz J. 1999. The human RNA helicase A (DDX9) gene maps to the prostate cancer susceptibility locus at chromosome band 1q25 and its pseudogene (DDX9P) to 13q22, respectively. Somat Cell Mol Genet 25:33–39. doi: 10.1023/B:SCAM.0000007138.44216.3a. [DOI] [PubMed] [Google Scholar]
- 40.MacGrogan D, Bookstein R. 1997. Tumour suppressor genes in prostate cancer. Semin Cancer Biol 8:11–19. doi: 10.1006/scbi.1997.0048. [DOI] [PubMed] [Google Scholar]
- 41.Zhang S, Hemmerich P, Grosse F. 2007. Werner syndrome helicase (WRN), nuclear DNA helicase II (NDH II) and histone gammaH2AX are localized to the centrosome. Cell Biol Int 31:1109–1121. doi: 10.1016/j.cellbi.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 42.Zhang S, Schlott B, Gorlach M, Grosse F. 2004. DNA-dependent protein kinase (DNA-PK) phosphorylates nuclear DNA helicase II/RNA helicase A and hnRNP proteins in an RNA-dependent manner. Nucleic Acids Res 32:1–10. doi: 10.1093/nar/gkg933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neplioueva V, Dobrikova EY, Mukherjee N, Keene JD, Gromeier M. 2010. Tissue type-specific expression of the dsRNA-binding protein 76 and genome-wide elucidation of its target mRNAs. PLoS One 5:e11710. doi: 10.1371/journal.pone.0011710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL, Zambetti GP. 2003. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4:321–328. doi: 10.1016/S1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 45.Garrison SP, Phillips DC, Jeffers JR, Chipuk JE, Parsons MJ, Rehg JE, Opferman JT, Green DR, Zambetti GP. 2012. Genetically defining the mechanism of Puma- and Bim-induced apoptosis. Cell Death Differ 19:642–649. doi: 10.1038/cdd.2011.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. 2010. The essence of senescence. Genes Dev 24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MacInnes AW, Amsterdam A, Whittaker CA, Hopkins N, Lees JA. 2008. Loss of p53 synthesis in zebrafish tumors with ribosomal protein gene mutations. Proc Natl Acad Sci U S A 105:10408–10413. doi: 10.1073/pnas.0805036105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu L, Ma W, Benchimol S. 1999. A translation repressor element resides in the 3′ untranslated region of human p53 mRNA. Oncogene 18:6419–6424. doi: 10.1038/sj.onc.1203064. [DOI] [PubMed] [Google Scholar]
- 49.Chen J, Kastan MB. 2010. 5′-3′-UTR interactions regulate p53 mRNA translation and provide a target for modulating p53 induction after DNA damage. Genes Dev 24:2146–2156. doi: 10.1101/gad.1968910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mazan-Mamczarz K, Galban S, Lopez de Silanes I, Martindale JL, Atasoy U, Keene JD, Gorospe M. 2003. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci U S A 100:8354–8359. doi: 10.1073/pnas.1432104100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abdelmohsen K, Panda AC, Kang MJ, Guo R, Kim J, Grammatikakis I, Yoon JH, Dudekula DB, Noh JH, Yang X, Martindale JL, Gorospe M. 2014. 7SL RNA represses p53 translation by competing with HuR. Nucleic Acids Res 42:10099–10111. doi: 10.1093/nar/gku686. [DOI] [PMC free article] [PubMed] [Google Scholar]