

Abstract
We are beginning to understand the molecular biology of hydrocephalus and its related diseases. X-linked hydrocephalus (XLH), holoprosencephaly (HPE), Dandy–Walker malformation (DWM), and neural tube defect (NTD) can all be discussed with respect to their available molecular genetics knowledge base and its clinical applications. XLH is single gene disorder caused by mutations in the neural cell adhesion molecule-encoding L1CAM (L1) gene. Our knowledge of the molecular basis of XLH is already being applied clinically in disease diagnosis, disease classification, and prenatal diagnosis. However, the molecular mechanism underlying XLH-related hydrocephalus still needs to be clarified. Sixteen causative genes for HPE have been identified, of which mutations are most often found in SHH, ZIC2, SIX3, and TGIF. Genetic interactions, gene complexity, and the wide variety of HPE phenotypes and genotypes are topics for future study. For DWM, two important loci, 3q24, which includes the FOXC1 gene, and 6q25.3, which includes the ZIC1 and ZIC4 genes, were recently identified as causative areas. The planar cell polarity (PCP) genes CELSR1, CELSR2, VANGL1, and VANGL2 have been implicated in NTD; these genes have roles in neural tube closure and ependymal ciliary movement.
Keywords: molecular biology, X-linked hydrocephalus, holoprosencephaly, Dandy–Walker syndrome, neural tube defect
Introduction
We live in a period where molecular biology is being widely and effectively applied in every field of medicine. Technical advances have led to the development of new molecular mechanism-based target drugs for several diseases. In the field of pediatric neurosurgery, the causative genes for some diseases have been identified, and this information has enabled genetic diagnosis and improved disease classifications.
Diseases treated by pediatric neurosurgeons, including the various forms of hydrocephalus, can be classified according to the available molecular genetics knowledge base, and its clinical applications, as follows. In the first group, genetic testing has been clinically established and is already used in disease diagnosis, disease classification, carrier detection, and prenatal diagnosis. In the second group, the molecular cause has not been firmly established and clinical tools are not yet available, but some causative genes have been identified, and/or there are recent molecular findings regarding the pathophysiology and classification. In the third group, the molecular basis of the disease is still uncertain, and the clinical significance of current research findings has not been established.
Of hydrocephalus and its related diseases, X-linked hydrocephalus (XLH) is categorized in the first group, holoprosencephaly (HPE) and porencephaly in the second, and Dandy–Walker syndrome and myelomeningocele in the third.
XLH
XLH was first described by Bickers and Adams in 1949, as HSAS, the acronym for Hydrocephalus due to Stenosis of the Aqueduct of Sylvius (MIM 307000).1) Since the first family of XLH carriers with a gene mutation in neural cell adhesion molecule L1CAM (L1) was reported in 1992,2) there have been many advances in the genetics of XLH. L1 is a member of the immunoglobulin (Ig) superfamily of cell adhesion molecules, and is expressed predominantly in developing neurons. Mutations in the L1 gene were found to be responsible for many cases of XLH, mental retardation, adducted thumbs, shuffling gait and aphasia (MASA) syndrome, certain forms of X-linked spastic paraplegia (SPG1), and X-linked agenesis of the corpus callosum (ACC). Therefore, these syndromes have been reclassified and grouped together as L1 syndrome.3)
The L1 gene is located on the X chromosome in humans, and is composed of 28 exons. The open reading frame has 3,825 base pairs (bps), and encodes a protein of 1,257 amino acids. According to an updated (May, 2014) database of L1 gene mutations (web site maintained by Yvonne Vos from the Department of Genetics, University Medical Center Groningen, Groningen), 211 mutations have been found in 254 unrelated families with L1 syndrome.4–6) In Japan, Kanemura et al. conducted a nation-wide investigation of L1 gene mutations and identified L1 mutations in 90 unrelated families.7)
The sites and types of the LI gene mutations in families with XLH are almost always different, regardless of race. Genotype and phenotype correlations have been reported.8,9) Yamasaki et al. revealed a striking correlation between the mutation class and the severity of ventricular dilatation. Class I mutations affect only the cytoplasmic domain (CD) of L1. Class II mutations consist of missense point mutations and deletions in the extracellular domain (ED) that result in a predicted protein that should remain associated with the plasma membrane. Class III mutations include nonsense or frame-shift mutations that produce a premature stop codon in the L1ED. The mutant molecules in this class do not remain associated with the cell membrane and therefore lose all the normal functions of L1. Mutations in the non-coding region are divided into splice-site mutations and others. Splice-site mutations result in the same L1 protein structure as Class III mutations and cause loss of function.10)
Class II mutations can be divided into two subclasses based on molecular modeling studies. One subgroup includes mutations affecting the key residues in L1ED that are responsible for maintaining the conformation of the domains, and the other subgroup includes mutations that affect residues on the protein’s surface.11) Patients whose ventricles showed severe dilatation had Class III mutations, exon 1–26 splice-site mutations, or Class II L1ED key-residue mutations. All of these mutations cause the loss of L1ED function, and are therefore referred to as L1-LF mutations.
All of the patients with L1-LF mutations had severe ventricular dilatation (Fig. 1a), required a ventriculoperitoneal (VP) shunt in the early days of life; however, most of these patients show severe developmental delays, including lack of independent locomotion and an undetectably low intelligence quotient (IQ). To look at the characteristics of radiological findings, they showed a rippled ventricular wall after shunting (Fig. 1b), which is not seen in other kinds of hydrocephalus, and is an absolute characteristic of the severe hydrocephalus that accompanies L1-LF mutations. This unique neuro-radiological finding can be used as one of the clinical diagnostic criteria of XLH with an L1-LF mutation.
Fig. 1.

X-linked hydrocephalus with L1 gene mutation. a: MRI (T1) shows severe ventricular dilatation. b: MRI (T2) after VP shunt shows rippled ventricular wall. c: MRI (T2) shows an enlarged massa intermedia ( ) anterior vermis hypoplasia (←), and a large quadrigeminal plate (⇒), callosal dysplasia was observed (
) anterior vermis hypoplasia (←), and a large quadrigeminal plate (⇒), callosal dysplasia was observed ( ). MRI: magnetic resonance imaging, VP: ventriculoperitoneal.
). MRI: magnetic resonance imaging, VP: ventriculoperitoneal.
Notably, most of the patients with L1-LF mutations also had an enlarged massa intermedia, enlarged quadrigeminal plate, and hypoplasia of the cerebellar vermis (Fig. 1c). These results are consistent with reported pathological findings of XLH, which include an enlarged massa intermedia or fused thalami (Fig. 1c). Hypoplasia of the cerebellar vermis has also been reported in XLH patients12) and observed in L1-knockout mice.13)
There are two possible explanations for the ventricular dilatation caused by L1 mutations. First, a decrease in elasticity in the white matter might increase the vulnerability of the ventricular system to alteration by cerebrospinal fluid (CSF) pressure. Second, maldevelopment of the midline structure might cause narrowing of the CSF pathway. Both mechanisms may be at work, and both are compatible with the loss of L1 functions, such as L1-mediated cell adhesion and cell migration, being responsible for the ventricular dilatation.
These characteristic radiological findings could be explained by a decrease in white-matter elasticity, resulting from the loss of axons and axon adhesion, which could also explain the rippled appearance of the ventricular wall. In addition, maldevelopment of the midline structure could be induced by the disturbance of L1-mediated cell migration, which could cause the enlargement of the massa intermedia and quadrigeminal plate. In support of this mechanism, Kamiguchi et al. reported in vitro findings that neuronal migration from the subependymal zone in the songbird brain depends on a heterophilic interaction between neuronal L1 and a radial glial cell receptor.14) The enlarged massa intermedia would cause narrowing of the third ventricle, and the enlarged quadrigeminal plate would cause narrowing of the aqueduct.
L1 genetic testing has been clinically established and is already used in prenatal and postnatal diagnosis of the disease and in carrier detection.15) Renier reported that XLH shows the poorest outcome of any prenatal non-communicating hydrocephalus.16) For a mother carrying an L1 mutation, 50% of the male fetuses could have severe hydrocephalus; therefore, prenatal molecular genetic diagnosis with genetic counseling is extremely beneficial for a family with XLH. The prenatal molecular genetic diagnosis of L1 syndrome was first reported by Jouet et al. in 1995.17)
Later, prenatal L1 gene analysis was systematically organized by Yamasaki et al.15) In 2003, new clinical guidelines for genetic testing were established by 10 Japanese genetic-medicine-related societies (Guideline 2003). This guideline recommends that carriers be tested voluntarily with informed consent, which means that carrier testing in childhood is prohibited. Therefore, since the release of Guideline 2003, only male fetuses have been tested. Nine obligate carriers have requested prenatal testing a total of 14 times.15) The fetuses were tested for sex, L1 mutation, or both using chorionic villus sampling (CVS) or amniocentesis (AC). Of the 14 fetuses, 4 were male and 10 were female. Prenatal genetic testing was performed on seven of the fetuses, three females (prior to 2003) and four males. Of the four male fetuses tested, only one had an L1 gene mutation. The mother terminated the pregnancy. In 13 of the 14 cases, the mothers continued their pregnancies, and delivered healthy babies that lacked an XLH phenotype. Thus, the diagnoses were made with perfect accuracy. In 12 of the 14 cases, DNA was obtained by CVS between 10 weeks and 12 weeks gestation, and in two it was obtained by AC between 15 weeks and 16 weeks gestation. No maternal or fetal complications occurred during either CVS or AC.15)
HPE
HPE was once thought to be caused by an abnormal separation of the prosencephalon occurring after the completion of neural tube closure. However, molecular genetic research revealed that HPE starts before closure of the neural tube and is associated with a molecular abnormality manifested at the prochordal plate during the preneurulation period. Neural differentiation abnormalities caused by a mesenchyme defect are thought to lead to a lack of separation of the left and right cerebral hemispheres, resulting in various symptoms.18) DeMyer’s classical classification of HPE into three types—the alobar (Fig. 2), semilobar, and lobar types—according to the degree of unseparation between the left and right cerebral hemispheres, is still used today.19)
Fig. 2.
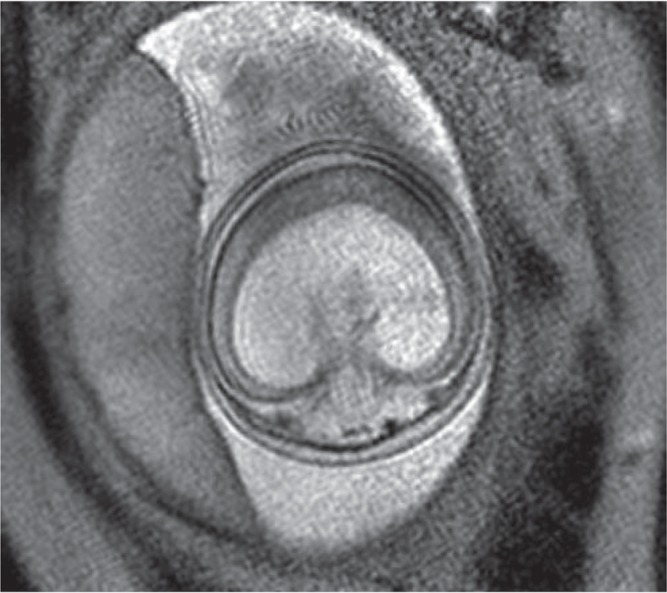
Fetal magnetic resonance imaging of the patient with 13 trisomy shows alobar type holoprosencephaly.
The incidence of HPE was previously reported as 1 in 10,000 births, because although it occurs in 1 in 250 pregnancies, only 3% of the affected fetuses are delivered. In the West Midland Congenital Abnormality Register WMCAR), for which the data were collected in 1995–2004, after the technology for prenatal diagnosis was established, the incidence is reported as 1.7 in 10,000 births, essentially the same as reported previously. HPE is rarest among whites and most common among Africans and Pakistanis.20)
Environmental and genetic factors are interrelated, and both contribute to HPE. One report maintains that maternal diabetes is an important risk factor, increasing the frequency of HPE 100-fold. Other factors are alcohol, anticonvulsant drugs, retinoic acid, smoking, fetal cytomegalovirus infection, and hypocholesterolemia. The relationship between HPE and cholesterol has been studied. Patients with Smith-Lemli-Opitz syndrome, a common complication of HPE, have a defect in 7-dehydrocholesterol reductase, which is involved in cholesterol metabolism. Cholesterol is necessary to activate the sonic hedgehog (Shh) signaling pathway, the malfunction of which is thought to cause HPE.21)
Chromosomal abnormalities are seen in 40–45% of HPE cases. The chromosomal abnormalities include trisomy 13 and trisomy 18, but trisomy 13 is the most common, accounting for 30–70% of the chromosomal abnormalities in reports on HPE Cases with known chromosomal abnormalities are classified as HPE 1 to HPE 12. In addition, there are 16 known HPE-associated genes, and the causal genes for HPE2–5,7,9,10,11 and 12 have been identified (Table 1). Of the 16 known HPE-associated genes, mutations are found most often in SHH, ZIC2, SIX3, and TGIF. A genetic study of 205 patients with HPE revealed SHH mutations in 47 (23%), ZIC2 mutations in 88 (43%), SIX3 mutations in 59 (29%), and TGIF mutations in 11 (5%).22)
Table 1.
Causative genes of hydrocephalus
| HPE | Location of chromosome | Casusal genes | Authors | References23,24) |
|---|---|---|---|---|
| HPE2 | 2p21 | SIX3 | Wallis DE | Nature Genet 22: 196–198, 1999 |
| HPE3 | 7p36 | Sonic hedgehog (SHH) | Roessler E | Nature Genet 14: 357–360, 1996 |
| HPE4 | 18p11.3 | TGIF1 | Gripp KW | Nature Genet 25: 205–208, 2000 |
| HPE5 | 13q32 | ZIC2 | Brown SA | Nature Genet 20: 180–183, 1998 |
| HPE7 | 9q22.3 | PATCHED-1(PTCH1) | Ming JE | Am J Hum Genet 71: 1017–1032, 2002 |
| HPE9 | 2q14 | GLI2 | Roessler E | Proc Natl Acad Sci USA 100: 13424–13429, 2003 |
| HPE10 | 1q42 | DISP1 | Roessler E | Hum Genet 125: 393–340, 2009 |
| HPE11 | 11q24.2 | CDON | Bae GU | Am J Hum Genet 89: 231–240, 2011 |
| HPE12 | 6q27 | DLL1 | Dupe V | Hum Mol Genet 20: 1122–1131, 2011 |
| 3p21–p23 | TDGF1 | De la Cruz JM | Hum Genet 110: 422–428, 2002 | |
| 6q22.31–q23.2 | EYA4 | Abe Y | Hum Mutat 30: E946–E955, 2009 | |
| 8q24.3 | FAST1(FOXH1) | Cohen MM Jr | Am J Med Genet 123A: 5–28, 2003 | |
| 9q21.33 | GAS1 | Ribeiro LA | Am J Med Genet 152A: 1688–1694, 2010 | |
| 10q22.4 | NODAL | Roessler E | Mol Genet Metab 98: 225–234, 2009 | |
| 10q24 | FGF8 | Arauz RF | Mol Syndromol 1: 59–66, 2010 | |
| 11q13.4 | DHCR7 | |||
| HPE1 | 21q22 | |||
| HPE6 | 2p37.1 | |||
| HPE8 | 14q13 |
HPE: hydrocephalus.
A mutation in SHH was first identified as a cause of HPE in 1996.23) SHH encodes Shh, a secreted protein that is a key inductive signal for ventral patterning of neural tube. Since then, many genes for Shh signaling components have been shown to bear HPE-associated mutations. Of the patients with ZIC2 mutations, almost 90% have a structural brain anomaly. Patients with SIX3 mutations are most likely to have an alobar type of brain anomaly. However, in the study of 205 patients described above, 44 relatives who were phenotypically normal also had mutations in SHH, ZIC2, SIX3, or TGIF. Therefore, more genotype and phenotype correlation studies are needed to improve the genetic analysis of HPE for clinical applications.24)
Notably, however, only about 5–10% of HPE cases are due to mutations in established HPE genes, and 30–50% of HPE cases are due to a chromosomal anomaly, including deletions and duplications. Therefore, a comprehensive cytogenetic study of patients with HPE should begin with a high-resolution karyotype. Finally, for nonsyndromic HPE patients and those without a chromosomal abnormality, a molecular analysis of the most common implicated genes (SHH, ZIC2, and SIX3)25) should be performed.
Dandy–Walker Malformation (DWM)
DWM is a congenital disease whose features are aplasia or dysplasia of the cerebellar vermis, a midline cyst connecting with the IVth ventricle, and elevation of the tentorium, torcular herophili, and transverse sinus (Fig. 3). Epidemiologically, DWM occurs with a frequency of one in 25,000–35,000 births, and according to a nationwide survey in Japan, with equal frequency in both sexes.
Fig. 3.

Magnetic resonance imaging (T1) after birth shows Dandy–Walker malformation.
The etiology of DWM is unknown, but one well-accepted idea is that it is caused by the suspension of rhombencephalic development, accompanied by dysraphism of the cerebellum at the midline. DWM is accompanied by more than 18 types of chromosomal abnormality; for example, abnormalities of 2q, 5p, 8p, 9p, 13q, 16q, and 17q are reported. More than 40 genetic syndromes co-occur with DWM. In addition, DWM can arise as a result of maternal diabetes, maternal use of warfarin, or fetal infection by cytomegalovirus or rubella.
Recently, two important loci, 3q24 and 6q25.3, were identified as causative areas of DWM. In these areas, ZIC1 and ZIC4 located in 6q25.3 and FOXC1 in 3q24 were found to be important genes. More than 10 cases of DWM with deletions in 6q25.3, including the ZIC1 or ZIC4 gene, have been identified.
Neural Tube Defect (NTD)
NTD is a situation in which the closure of neural tube does not occur properly. When it occurs in the caudal portion of the neural tube, it becomes a myelomeningocele. Tissues that normally cover the dorsal spinal cord, such as the meninges, the dorsal part of the spinal arch, and the skin do not form, causing the neural placode to be exposed dorsally. Of myelomeningocele cases, 80–90% are accompanied by hydrocephalus, requiring treatment.
It has been discovered that disruption of the planar cell polarity (PCP) signaling pathway causes NTD in mice. The PCP signaling pathway is important for the uniform orientation of cell polarity in tissues. The NTD phenotype of a classic mutant mouse, Looptail, is caused by a mutation of the PCP gene Vangle2. Recently, PCP genes were strongly implicated as causal in human NTDs. Single-nucleotide variants, predominantly heterozygous missense genomic alterations, have been found in the coding region of the core PCP genes CELSR1, FZD6, PRICKLE1, DVL2, VANGL1, and VANGL2 of patients with NTD.26) PCP signaling is also essential for the directional beating of motile cilia. The apical surface of ependymal cells of the cerebral ventricle is covered with cilia, whose beating facilitates the circulation of CSF. Tissir et al. reported that PCP-related cadherins Celsr2 and Celsr3 control this process. Celsr2-deficient mice develop defective CSF dynamics and hydrocephalus.27) Celsr2 and Celsr3 double mutants show impaired ciliogenesis, resulting in lethal hydrocephalus. Therefore, PCP genes are now among the genes investigated as likely candidates for the molecular basis of hydrocephalus and hydrocephalus-related disease.
Conclusion
Hydrocephalus and hydrocephalus-related diseases are considered to be multifactorial diseases, caused by both genetic and environmental factors. For single gene disorders like XLH, clinical genetic testing is already available. For these diseases, future research will focus on developing therapies based on their molecular mechanisms. On the other hand, while genetic factors are mainly responsible for HPE and DWM, these diseases involve interactions of multiple genes and their phenotypes are heterogeneous. The molecular biology of these disorders is still not fully understood. The role of PCP genes in the genesis of hydrocephalus is becoming clear, although more analysis is needed before we can fully understand the molecular biology in detail and develop appropriate clinical applications.
References
- 1). Bickers DS, Adams RD: Hereditary stenosis of the aqueduct of Sylvius as a cause of congenital hydrocephalus. Brain 72: 246– 262, 1949. [DOI] [PubMed] [Google Scholar]
- 2). Rosenthal A, Jouet M, Kenwrick S: Aberrant splicing of neural cell adhesion molecule L1 mRNA in a family with X-linked hydrocephalus. Nat Genet 2: 107– 112, 1992. [DOI] [PubMed] [Google Scholar]
- 3). Jouet M, Rosenthal A, Armstrong G, MacFarlane J, Stevenson R, Paterson J, Metzenberg A, Ionasescu V, Temple K, Kenwrick S: X-linked spastic paraplegia (SPG1), MASA syndrome and X-linked hydrocephalus result from mutations in the L1 gene. Nat Genet 7: 402– 407, 1994. [DOI] [PubMed] [Google Scholar]
- 4). Gu SM, Orth U, Zankl M, Schröder J, Gal A: Molecular analysis of the L1CAM gene in patients with X-linked hydrocephalus demonstrates eight novel mutations and suggests non-allelic heterogeneity of the trait. Am J Med Genet 71: 336– 340, 1997. [DOI] [PubMed] [Google Scholar]
- 5). Finckh U, Schröder J, Ressler B, Veske A, Gal A: Spectrum and detection rate of L1CAM mutations in isolated and familial cases with clinically suspected L1-disease. Am J Med Genet 92: 40– 46, 2000. [DOI] [PubMed] [Google Scholar]
- 6). Vos YJ, de Walle HE, Bos KK, Stegeman JA, Ten Berge AM, Bruining M, van Maarle MC, Elting MW, den Hollander NS, Hamel B, Fortuna AM, Sunde LE, Stolte-Dijkstra I, Schrander-Stumpel CT, Hofstra RM: Genotype-phenotype correlations in L1 syndrome: a guide for genetic counselling and mutation analysis. J Med Genet 47: 169– 175, 2010. [DOI] [PubMed] [Google Scholar]
- 7). Kanemura Y Okamoto N, Sakamoto H, Kamiguchi H, Yamasaki M: Molecular mechanisms and neuroimaging criteria for severe L1 syndrome with X-linked hydrocephalus. J Neurosurg 105 (5 Suppl): 403– 412, 2006. [DOI] [PubMed] [Google Scholar]
- 8). Yamasaki M, Thompson P, Lemmon V: CRASH syndrome: mutations in L1CAM correlate with severity of the disease. Neuropediatrics 28: 175– 178, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9). Fransen E, Van Camp G, D’Hooge R, Vits L, Willems PJ: Genotype-phenotype correlation in L1 associated diseases. J Med Genet 35: 399– 404, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10). Kamiguchi H, Hlavin ML, Yamasaki M, Lemmon V: Adhesion molecules and inherited diseases of the human nervous system. Annu Rev Neurosci 21: 97– 125, 1998. [DOI] [PubMed] [Google Scholar]
- 11). Bateman A, Jouet M, MacFarlane J, Du JS, Kenwrick S, Chothia C: Outline structure of the human L1 cell adhesion molecule and the sites where mutations cause neurological disorders. EMBO J 15: 6050– 6059, 1996. [PMC free article] [PubMed] [Google Scholar]
- 12). Yamasaki M, Arita N, Hiraga S, Izumoto S, Morimoto K, Nakatani S, Fujitani K, Sato N, Hayakawa T: A clinical and neuroradiological study of X-linked hydrocephalus in Japan. J Neurosurg 83: 50– 55, 1995. [DOI] [PubMed] [Google Scholar]
- 13). Fransen E, D’Hooge R, Van Camp G, Verhoye M, Sijbers J, Reyniers E, Soriano P, Kamiguchi H, Willemsen R, Koekkoek SK, De Zeeuw CI, De Deyn PP, Van der Linden A, Lemmon V, Kooy RF, Willems PJ: L1 knockout mice show dilated ventricles, vermis hypoplasia and impaired exploration patterns. Hum Mol Genet 7: 999– 1009, 1998. [DOI] [PubMed] [Google Scholar]
- 14). Barami K, Kirschenbaum B, Lemmon V, Goldman SA: N-cadherin and Ng-CAM/8D9 are involved serially in the migration of newly generated neurons into the adult songbird brain. Neuron 13: 567– 582, 1994. [DOI] [PubMed] [Google Scholar]
- 15). Yamasaki M, Nonaka M, Suzumori N, Nakamura H, Fujita H, Namba A, Kamei Y, Yamada T, Pooh RK, Tanemura M, Sudo N, Nagasaka M, Yoshioka E, Shofuda T, Kanemura Y: Prenatal molecular diagnosis of a severe type of L1CAM syndrome (X-linked hydrocephalus) J Neurosurg Pediatrics 8: 411–416, 2011. [DOI] [PubMed] [Google Scholar]
- 16). Renier D, Sainte-Rose C, Pierre-Kahn A, Hirsch JF: Prenatal hydrocephalus: outcome and prognosis. Child Nerv Syst 4: 213– 222, 1988. [DOI] [PubMed] [Google Scholar]
- 17). Jouet M, Kenwrick S: Gene analysis of L1 neural cell adhesion molecule in prenatal diagnosis of hydrocephalus. Lancet 345: 161– 162, 1995. [DOI] [PubMed] [Google Scholar]
- 18). Roessler E, Muenke M: How a Hedgehog might see holoprosencephaly. Hum Mol Genet 12: 15– 25, 2003. [DOI] [PubMed] [Google Scholar]
- 19). DeMyer WE, Zeman W: Alobar holoprosencephaly (arhinencephaly) with median cleft lip and palate: clinical, electroencephalographic and nosologic considerations. Confin Neurol 23: 1– 36, 1963. [DOI] [PubMed] [Google Scholar]
- 20). Ong S, Tonks A, Woodward ER, Wyldes MP, Kilby MD: An epidemiological study of holoprosencephaly from a regional congenital anomaly register: 1995–2004. Prenat Diagn 27: 340– 347, 2007. [DOI] [PubMed] [Google Scholar]
- 21). Hahn JS, Pinter JD: Holoprosencephaly: genetic, neuroradiological, and clinical advances. Semin Pediatr Neurol 9: 309– 319, 2002. [DOI] [PubMed] [Google Scholar]
- 22). Solomon BD, Mercier S, Velez JI, Pineda-Alvarez DE, Wyllie A, Zhou N, Dubourg C, David V, Odent S, Roessler E, Muenke M: Analysis of genotype-phenotype correlations in human holoprosencephaly. Am J Med Genet C Semin Med Genet 154C: 133– 141, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23). Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M: Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet 14: 357– 360, 1996. [DOI] [PubMed] [Google Scholar]
- 24). Solomon BD, Gropman A, Muenke M: Holoprosencephaly overview. In: Gene Reviews (http://www.nebi.nlm.nih.gov/books/NBK1530)
- 25). Pineda-Alvarez DE, Dubourg C, David V, Roessler E, Muenke M: Current recommendations for the molecular evaluation of newly diagnosed holoprosencephaly patients. Am J Med Genet C Semin Med Genet 154C: 93–101, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26). Murdoch JN, Damrau C, Paudyal A, Bogani D, Wells S, Greene ND, Stanier P, Copp AJ: Genetic interactions between planar cell polarity genes cause diverse neural tube defects in mice. Dis Model Mech 7: 1153– 1163, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27). Tissir F, Qu Y, Montcouquiol M, Zhou L, Komatsu K, Shi D, Fujimori T, Labeau J, Tyteca D, Courtoy P, Poumay Y, Uemura T, Goffinet AM: Lack of cadherins Celsr2 and Celsr3 impairs ependymal ciliogenesis, leading to fatal hydrocephalus. Nat Neurosci 13: 700– 707, 2010. [DOI] [PubMed] [Google Scholar]