

Abstract
Background
Leishmaniasis is a neglected parasitic disease with diverse clinical manifestations and a complex epidemiology. It has been shown that its parasite-related traits vary between species and that they modulate infectivity, pathogenicity, and virulence. However, understanding of the species-specific adaptations responsible for these features and their evolutionary background is limited. To improve our knowledge regarding the parasite biology and adaptation mechanisms of different Leishmania species, we conducted a proteome-wide phylogenomic analysis to gain insights into Leishmania evolution.
Results
The analysis of the reconstructed phylomes (totaling 45,918 phylogenies) allowed us to detect genes that are shared in pathogenic Leishmania species, such as calpain-like cysteine peptidases and 3'a2rel-related proteins, or genes that could be associated with visceral or cutaneous development. This analysis also established the phylogenetic relationship of several hypothetical proteins whose roles remain to be characterized. Our findings demonstrated that gene duplication constitutes an important evolutionary force in Leishmania, acting on protein families that mediate host-parasite interactions, such as amastins, GP63 metallopeptidases, cathepsin L-like proteases, and our methods permitted a deeper analysis of their phylogenetic relationships.
Conclusions
Our results highlight the importance of proteome wide phylogenetic analyses to detect adaptation and evolutionary processes in different organisms and underscore the need to characterize the role of expanded and species-specific proteins in the context of Leishmania evolution by providing a framework for the phylogenetic relationships of Leishmania proteins.
Phylogenomic data are publicly available for use through PhylomeDB (http://www.phylomedb.org).
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2091-2) contains supplementary material, which is available to authorized users.
Keywords: Phylogenomics, Leishmania, Homology prediction
Background
Leishmaniasis is a group of neglected tropical diseases caused by protozoan parasites belonging to the genus Leishmania. The disease is present in 98 countries causing more than 1.5 million cases per year [1, 2] and posing 350 million people at risk of infection [3].
Leishmania belongs to the Trypanosomatidae family that is composed of obligatory parasitic organisms. Members of this family can parasitize insects as their hosts, including monoxenic organisms such as Crithidia, Leptomonas, Herpetomonas and Blastocrithidia, whereas others can also parasitize vertebrates, such as in the digenetic genera Trypanosoma and Leishmania, or plants in the genera Phytomonas [4].
The Leishmania genus presents great phenotypic diversity represented by more than 30 different species, of which at least 20 are pathogenic to humans [5]. Phylogenetic analyses of the genus has further divided it into three subgenera named Leishmania, Viannia and Sauroleishmania [6–8].
The Leishmania subgenus is distributed throughout the Old and New Worlds, and it is transmitted by the bite of infected female sand flies of the genus Phlebotomus (Old World) and Lutzomyia (New World). The Viannia subgenus is exclusively found in the New World and is only transmitted by Lutzomyia sand flies [6]. In both subgenera, parasites are present as intracellular amastigotes inside phagolysosomes of phagocytes in the vertebrate host or as promastigote forms in the insect vector.
The Sauroleishmania subgenus that is present in the Old World is composed of non-human pathogenic Leishmania and it is assumed that it infects lizards through ingestion of infected Sergentomya sand flies [9]. Parasites of this subgenus are found as extracellular promastigotes or amastigote-like forms infecting monocyte-like cells or erythrocytes [6, 7, 10].
Leishmania parasites cause a wide spectrum of clinical manifestations that are classified into cutaneous (CL), mucosal (ML) and visceral leishmaniasis (VL). Previous studies have shown that clinical manifestation and treatment needs are associated with the infecting Leishmania species and the host immune response [11].
CL is primarily caused by Leishmania (Leishmania) major, L. (Leishmania) mexicana, L. (Viannia) braziliensis and other species of the Viannia subgenus. ML occurs in approximately 5 % of individuals with previous CL, most of who were infected with L. (Viannia) braziliensis [12]. VL is caused by L. (Leishmania) infantum and L. (Leishmania) donovani, which are included within the L. donovani complex [2].
Parasite-related factors modulate infectivity, pathogenicity, and virulence [2]. Promastigote virulence factors mediate invasion during the initial steps of an infection. For instance, lipophosphoglycan affects macrophage and dendritic cell functions and gp63 protects against complement mediated lysis and facilitates invasion [2, 13].
Candidate virulence factors in visceralizing parasites include the A2 gene family. This family has been detected in L. (Leishmania) infantum, L. (Leishmania) donovani and, as a non-expressed pseudogene, in the L. (Leishmania) major genome. All members of the A2 gene family are highly expressed during the amastigote stage, potentially allowing parasite survival at higher temperatures in visceral organs [14].
Over the last decade, Leishmania genome sequencing projects have resulted in the availability of a great amount of molecular data, including the genomes of L. (Leishmania) major Friedlin [15], L. (Leishmania) infantum JPCM5, L. (Viannia) braziliensis M2904 [16], L. (Leishmania) amazonensis M2269 [7] and several others draft assemblies that are available to the scientific community [17].
Comparative genomic studies have reported high synteny across Leishmania species despite a breach of 36–46 million years divergence between New World and Old World species [18]. Only 200 genes with differential distributions across L. (Leishmania) major, L. (Leishmania) infantum, and L. (Viannia) braziliensis have been described based on sequence similarity [16].
The identification of homologous genes is a critical step to understand the evolutionary history of an organism. Homologs can be divided into two types: orthologs, which originated through a speciation event from a common ancestor and paralogs, which resulted from a duplication event [19–21]. This classification is critical to understanding the diversification processes because duplication events are often related to a posterior functional divergence [22, 23].
Accurate predictions of homology relationships can be used to infer gene functionality [22], reconstruct species phylogenies, and characterize genomes based on their encoded genes [19]. For these purposes, different methods have been proposed. Most of them rely on sequence similarity between genes where function and homology are assessed from the most similar sequences [22]. These methods are fast; however, they have drawbacks because sequence similarity does not always have a direct relationship to functionality [22].
Phylogenomics, which analyzes genomic information in the context of its evolution, is a promising method for inferring homology relationships [24, 25]. This method establishes homology from an evolutionary perspective rather than relying only on sequence similarity [22]. It has also been previously used to reveal the origin and evolution of phenotypic characteristics and further our knowledge of metabolism, pathogenicity, and adaptation of an organism to its surroundings [24, 26–28].
In the current study, we employed a phylogenomics-based approach to analyze the phylomes of six Leishmania species to study their evolution and provide a comprehensive survey of the phylogenetic history of all proteins in Leishmania.
Methods
Sequence data
Predicted proteomes from six Leishmania species Predicted proteomes from six Leishmania species (L. (Viannia) braziliensis, L. (Leishmania) mexicana, L. (Leishmania) major, L. (Leishmania) infantum, L. (Leishmania) donovani, Leishmania (Sauroleishmania) tarentolae) and Trypanosoma brucei were downloaded from the TritrypDB V5 [17] (Table 1). Prior to the analysis, proteome data were filtered with a customized Perl script to select proteins starting with methionine, lacking internal stop codons, represented by the 20 IUPAC amino acid codes, and longer than 100 amino acids.
Table 1.
Proteomes selected for the construction of Leishmania phylomes
| Species | NCBI ID | Total proteins | Valid proteins | Trees generated | Proteome coverage (%) | |
|---|---|---|---|---|---|---|
| # | % | |||||
| L. (Viannia) braziliensis | 420245 | 8357 | 7942 | 95.0 | 7712 | 97.1 |
| L. (Leishmania) donovani | 981087 | 8033 | 7736 | 96.3 | 7550 | 97.6 |
| L. (Leishmania) infantum | 435258 | 8238 | 7974 | 96.8 | 7808 | 97.9 |
| L. (Leishmania) major | 347515 | 8400 | 8170 | 97.3 | 7849 | 96.1 |
| L. (Leishmania) mexicana | 929439 | 8250 | 7953 | 96.4 | 7796 | 98.0 |
| L. (Sauroleishmania) tarentolae | 5689 | 8452 | 7465 | 88.3 | 7203 | 96.5 |
Phylome reconstruction
Phylome reconstruction for all species was done following an automated pipeline that was previously described [29] (Fig. 1). Briefly, a local database was created comprising all proteomic data. For each protein sequence (seed), a Smith-Waterman search [30] was performed against the aforementioned database to retrieve highly similar proteins with a continuous alignment length of more than 50 % of the query sequence and e-value ≤ 1e-05.
Fig. 1.
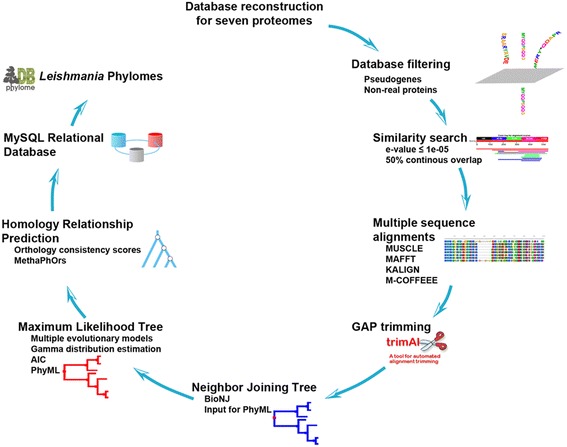
Phylogenomics pipeline. Each protein was treated as a seed and compared against all proteins encoded in the database. Groups of similar proteins were aligned and trimmed to remove gap-enriched regions. The trimmed alignment was used to build a NJ tree, which was then employed to create a maximum likelihood tree using the two best evolutionary models selected by AIC. Lineage specific duplications and homology relationships were determined and a relational database was created to store and analyze phylomic data
Sets of similar protein sequences were aligned using MUSCLE v3.8 [31], MAFFT v6.712b [32] and KALIGN v2.04 [33]. Alignments were performed in the forward and reverse directions and combined using M-COFFEE [34]. Gaps were removed from the final alignment using trimAl v1.3 [35] with a consistency and gap score cutoffs of 0.1667 and 0.1, respectively.
Neighbor-joining trees were constructed for each trimmed alignment as implemented in BioNJ [36], and T. brucei protein sequences were used as the out-group. The resulting NJ tree was used as input for PhyML v3.0 [37] to create a maximum likelihood tree, allowing branch length optimization using different evolutionary models (JTT, LG, WAG, Blosum62, MtREV, VT and Dayhoff).
The two evolutionary models that better modeled the data were determined according to the Akaike Information Criterion (AIC) [38]. Maximum likelihood trees were derived using the two selected models. In all cases, we used a discrete gamma-distribution model with four rate categories plus invariant positions; the gamma parameter and fraction of invariant positions were estimated from the data. Tree support values were calculated with an approximate likelihood ratio test (aLTR) in PhyML [37].
All phylome-related data, including trees and alignments, can be downloaded and browsed through PhylomeDB [39] (www.phylomedb.org).
Detection of homology relationships
To identify orthologs and paralogs, we used a species-overlap algorithm [27] as implemented in the environment for tree exploration (ETE) v2 [40]. Shortly, this algorithm starts at each seed protein used for generating the tree and traverses it until reaching the root. Each internal node was labeled as a duplication or speciation event, depending on whether their daughter partitions showed genes from the same or different species.
Orthology and paralogy relationships derived from the analyses of each phylome were combined into a single prediction using the MetaPhOrs algorithm [41] with a cutoff consistency score of 0.5, meaning that orthology relationship between two genes is called if the majority of examined trees containing these two sequences are consistent with this prediction.
Detection of species-specific expansions
We analyzed the Leishmania methaphylome using ETE to identify families that were specifically expanded in each species since their diversification. For this purpose, we considered those duplications detected by the species overlap algorithm that only comprised paralogs as species-specific expansions. An in-house Perl script was subsequently used to filter out redundant paralogous and orthologous proteins and load them into a MySQL relational database.
Gene Ontology codes that were significantly overrepresented in expanded families were detected using the hypergeometric distribution analysis in BiNGO [42] with Benjamini and Hochberg false discovery rate correction (corrected p value <0.05).
Results and discussion
Phylome reconstruction
The Leishmania metaphylome was derived from comparative analyses of all proteins encoded by six Leishmania species and Trypanosoma brucei, which was included as the out-group. The selected set of species includes causal agents of CL (L. (Viannia) braziliensis, L. (Leishmania) mexicana and L. (Leishmania) major), ML (L. (Viannia) braziliensis), VL (L. (Leishmania) infantum and L. (Leishmania) donovani) and a non-human pathogenic Leishmania (L. (Sauroleishmania) tarentolae).
From an initial set of 49,730 Leishmania proteins, 47,240 (94.9 %) were analyzed after filtering for valid sequences resulting in 45,918 phylogenetic trees summarizing the evolutionary relationships of 46,667 proteins (98.8 % of all valid proteins). This coverage is greater than the ones obtained for other phylomes such as the Schistosoma mansoni (70 %) [24] or the pea aphid Acyrthosiphon pisum (67 %) [26], thereby underscoring the high quality and sequence conservation of the datasets.
The absence of trees for the remaining 573 proteins could be due to high divergence between these proteins and their homologs in the dataset. Alternatively, this set of remaining proteins may include species-specific proteins that did not present homologs due to their uniqueness (Additional file 1: Table S1). Finally, another possibility is the presence of errors in the gene models as has been previously suggested [24].
Species-specific expansions
It has been shown that gene duplication plays an important role in evolution that results in increased expression or novel functionalization and/or sub-functionalization [43, 44]. Duplicated or diversified paralogs may be kept in the genome if they provide a selective advantage [27]. Therefore, inspecting the functions of expanded families may provide evidence of these processes in the evolution of Leishmania.
The Leishmania metaphylome provides an overview of protein evolutionary relationships that can be explored to reveal events related to Leishmania diversification and adaptation. Using the species-overlap algorithm [40], we analyzed species-specific protein expansions in all Leishmania proteomes and reported the most expanded proteins for each species (Table 2, Additional file 1: Table S2).
Table 2.
Top Leishmania species-specific protein expansions using the species-overlap algorithm
| Species | Seed | Seed annotation | Expansions |
|---|---|---|---|
| L. (Viannia) braziliensis | LbrM.34.0020 | TATE DNA Transposon | 30 |
| L. (Viannia) braziliensis | LbrM.08.1140 | amastin-like protein | 28 |
| L. (Viannia) braziliensis | LbrM.10.0520 | GP63, leishmanolysin,metallo-peptidase, Clan MA(M), Family M8 | 25 |
| L. (Leishmania) major | LmjF.12.0755 | surface antigen protein 2, putative | 20 |
| L. (Leishmania) mexicana | LmxM.08.0750 | amastin-like protein, putative | 19 |
| L. (Sauroleishmania) tarentolae | LtaPcontig05711-1 | Hypothetical protein, unknown function | 14 |
| L. (Sauroleishmania) tarentolae | LtaP10.0670 | Major surface protease gp63, putative;GP63, leishmanolysin | 12 |
| L. (Leishmania) major | LmjF.34.1720 | amastin-like surface protein, putative | 12 |
| L. (Leishmania) major | LmjF.12.0950 | hypothetical protein, conserved | 11 |
| L. (Viannia) braziliensis | LbrM.30.0450 | histone H4 | 10 |
| L. (Leishmania) mexicana | LmxM.08.1080 | cathepsin L-like protease, putative | 8 |
| L. (Viannia) braziliensis | LbrM.19.1530 | glycerol uptake protein, putative | 7 |
| L. (Viannia) braziliensis | LbrM.02.0550 | Retrotransposable element SLACS | 7 |
| L. (Leishmania) mexicana | LmxM.12.0870partial | surface antigen protein 2, putative | 7 |
| L. (Leishmania) major | LmjF.09.0156 | ATG8/AUT7/APG8/PAZ2, putative (ATG8C.4) | 7 |
| L. (Leishmania) major | LmjF.08.1030 | cathepsin L-like protease | 7 |
| L. (Leishmania) donovani | LdBPK_100380.1 | folate/biopterin transporter, putative | 5 |
| L. (Leishmania) infantum | LinJ.10.0520 | GP63, leishmanolysin,metallo-peptidase, Clan MA(M), Family M8 (GP63-3) | 5 |
| L. (Leishmania) infantum | LinJ.36.0010 | phosphoglycan beta 1,3 galactosyltransferase 4 (SCG4) | 5 |
Our results show that species-specific expansions vary greatly between species with L. (Viannia) braziliensis and L. (Leishmania) donovani accumulating the highest and lowest number of expansions, respectively (Fig. 2). Expanded proteins include well characterized families such as amastins, metalloproteinases, cysteine proteases and surface antigen proteins (Additional file 1: Table S2). These families are important virulence factors in Leishmania and reveal an evolutionary trend towards parasitism.
Fig. 2.
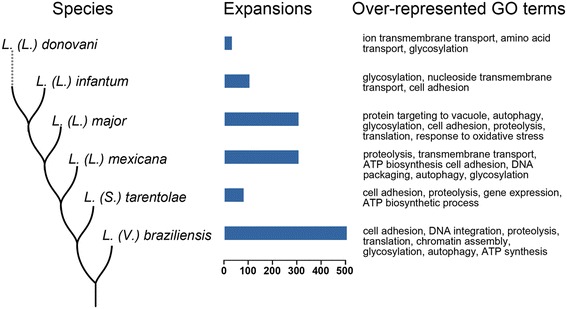
Estimates of expansions in Leishmania. Horizontal bars indicate the number of genes in expanded families per species. On the right we show significantly over-represented GO terms (corrected p value <0.05) compared to the rest of the genome in the set of expanded gene families
Over-represented Gene Ontology terms in expanded families also show species-specific adaptations (Fig. 2). However, common over-represented terms such as “glycosylation,” “proteolysis,” “cell adhesion” and “autophagy” are consistent with adaptation towards a parasitic lifestyle.
Glycosylation appears as an important mechanism of protein modification and may play a role in protein maturation and protein function in Leishmania [45]. Promastigote and amastigote stages express different types of proteophosphoglycans (PPGs) on their surfaces, and changes in the glycosylation of these proteins have resulted in striking reductions in promastigote and amastigote virulence in L. (Leishmania) major [46].
Proteolysis is a key component of pathogenesis in Leishmania, acting on several host intracellular proteins such as cytoskeleton regulators, transcription factors or protein phosphatases [47, 48]. It has also been suggested that the direction of proteolytic activities towards degradative enzymes in phagolysosomes and major histocompatibility complex molecules may promote parasite survival by impairing host response and proper antigen presentation [49].
Autophagy has been shown to play an important function during Leishmania differentiation from procyclic to metacyclic promastigotes and into amastigotes with an increase in autophagosomes and protein degradation levels [50]. Additionally, degradation of glycosomes allows organelle renewal and enables the parasites to rapidly adapt to the new conditions within their various hosts [51].
Among the most expanded proteins in L. (Viannia) braziliensis, we detected the presence of TATE DNA transposons (Telomere-Associated Transposable Element) and SLACS (Spliced Leader Associated Conserved Sequence). SLACS are specific retroposons that are located between tandem arrays of spliced leader RNA genes while TATE transposons tend to be located at telomeres. These transposable elements are the source of most siRNA in L. (Viannia) braziliensis [52] that are generated by the RNAi machinery, which appears to be specific to the Viannia subgenus to downregulate the expression of mobile elements that can affect genome integrity [52].
Another expanded protein family in L. (Viannia) braziliensis is amastin. This family of surface glycoproteins is highly expressed in amastigotes and, while their exact function is not known, they appear to mediate host-parasite interactions, allowing parasite infection and survival [53]. It has been previously shown that amastins are expanded in all Leishmania species compared to Trypanosoma, suggesting a functional adaptation [53]. The corresponding amastin phylogeny of our analysis comprises only proteins that originated after Leishmania diversification [53] and revealed that L. (Viannia) braziliensis, L. (Leishmania) mexicana, and L. (Leishmania) major have greatly expanded their delta-amastin repertoire compared to the visceral species included in the phylogenomic analyses (Fig. 3). (For an extensive evolutionary analysis of amastins in Trypanosomatids see Jackson [53]).
Fig. 3.
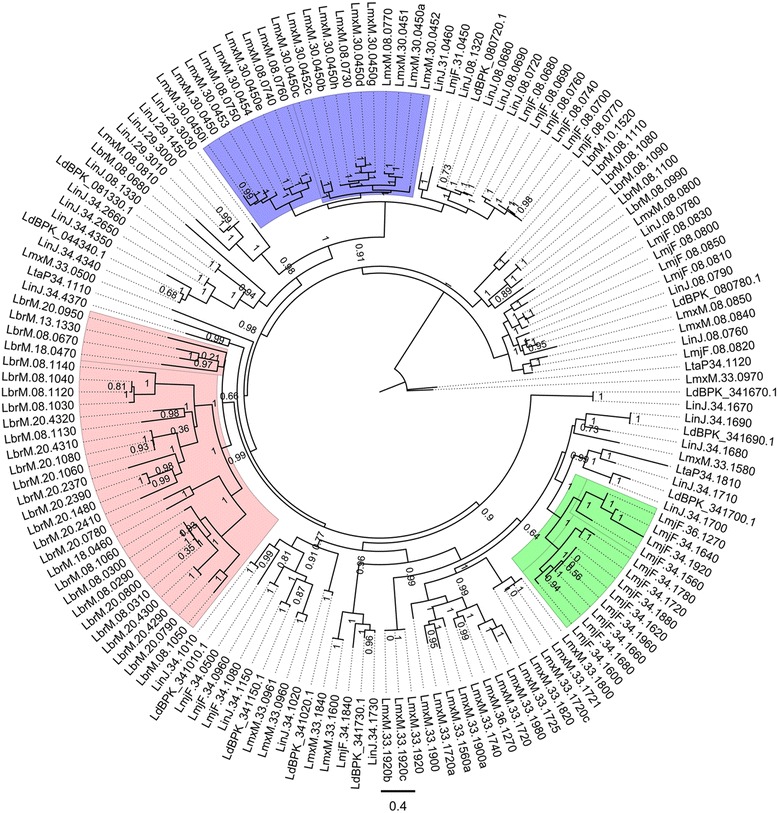
Amastin phylogenetic tree. Phylogenetic relationships of 150 Amastin protein members using L. (Viannia) braziliensis LbrM.08.1140 as seed protein with JTT as the best-fit model. Numbers indicate support values computed by the approximate likelihood ratio test (aLTR). Colored regions show species-specific expansions as follows: Rose: L. (Viannia) braziliensis; Green: L. (Leishmania) major; Blue: L. (Leishmania) mexicana. Gene codes indicate the following species: LinJ: L. (Leishmania) infantum; LmxM: L. (Leishmania) mexicana; LmjF: L. (Leishmania) major; LdBPK: L. (Leishmania) donovani; LbrM: L. (Viannia) braziliensis; Lta: L. (Sauroleishmania) tarentolae
We have also detected expansions in the GP63 protein family in L. (Viannia) braziliensis and L. (Sauroleishmania) tarentolae (Table 2). These metallopeptidases participate in parasite interactions with both invertebrate and vertebrate hosts, ensuring parasite invasion and survival [54–57].
To perform a deeper analysis of the GP63 phylogenomic data, we conducted a case analysis using the tree that retrieved the largest number of homologs across all species (seed “LbrM.31.2240”) (Fig. 4). This tree recovered 63 GP63 proteins from an initial set of 74 annotated GP63 plus 11 additional proteins that were annotated as hypothetical proteins in the input dataset. Proteins annotated as GP63 that were not present in this tree are shorter in length, lack a peptidase domain, or have an incorrect annotation in the proteome dataset.
Fig. 4.
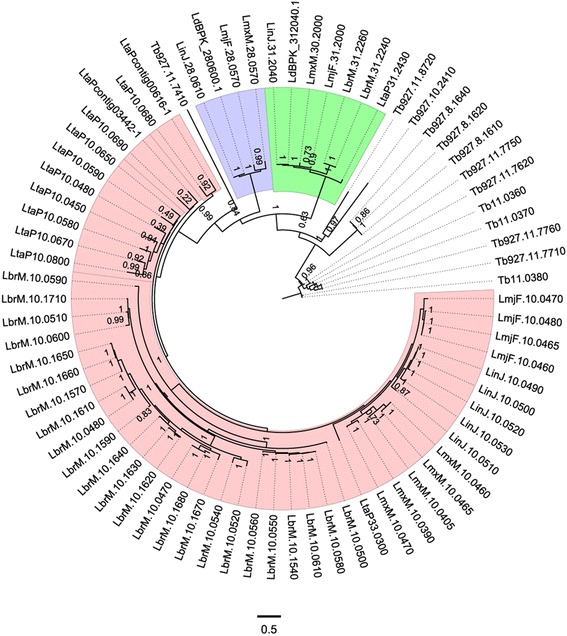
GP63 phylogenetic tree. Phylogenetic tree for GP63 using L. (Viannia) braziliensis seed protein LbrM.31.2240 and WAG as the best-fit model. Numbers indicate support values computed by the approximate likelihood ratio test (aLTR). Colored regions denote GP63 distribution as follows: Green: Chromosome 31 GP63; Blue: Chromosome 28 GP63; Rose: Chromosome 10 GP63. Gene codes indicate the following species: LinJ: L. (Leishmania) infantum; LmxM: L. (Leishmania) mexicana; LmjF: L. (Leishmania) major; LdBPK: L. (Leishmania) donovani; LbrM: L. (Viannia) braziliensis; Lta: L. (Sauroleishmania) tarentolae; Tb: T. brucei
GP63 genes in the Leishmania subgenus range from two genes in L. (Leishmania) donovani to seven in L. (Leishmania) infantum and GP63. On the contrary, the GP63 repertoire has greatly expanded in L. (Viannia) braziliensis and L. (Sauroleishmania) tarentolae reaching up to 26 and 13 genes, respectively (Fig. 4).
Our analysis shows that the GP63 family appears to have suffered expansion events at different times during Trypanosomatids’ evolution and can be divided in three distinct subfamilies located on chromosomes 31, 28, and 10 (Fig. 5). GP63 of chromosome 31 consists of a single GP63 gene present in all Leishmania species except L. (Viannia) braziliensis, where it is composed of two distinct isoforms that are located in an array (Figs. 4 and 5).
Fig. 5.
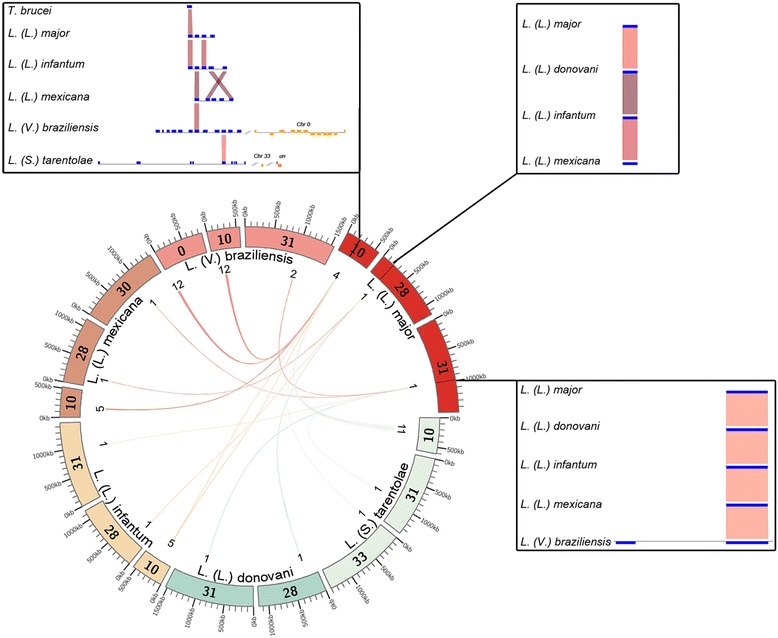
GP63 chromosome distribution. Location of GP63 genes in Leishmania genomes. Boxes represent each GP63 gene per species for each chromosome, while inner links show best reciprocal blast-best hit, and numbers indicate GP63 genes on the respective chromosome; Chr 0: Chromosome 0; un: undetermined. Inner lines indicate blast results against the L. (Leishmania) major GP63 genes and show the position of each gene within its respective chromosome
GP63 of chromosome 28 is present only in the Leishmania subgenus and is represented by one gene in L. (Leishmania) major, L. (Leishmania) mexicana, L. (Leishmania) donovani and L. (Leishmania) infantum, sharing more than 93 % similarity at the protein level.
GP63 of chromosome 10 constitutes a set of gene arrays in all Leishmania species except L. (Leishmania) donovani, where it is completely absent. The phylogeny shows that this subfamily branches with T. brucei GP63, supporting a common origin with subsequent gains and losses in Leishmania (Fig. 4). Among chromosome 10 GP63s, L. (Sauroleishmania) tarentolae and L. (Viannia) braziliensis stand out as the species with the highest number of expansions.
Alignment data for the Chr 10 subfamily revealed that L. (Sauroleishmania) tarentolae Chr 10 GP63 proteins are shorter than those of L. (Viannia) braziliensis (291 versus 560 amino acids), lack predicted extracellular regions, and have a shorter peptidase domain. These characteristics may affect parasite host interaction and limit GP63 protease activity in L. (Sauroleishmania) tarentolae, as has been previously suggested [7]. Another possibility could be assembly completeness of the L. (Sauroleishmania) tarentolae genome, which may result in partial GP63 sequences [7].
Given that the long arrays in L. (Viannia) braziliensis are absent from the other Leishmania species, it is highly possible that this expansion occurred after the origin of the Viannia subgenus. Interestingly, it has been previously shown that GP63 is also present in high copy number in L. (Viannia) peruviana and L. (Viannia) guyanensis [58, 59].
This information suggests that large GP63 expansions in chromosome 10 are characteristic of the Viannia subgenus and could respond to an adaptation mechanism to the wider range of reservoirs and vectors that the species of this subgenus infect. In the case of L. (Sauroleishmania) tarentolae, GP63 expansions could be related to interactions with a different genus that serves as vector (Sergentomya) and the lizard host.
Histone 4 has also been shown to be differentially expanded in L. (Viannia) braziliensis with 10 genes. In the Leishmania subgenus, Histone 4 is reduced to three or less genes and is completely absent in Sauroleishmania (Fig. 6). However, the lack of Histone 4 in Sauroleishmania could likely result from the limitations in the current genome assembly of this species.
Fig. 6.
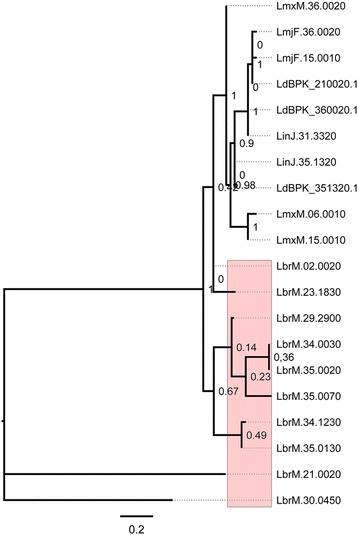
Histone 4 phylogenetic relationship. Phylogenetic relationships for Histone 4 using L. (Viannia) braziliensis seed protein LbrM.30.0450 and JTT as the best-fit model. Numbers indicate support values computed by the approximate likelihood ratio test (aLTR). Due to their high sequence similarity, there is a large inconsistency in most nodes as reflected in the tree. Rose-colored regions indicate L. (Viannia) braziliensis expansions. Gene codes indicate the following species: LinJ: L. (Leishmania) infantum; LmxM: L. (Leishmania) mexicana; LmjF: L. (Leishmania) major; LdBPK: L. (Leishmania) donovani; LbrM: L. (Viannia) braziliensis
H4 expansions in L. (Viannia) braziliensis are not restricted to a single chromosome, suggesting derivation of novel loci through transposition. Sequence alignment of these expansions showed a conserved core with more than 80 % sequence similarity among all sequences and the presence of variable regions at the N and C terminal ends.
Post-translational modification analysis in histones of Trypanosomatids has revealed that H4 and H3 are heavily acetylated and methylated on the N-terminal tails in Trypanosoma, and these modifications change during parasite development [60]. Whether expansions and diversification in histone 4 of L. (Viannia) braziliensis have a role in transcriptional regulation in Leishmania remains to be investigated.
Our results also revealed species-specific expansions in cysteine peptidases (CPs) in L. (Leishmania) mexicana, L. (Leishmania) major and L. (Viannia) braziliensis. These expansions are located in tandem arrays in chromosome 8 (Fig. 7). Previous studies on Cathepsin-B have shown immunomodulatory roles suppressing the Th1 response, ensuring parasite survival in L. (Leishmania) mexicana and L. (Leishmania) major and that their activity could result in different disease phenotypes in both species [61, 62]. The corresponding phylogeny of cysteine peptidases showed that cathepsin-L genes are exclusively located in chromosome 8, cysteine peptidases A in chromosome 19, and cathepsin-B in chromosome 29.
Fig. 7.
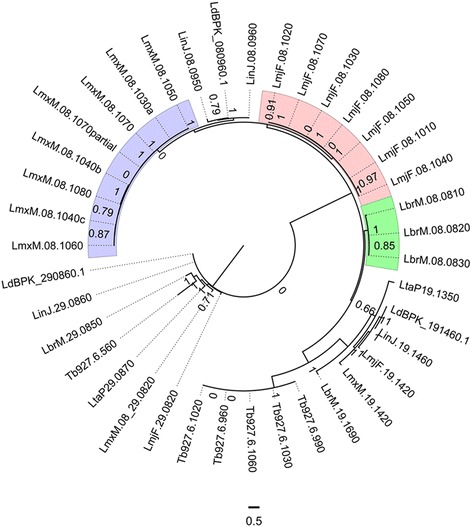
Cysteine peptidases phylogenetic relationship. Phylogenetic relationships for cysteine peptidases using L. (Leishmania) major seed protein LmjF.08.1070 under the WAG model. Numbers indicate support values computed by the approximate likelihood ratio test (aLTR). Blue, rose and green colored regions represent L. (Leishmania) mexicana, L. (Leishmania) major and L. (Viannia) braziliensis specific cathpesin-L expansions, respectively. Gene codes indicate the following species: LinJ: L. (Leishmania) infantum; LmxM: L. (Leishmania) mexicana; LmjF: L. (Leishmania) major; LdBPK: L. (Leishmania) donovani; LbrM: L. (Viannia) braziliensis; Lta: L. (Sauroleishmania) tarentolae
L. (Leishmania) mexicana, L. (Leishmania) major and L. (Viannia) braziliensis present eight, seven and three expansions of Cathepsin-L, respectively (Fig. 7). These expansions are organized into gene arrays and share more than 70 % similarity at the protein level.
RNA-expression data for L. (Leishmania) major retrieved from the Trytrip database [17] shows that these Cathepsin-L genes have, on average, a 1.7-fold increase in amastigotes versus procyclic promastigotes and up to a 1.8-fold increase between metacyclic versus procyclic promastigotes, which suggests that Cathepsin-L expression is modulated during parasite development with expression increasing towards the infective and intracellular stages.
Orthology relationships in Leishmania
Using BioPerl:Trees, we extracted orthologs and paralogs for each seed protein to analyze the ones that are unique in each species and to look at their respective homologs.
A total of 28 trees summarizing the relationships of 72 genes were species-unique (Additional file 1: Table S3). From these, 25 trees belonged to L. (Viannia) braziliensis and comprised TATE DNA transposons, SLACS, a phosphatidic acid phosphatase, and hypothetical proteins. The remaining trees belonged to a folate biopterin transporter, an oligosaccharyl transferase in L. (Leishmania) donovani, and a hypothetical protein in L (Leishmania) major. The absence of a greater number of species-specific trees reflects the high conservation between Leishmania proteomes and underscores the importance of species-specific expansions. Another possibility is the variance in assembly completeness of Leishmania genomes that can limit an accurate assessment of orthology and paralogy relationships.
We found 299 trees comprising 1519 genes across five human pathogenic Leishmania species without orthologs in L. (Sauroleishmania) tarentolae. Protein families in these trees include histone 4, k39 kinesin, calpain-like cysteine peptidases, a2-rel and hypothetical proteins (Additional file 1: Table S4).
Calpain-like cysteine peptidases are predicted to encode large proteins with potential functions in signal transduction, cytoskeletal remodeling and membrane attachment during Leishmania differentiation [63, 64].
Previous studies have shown that disruption by gene targeting of a2-rel-related genes in L. (Leishmania) donovani generated mutants with reduced infectivity in mice and limited their proliferation in culture [65]; however, their specific function has not been elucidated yet.
We found a total of 11 trees that were shared by species of the Leishmania donovani complex without orthologs in L. (Leishmania) major, L. (Viannia) braziliensis nor L. (Sauroleishmania) tarentolae (Additional file 1: Table S5). Among these genes we found the presence of the A2 gene family that is the prototype of genes involved in visceralization [66] and hypothetical proteins that remain to be characterized.
Leishmania species that are associated with CL include L. (Viannia) braziliensis, L. (Leishmania) mexicana, L. (Leishmania) major and occasionally L. (Leishmania) infantum [2]. We found a total of 15 trees specific for all these species comprising of 72 proteins, most of which are annotated as hypothetical (Additional file 1: Table S6).
Conclusions
Our results indicate that gene expansions are a common trait in Leishmania genomes and represent an important force in the evolution of these parasites. Major species-specific expansions in genes mediating host-parasite interactions reflect genome complexity and evolutionary processes that influence the wide spectrum of diseases that are caused by different Leishmania species.
An important limitation of the current study is the different assembly completeness across the Leishmania genomes analyzed. It is known that repetitions and head-to-tail duplicated genes are likely to suffer from assembly and annotation errors leading to partial sequences that could have been excluded during the filtering steps. In this sense, it might be possible that the exact number of expanded genes may vary with subsequent improvements of the current genome assemblies.
The Leishmania metaphylome appears as a promising resource to aid the scientific community in understanding the complexity of host-parasite relationships and highlighting areas of interest for additional experimentation. Further studies are needed to determine the function of relevant hypothetical proteins that were identified here, characterize species-specific expansions, and employ transcriptomic data to complement our results.
Acknowledgements
We thank Leszek P. Pryszcz for his assistance with MetaPhOrs. DB group is funded by The National Institute of Science and Technology for Vaccines (Brazil) (MCT/CNPq, grant CNPq 573547/2008-4), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG, grant # APQ-04073-10, PPM-00219-13) and Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior (CAPES, grant # 051/2013). TG group research is funded in part by a grant from the Spanish ministry of Economy and Competitiveness (BIO2012-37161), a Grant from the Qatar National Research Fund grant (NPRP 5-298-3-086), and a grant from the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013) / ERC (Grant Agreement n. ERC-2012-StG-310325). GO group was funded by NIH-Fogarty (TW007012), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (REDE-56/11, RED-00014-14) and Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior (309312/2012-4).
Disclaimer
The views expressed in this article are those of the authors only and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the U.S. Government.
Copyright statement
Some authors of this manuscript are employees of the U.S. Government. This work was prepared as part of their duties. Title 17 U.S.C. § 105 provides that ‘Copyright protection under this title is not available for any work of the United States Government.’ Title 17 U.S.C. § 101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person’s official duties.
Additional file
Supplementary tables. (XLSX 148 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HOV carried out bioinformatics analysis, participated in study conception, design and drafted the manuscript. LLSS participated in study design, provided support with phylogenies and manuscript writing. GO participated in study design and manuscript writing. TG carried out bioinformatics analysis, contributed with data storage and publication on Phylome DB, manuscript writing. DCB participated in study design, coordination and manuscript writing. All authors read and approved the final manuscript.
Contributor Information
Hugo O. Valdivia, Email: hvalrod@hotmail.com
Larissa L. S. Scholte, Email: larissa@cpqrr.fiocruz.br
Guilherme Oliveira, Email: oliveira@cpqrr.fiocruz.br.
Toni Gabaldón, Email: toni.gabaldon@crg.eu.
Daniella C. Bartholomeu, Email: daniella@icb.ufmg.br
References
- 1.Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M. Leishmaniasis worldwide and global estimates of its incidence. PLoS One. 2012;7(5) doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray HW, Berman JD, Davies CR, Saravia NG. Advances in leishmaniasis. Lancet. 2005;366(9496):1561–1577. doi: 10.1016/S0140-6736(05)67629-5. [DOI] [PubMed] [Google Scholar]
- 3.Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis. 2004;27(5):305–318. doi: 10.1016/j.cimid.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Maslov DA, Podlipaev SA, Lukes J. Phylogeny of the kinetoplastida: taxonomic problems and insights into the evolution of parasitism. Mem Inst Oswaldo Cruz. 2001;96(3):397–402. doi: 10.1590/S0074-02762001000300021. [DOI] [PubMed] [Google Scholar]
- 5.Banuls AL, Hide M, Prugnolle F. Leishmania and the leishmaniases: a parasite genetic update and advances in taxonomy, epidemiology and pathogenicity in humans. Adv Parasitol. 2007;64:1–109. doi: 10.1016/S0065-308X(06)64001-3. [DOI] [PubMed] [Google Scholar]
- 6.Bates PA. Transmission of Leishmania metacyclic promastigotes by phlebotomine sand flies. Int J Parasitol. 2007;37(10):1097–1106. doi: 10.1016/j.ijpara.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Real F, Vidal RO, Carazzolle MF, Mondego JM, Costa GG, Herai RH, Wurtele M, de Carvalho LM, Carmona e Ferreira R, Mortara RA, et al. The genome sequence of Leishmania (Leishmania) amazonensis: functional annotation and extended analysis of gene models. DNA Res. 2013;20(6):567–581. doi: 10.1093/dnares/dst031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Croan DG, Morrison DA, Ellis JT. Evolution of the genus Leishmania revealed by comparison of DNA and RNA polymerase gene sequences. Mol Biochem Parasitol. 1997;89(2):149–159. doi: 10.1016/S0166-6851(97)00111-4. [DOI] [PubMed] [Google Scholar]
- 9.LUMSDEN WHR, Evans D. Biology of the Kinetoplastida. Vol. 2. London: Academic Press Inc. (London) Ltd.; 1979.
- 10.Paperna I, Boulard Y, Hering-Hagenbeck SH, Landau I. Description and ultrastructure of Leishmania zuckermani n. sp. amastigotes detected within the erythrocytes of the South African gecko Pachydactylus turneri Gray, 1864. Parasite. 2001;8(4):349–353. doi: 10.1051/parasite/2001084349. [DOI] [PubMed] [Google Scholar]
- 11.Kaye P, Scott P. Leishmaniasis: complexity at the host-pathogen interface. Nat Rev Microbiol. 2011;9(8):604–615. doi: 10.1038/nrmicro2608. [DOI] [PubMed] [Google Scholar]
- 12.Queiroz A, Sousa R, Heine C, Cardoso M, Guimaraes LH, Machado PR, Carvalho EM, Riley LW, Wilson ME, Schriefer A. Association between an emerging disseminated form of leishmaniasis and Leishmania (Viannia) braziliensis strain polymorphisms. J Clin Microbiol. 2012;50(12):4028–4034. doi: 10.1128/JCM.02064-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMahon-Pratt D, Alexander J. Does the Leishmania major paradigm of pathogenesis and protection hold for New World cutaneous leishmaniases or the visceral disease? Immunol Rev. 2004;201:206–224. doi: 10.1111/j.0105-2896.2004.00190.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhang WW, Matlashewski G. Loss of virulence in Leishmania donovani deficient in an amastigote-specific protein, A2. Proc Natl Acad Sci U S A. 1997;94(16):8807–8811. doi: 10.1073/pnas.94.16.8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R, et al. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309(5733):436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peacock CS, Seeger K, Harris D, Murphy L, Ruiz JC, Quail MA, Peters N, Adlem E, Tivey A, Aslett M, et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet. 2007;39(7):839–847. doi: 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, Gao X, et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010;38(Database issue):D457–462. doi: 10.1093/nar/gkp851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukes J, Mauricio IL, Schonian G, Dujardin JC, Soteriadou K, Dedet JP, Kuhls K, Tintaya KW, Jirku M, Chocholova E, et al. Evolutionary and geographical history of the Leishmania donovani complex with a revision of current taxonomy. Proc Natl Acad Sci U S A. 2007;104(22):9375–9380. doi: 10.1073/pnas.0703678104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabaldon T, Dessimoz C, Huxley-Jones J, Vilella AJ, Sonnhammer EL, Lewis S. Joining forces in the quest for orthologs. Genome Biol. 2009;10(9):403. doi: 10.1186/gb-2009-10-9-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fitch WM. Distinguishing homologous from analogous proteins. Syst Zool. 1970;19(2):99–113. doi: 10.2307/2412448. [DOI] [PubMed] [Google Scholar]
- 21.Descorps-Declere S, Lemoine F, Sculo Q, Lespinet O, Labedan B. The multiple facets of homology and their use in comparative genomics to study the evolution of genes, genomes, and species. Biochimie. 2008;90(4):595–608. doi: 10.1016/j.biochi.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 22.Eisen JA, Wu M. Phylogenetic analysis and gene functional predictions: phylogenomics in action. Theor Popul Biol. 2002;61(4):481–487. doi: 10.1006/tpbi.2002.1594. [DOI] [PubMed] [Google Scholar]
- 23.Gabaldon T, Koonin EV. Functional and evolutionary implications of gene orthology. Nat Rev Genet. 2013;14(5):360–366. doi: 10.1038/nrg3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silva LL, Marcet-Houben M, Nahum LA, Zerlotini A, Gabaldon T, Oliveira G. The Schistosoma mansoni phylome: using evolutionary genomics to gain insight into a parasite's biology. BMC Genomics. 2012;13:617. doi: 10.1186/1471-2164-13-617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gabaldon T. Large-scale assignment of orthology: back to phylogenetics? Genome Biol. 2008;9(10):235. doi: 10.1186/gb-2008-9-10-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huerta-Cepas J, Marcet-Houben M, Pignatelli M, Moya A, Gabaldon T. The pea aphid phylome: a complete catalogue of evolutionary histories and arthropod orthology and paralogy relationships for Acyrthosiphon pisum genes. Insect Mol Biol. 2010;19(Suppl 2):13–21. doi: 10.1111/j.1365-2583.2009.00947.x. [DOI] [PubMed] [Google Scholar]
- 27.Huerta-Cepas J, Dopazo H, Dopazo J, Gabaldon T. The human phylome. Genome Biol. 2007;8(6):R109. doi: 10.1186/gb-2007-8-6-r109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jackson AP, Allison HC, Barry JD, Field MC, Hertz-Fowler C, Berriman M. A cell-surface phylome for African trypanosomes. PLoS Negl Trop Dis. 2013;7(3) doi: 10.1371/journal.pntd.0002121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huerta-Cepas J, Capella-Gutierrez S, Pryszcz LP, Denisov I, Kormes D, Marcet-Houben M, Gabaldon T. PhylomeDB v3.0: an expanding repository of genome-wide collections of trees, alignments and phylogeny-based orthology and paralogy predictions. Nucleic Acids Res. 2011;39(Database issue):D556–560. doi: 10.1093/nar/gkq1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147(1):195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 31.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lassmann T, Sonnhammer EL. Kalign--an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics. 2005;6:298. doi: 10.1186/1471-2105-6-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallace IM, O'Sullivan O, Higgins DG, Notredame C. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 2006;34(6):1692–1699. doi: 10.1093/nar/gkl091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol. 1997;14(7):685–695. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- 37.Guindon S, Delsuc F, Dufayard JF, Gascuel O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol. 2009;537:113–137. doi: 10.1007/978-1-59745-251-9_6. [DOI] [PubMed] [Google Scholar]
- 38.Akaike H. A new look at the statistical model identification. Automatic Control IEEE Trans. 1974;19(6):716–723. doi: 10.1109/TAC.1974.1100705. [DOI] [Google Scholar]
- 39.Huerta-Cepas J, Capella-Gutierrez S, Pryszcz LP, Marcet-Houben M, Gabaldon T. PhylomeDB v4: zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res. 2014;42(Database issue):D897–902. doi: 10.1093/nar/gkt1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huerta-Cepas J, Dopazo J, Gabaldon T. ETE: a python Environment for Tree Exploration. BMC Bioinformatics. 2010;11:24. doi: 10.1186/1471-2105-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pryszcz LP, Huerta-Cepas J, Gabaldon T. MetaPhOrs: orthology and paralogy predictions from multiple phylogenetic evidence using a consistency-based confidence score. Nucleic Acids Res. 2011;39(5) doi: 10.1093/nar/gkq953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 43.Vogel C, Chothia C. Protein family expansions and biological complexity. PLoS Comput Biol. 2006;2(5) doi: 10.1371/journal.pcbi.0020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roth C, Rastogi S, Arvestad L, Dittmar K, Light S, Ekman D, Liberles DA. Evolution after gene duplication: models, mechanisms, sequences, systems, and organisms. J Exp Zool B Mol Dev Evol. 2007;308(1):58–73. doi: 10.1002/jez.b.21124. [DOI] [PubMed] [Google Scholar]
- 45.Rosenzweig D, Smith D, Myler PJ, Olafson RW, Zilberstein D. Post-translational modification of cellular proteins during Leishmania donovani differentiation. Proteomics. 2008;8(9):1843–1850. doi: 10.1002/pmic.200701043. [DOI] [PubMed] [Google Scholar]
- 46.Spath GF, Lye LF, Segawa H, Sacks DL, Turco SJ, Beverley SM. Persistence without pathology in phosphoglycan-deficient Leishmania major. Science. 2003;301(5637):1241–1243. doi: 10.1126/science.1087499. [DOI] [PubMed] [Google Scholar]
- 47.Halle M, Gomez MA, Stuible M, Shimizu H, McMaster WR, Olivier M, Tremblay ML. The Leishmania surface protease GP63 cleaves multiple intracellular proteins and actively participates in p38 mitogen-activated protein kinase inactivation. J Biol Chem. 2009;284(11):6893–6908. doi: 10.1074/jbc.M805861200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Contreras I, Gomez MA, Nguyen O, Shio MT, McMaster RW, Olivier M. Leishmania-induced inactivation of the macrophage transcription factor AP-1 is mediated by the parasite metalloprotease GP63. PLoS Pathog. 2010;6(10) doi: 10.1371/journal.ppat.1001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silverman JM, Chan SK, Robinson DP, Dwyer DM, Nandan D, Foster LJ, Reiner NE. Proteomic analysis of the secretome of Leishmania donovani. Genome Biol. 2008;9(2):R35. doi: 10.1186/gb-2008-9-2-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Besteiro S, Williams RA, Coombs GH, Mottram JC. Protein turnover and differentiation in Leishmania. Int J Parasitol. 2007;37(10):1063–1075. doi: 10.1016/j.ijpara.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Michels PA, Bringaud F, Herman M, Hannaert V. Metabolic functions of glycosomes in trypanosomatids. Biochim Biophys Acta. 2006;1763(12):1463–1477. doi: 10.1016/j.bbamcr.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 52.Atayde VD, Shi H, Franklin JB, Carriero N, Notton T, Lye LF, Owens K, Beverley SM, Tschudi C, Ullu E. The structure and repertoire of small interfering RNAs in Leishmania (Viannia) braziliensis reveal diversification in the trypanosomatid RNAi pathway. Mol Microbiol. 2013;87(3):580–593. doi: 10.1111/mmi.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jackson AP. The evolution of amastin surface glycoproteins in trypanosomatid parasites. Mol Biol Evol. 2010;27(1):33–45. doi: 10.1093/molbev/msp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jecna L, Dostalova A, Wilson R, Seblova V, Chang KP, Bates PA, Volf P. The role of surface glycoconjugates in Leishmania midgut attachment examined by competitive binding assays and experimental development in sand flies. Parasitology. 2013;140(8):1026–1032. doi: 10.1017/S0031182013000358. [DOI] [PubMed] [Google Scholar]
- 55.Gomez MA, Contreras I, Halle M, Tremblay ML, McMaster RW, Olivier M. Leishmania GP63 alters host signaling through cleavage-activated protein tyrosine phosphatases. Sci Signal. 2009;2(90):ra58. doi: 10.1126/scisignal.2000213. [DOI] [PubMed] [Google Scholar]
- 56.Corradin S, Ransijn A, Corradin G, Roggero MA, Schmitz AA, Schneider P, Mauel J, Vergeres G. MARCKS-related protein (MRP) is a substrate for the Leishmania major surface protease leishmanolysin (gp63) J Biol Chem. 1999;274(36):25411–25418. doi: 10.1074/jbc.274.36.25411. [DOI] [PubMed] [Google Scholar]
- 57.Brittingham A, Morrison CJ, McMaster WR, McGwire BS, Chang KP, Mosser DM. Role of the Leishmania surface protease gp63 in complement fixation, cell adhesion, and resistance to complement-mediated lysis. J Immunol. 1995;155(6):3102–3111. [PubMed] [Google Scholar]
- 58.Victoir K, Dujardin JC, de Doncker S, Barker DC, Arevalo J, Hamers R, Le Ray D. Plasticity of gp63 gene organization in Leishmania (Viannia) braziliensis and Leishmania (Viannia) peruviana. Parasitology. 1995;111(Pt 3):265–273. doi: 10.1017/S0031182000081828. [DOI] [PubMed] [Google Scholar]
- 59.Steinkraus HB, Greer JM, Stephenson DC, Langer PJ. Sequence heterogeneity and polymorphic gene arrangements of the Leishmania guyanensis gp63 genes. Mol Biochem Parasitol. 1993;62(2):173–185. doi: 10.1016/0166-6851(93)90107-9. [DOI] [PubMed] [Google Scholar]
- 60.Kumar D, Rajanala K, Minocha N, Saha S. Histone H4 lysine 14 acetylation in Leishmania donovani is mediated by the MYST-family protein HAT4. Microbiology. 2012;158(Pt 2):328–337. doi: 10.1099/mic.0.050211-0. [DOI] [PubMed] [Google Scholar]
- 61.Buxbaum LU, Denise H, Coombs GH, Alexander J, Mottram JC, Scott P. Cysteine protease B of Leishmania mexicana inhibits host Th1 responses and protective immunity. J Immunol. 2003;171(7):3711–3717. doi: 10.4049/jimmunol.171.7.3711. [DOI] [PubMed] [Google Scholar]
- 62.Judice WA, Manfredi MA, Souza GP, Sansevero TM, Almeida PC, Shida CS, Gesteira TF, Juliano L, Westrop GD, Sanderson SJ, et al. Heparin modulates the endopeptidase activity of leishmania mexicana cysteine protease cathepsin L-Like rCPB2.8. PLoS One. 2013;8(11):e80153. doi: 10.1371/journal.pone.0080153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ono Y, Sorimachi H, Suzuki K. Structure and physiology of calpain, an enigmatic protease. Biochem Biophys Res Commun. 1998;245(2):289–294. doi: 10.1006/bbrc.1998.8085. [DOI] [PubMed] [Google Scholar]
- 64.Mottram JC, Coombs GH, Alexander J. Cysteine peptidases as virulence factors of Leishmania. Curr Opin Microbiol. 2004;7(4):375–381. doi: 10.1016/j.mib.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 65.Zhang WW, Matlashewski G. Characterization of the A2-A2rel gene cluster in Leishmania donovani: involvement of A2 in visceralization during infection. Mol Microbiol. 2001;39(4):935–948. doi: 10.1046/j.1365-2958.2001.02286.x. [DOI] [PubMed] [Google Scholar]
- 66.McCall LI, Zhang WW, Matlashewski G. Determinants for the development of visceral leishmaniasis disease. PLoS Pathog. 2013;9(1) doi: 10.1371/journal.ppat.1003053. [DOI] [PMC free article] [PubMed] [Google Scholar]