

Abstract
Background
Genetic aberrations in PAFAH1B1 result in isolated lissencephaly sequence (ILS), a neuronal migration disorder associated with severe mental retardation and intractable epilepsy. Approximately 60 % of patients with ILS show a 17p13.3 deletion or an intragenic variation of PAFAH1B1 that can be identified by fluorescence in situ hybridization (FISH) analysis or gene sequencing. Using multiplex ligation-dependent probe amplification (MLPA), 40–80 % of the remaining patients show small genomic deletions or duplications of PAFAH1B1. The intragenic duplications within PAFAH1B1 are predicted to abolish the PAFAH1B1 function, although a detailed characterization of the duplication regions have not been reported.
Results
Here we describe a female patient with ILS occurring predominantly in the posterior brain regions. MLPA was used to identify a small duplication within PAFAH1B1. This result was confirmed by array-based comparative genomic hybridization analysis, revealing a duplication of the 29-kb region encompassing putative regulatory elements and exon 2 of PAFAH1B1. The region was characterized as an intragenic tandem duplication by sequencing, revealing a 28-bp microhomology sequence at the breakpoint junctions. Parental genetic testing confirmed that the tandem duplication occurred de novo. Reverse transcription-PCR on RNA extracted from peripheral blood leukocytes revealed that the expression level of PAFAH1B1 decreased to that in a patient with Miller-Dieker syndrome, a contiguous gene-deletion disorder characterized by classical lissencephaly and a facial dysmorphism.
Conclusions
This study expanded the spectrum of PAFAH1B1 variants and identified a unique genomic architecture including microhomology sequences in PAFAH1B1 underlying an intragenic tandem duplication leading to ILS.
Keywords: Duplication, Lissencephaly, Microhomology, Neuronal migration, PAFAH1B1
Background
Classical lissencephaly is a brain malformation resulting from impaired neuronal migration. The condition is characterized by absent (agyria) or decreased (pachygyria) convolutions of the cerebral cortex, causing the brain surface to appear smooth. Lissencephaly occurs either as part of Miller–Dieker syndrome (MDS), a multiple malformation syndrome with severe grade lissencephaly and a characteristic facial dysmorphism, or as an isolated condition without peculiar facial features termed isolated lissencephaly sequence (ILS) [1]. Most cases of ILS are caused by variations in either the PAFAH1B1 gene on chromosome 17p13.3 or the DCX gene on chromosome Xp22.3 [2, 3]. PAFAH1B1 variations result in more severe abnormalities in the posterior brain regions, whereas DCX variations cause more severe abnormalities in the anterior brain regions [4, 5]. MDS is caused by a deletion of genes in chromosome 17p13.3, which at minimum includes the PAFAH1B1 and YWHAE genes [6].
The PAFAH1B1 gene (also known as LIS1) spans approximately 92-kb and contains 11 exons, the first of which is noncoding. The cDNA contains a 1230-bp-long coding sequence and encodes a 410-amino-acid polypeptide with a calculated molecular mass of 45 kDa. PAFAH1B1 participates in microtubule-dependent cell motility by controlling the function of the cytoplasmic dynein motor protein and plays an important role in neuronal migration during brain development [7].
Here, we describe a case of a Japanese patient with ILS who had an intragenic tandem duplication of the region encompassing the putative regulatory elements and exon 2 of the PAFAH1B1 gene. We characterized the boundary of the duplication region and found that each of the duplicated regions was flanked by an identical 28-bp sequence. The 28-bp microhomology potentially represents a molecular mechanism underlying the tandem duplication.
Case presentation
Case report
The patient, 4 years old at the time of this writing, is a Japanese female and the first child of nonconsanguineous parents. She was born at 40 weeks of gestation after an uneventful pregnancy with a birth weight of 2780 g, a length of 46 cm, and a head circumference of 32 cm (50th percentile). At the age of 2 months, she developed her first clonic seizures in the right arm. The tonic or clonic seizures also occurred in one or both limbs in the left side of her body. During the next 2 weeks, the polymorphous focal seizures became more frequent, occurring in clusters of 30–40 times a day. At the age of 3 months, she was referred to our hospital. Neurological examination revealed muscle hypotonia and a profound delay in psychomotor development. The patient had no eye contact and was not able to control her head. The hematological and chemical work-up was normal. Magnetic resonance imaging (MRI) of the brain demonstrated a thickened cortex and gyral malformations that were more severe in the posterior brain regions than in the anterior brain regions (Fig. 1). The patient did not show the characteristic facial appearance seen in patients with MDS. Therefore, on the basis of neuroradiological test results, she was diagnosed with ILS.
Fig. 1.
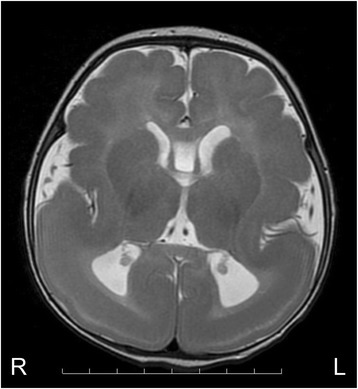
Brain MRI scan of a patient with an intragenic tandem duplication in the PAFAH1B1 gene. Axial T2-weighted brain image indicating the occurrence of predominant agyria in the posterior regions, and pachygyria in the anterior regions. The patient was diagnosed with grade 3 lissencephaly
The patient’s interictal electroencephalography (EEG) results indicated low-amplitude background activity over the posterior head regions and multifocal spikes. Ictal recordings revealed focal, rhythmic theta activities originating from the right or left parietal regions and eventually spreading to the ipsilateral hemisphere.
Valproic acid treatment was initiated but was unsuccessful in controlling her seizures. In the next 2 weeks, the spasms appeared in clusters, and her interictal EEG results showed hypsarrhythmia. The patient’s condition was found to evolve to West syndrome. Treatment with synthetic adrenocorticotropic hormone (ACTH) was started wherein a single daily dose of 0.0125 mg/kg was administrated for 14 consecutive days, with subsequent tapering. The patient’s spasms ceased by day 6 of the ACTH therapy but reappeared during the withdrawal stage. Her psychomotor development was severely delayed without head control, eye pursuit, or the use of meaningful words. Because of feeding problems resulting from difficulties in swallowing, she was fed via a gastrostomy tube.
Results
The patient exhibited an abnormal gyral pattern characterized by agyria in the posterior brain regions and pachygyria in the anterior brain regions. These characteristics are highly suggestive of PAFAH1B1 gene variations [4]. Fluorescence in situ hybridization (FISH) results for the 17p13.3 region and direct sequencing of the PAFAH1B1 gene revealed no abnormalities in this patient. Therefore, we analyzed the patient’s PAFAH1B1 gene via multiplex ligation-dependent probe amplification (MLPA) and identified a duplication of the region encompassing exon 2, but not exon 1 or 3 (Fig. 2). This result was confirmed by array-based comparative genomic hybridization (array CGH) analysis. The size of the duplication region was approximately 29 kb, spanning positions chr17:2,513,432 to 2,539,559 (GRCh37/hg19; Fig. 3a). To define the boundary of the duplication region, long-range polymerase chain reaction (PCR) was performed based on the array CGH and MLPA results. PCR performed using a forward primer in intron 2 and a reverse primer in intron 1 resulted in the amplification of a 1569-bp PCR product from the patient’s genomic DNA, but not from her parents’ genomic DNA (Fig. 3b), confirming that the duplication was intragenic and in tandem. These results further indicated that the duplication occurred de novo in the patient. Sequencing analysis of the PCR product revealed that each of the duplicated regions was flanked by an identical 28-bp sequence, indicating that the 28-bp microhomology was used to generate the tandem duplication (Fig. 3c).
Fig. 2.
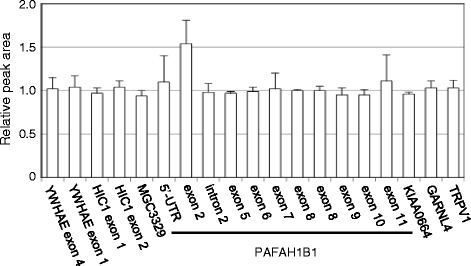
MLPA analysis performed on DNA from the patient revealed a small duplication in PAFAH1B1 that included exon 2. Results indicate the relative peak area of a probe target sequence with normalization against normal female samples and are shown as the means ± SD (n = 3)
Fig. 3.
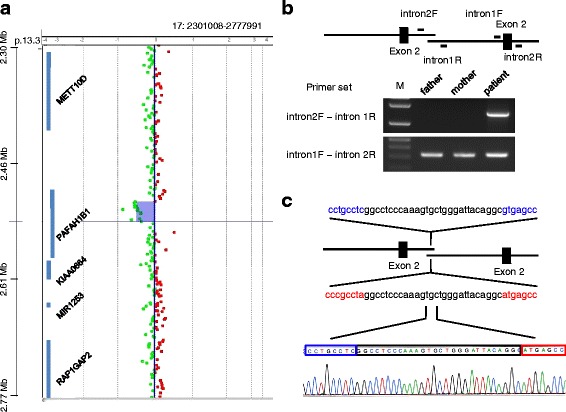
Intragenic tandem duplication in the PAFAH1B1 gene in a patient with isolated lissencephaly sequence. a Array-CGH results showing the log2 intensity ratios of the patient versus reference DNA. A 29-kb duplication (purple-shaded region with green dots) was detected within the PAFAH1B1 gene on chromosome 17p13.3. b Schematic representation of the duplicated region with the primer-binding sites. Long-range PCR was performed with the intron2F and intron1R primers, which yielded a 1569-bp PCR product from the patient’s genomic DNA, but not from her parents’ genomic DNA. These results indicated that the tandem duplication occurred de novo. c Sequencing analysis of the PCR product revealed region of identical sequences (28 nucleotides enclosed by black line) at the duplication junctions without additional sequence changes
Reverse transcription (RT)-PCR amplification with a forward primer in exon 1 and a reverse primer in exon 3 yielded two fragments with different lengths of 314-bp and 536-bp from the RNA of the patient, but only a 314-bp fragment from the control (Fig. 4a). Sequencing analysis of the novel 536-bp fragment confirmed that the transcript contained a tandem duplication of exon 2 (Fig. 4b). The PAFAH1B1 variant showed low-level expression (Fig. 4a). The expression level of the normal PAFAH1B1 transcript in the patient decreased to that in a patient with MDS (Fig. 4a).
Fig. 4.
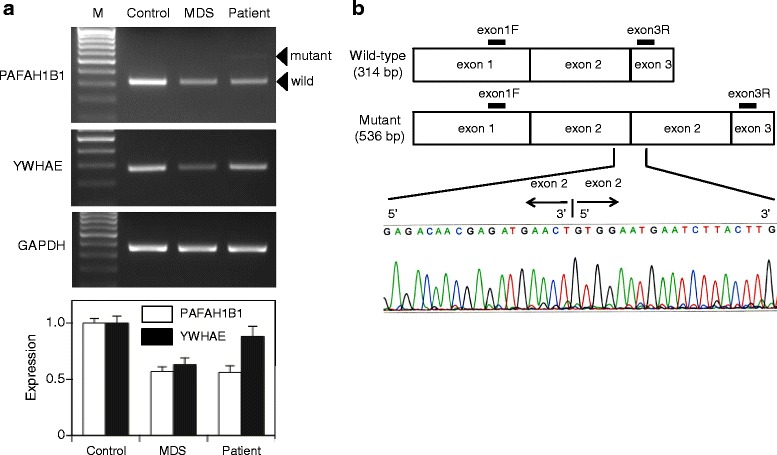
Decreased expression of PAFAH1B1 in a patient with isolated lissencephaly sequence (ILS). a RT-PCR on RNA extracted from peripheral blood leukocytes revealed that the expression levels of PAFAH1B1, but not YWHAE, were decreased in this patient, while the expression levels of both the genes were decreased in a patient with Miller-Dieker syndrome (MDS). Note that a longer PAFAH1B1 mRNA (536 bp) was present in the patient with ILS at a low level along with the normal mRNA (314 bp). The quantitative results in the bar graph indicate PAFAH1B1 and YWHAE mRNA expression levels, relative to those of controls, and are shown as the mean ± SD (n = 3). Data are expressed in arbitrary units. b Sequencing analysis of the longer PAFAH1B1 transcript found in the patient confirmed the tandem duplication of exon 2
Discussion
Previous studies using MLPA have identified 30 deletions and 7 duplications within the PAFAH1B1 gene among 57 patients with ILS occurring predominantly in the posterior brain regions, in whom FISH for 17p13.3 and gene sequencing gave negative results [8, 9]. These intragenic duplications are predicted to abolish the PAFAH1B1 function, although a detailed characterization of the duplication regions has not been reported. Here, we report our findings for a patient with posteriorly dominant ILS who had an intragenic tandem duplication in the PAFAH1B1 gene. The PAFAH1B1 expression level decreased to that in a patient with MDS. Analysis of the duplication boundary revealed a microhomology at the breakpoint, which suggested that the duplication might have been mediated in part by the microhomology sequence.
A recent report described the presence of microhomology sequences ranging from 2 to 75-bp in 79 % of the breakpoint junctions of rare pathogenic copy number variations (CNVs) [10]. These results indicate that rare pathogenic CNVs are caused by a replication-based mechanism of mitotic origin mediated by microhomology sequences in the context of local genomic architectures. Our investigation of the breakpoint region of the intragenic tandem duplication revealed a 28-bp microhomology sequence in introns 1 and 2 of the PAFAH1B1 gene. In addition, the observation of no additional sequence changes at the junction indicates that the duplication resulted from a single replication-based event. Genetic testing of the patient’s parents confirmed that the tandem duplication variant occurred de novo. Thus, a single event of microhomology-mediated rearrangement might be the potential molecular mechanism whereby the tandem duplication variant occurred.
A gradient of severity in lissencephaly can aid the planning of appropriate genetic testing. ILS occurring predominantly in the posterior brain regions is most frequently associated with PAFAH1B1 variants, but rarely with variants of the TUBA1A gene encoding tubulin alpha-1a [11]. We did not analyze the TUBA1A gene because the patient showed no additional malformations, such as cerebellar hypoplasia or the absence of the corpus callosum, which have been found in patients with TUBA1A-associated lissencephaly [11]. The duplication region identified in this patient includes putative noncoding regulatory elements of PAFAH1B1 and its second exon, which harbors the ATG translational start site. The mutant mRNA containing the tandem exon 2 duplication showed low-level expression. The 5′-untranslated region (5′-UTR) contains a variety of cis-regulatory elements which influences the expression of the downstream genes. Hence, this duplication potentially affected PAFAH1B1 expression through destabilizing the secondary structure of the 5′-UTR. A balanced chromosomal translocation that disrupted the 5′-UTR of the PAFAH1B1 gene has been found in a patient with lissencephaly [12]. The heterozygous deletion of YWHAE, together with PAFAH1B1 in patients with MDS, is known to increase the severity, usually with grade 1 lissencephaly (diffuse agyria) being observed [6]. The patient in this study showed decreased expression of PAFAH1B1, but not YWHAE, resulting in grade 3 lissencephaly (mixed agyria and pachygyria).
Conclusions
Our findings expanded the spectrum of PAFAH1B1 variants implicating in ILS. We observed a heterozygous intragenic tandem duplication involving the putative noncoding regulatory elements and exon 2 of PAFAH1B1, which resulted in decreased expression of this gene in a patient with ILS. We further found a unique genomic architecture including microhomology sequences in introns 1 and 2 of the PAFAH1B1 gene underlying an intragenic tandem duplication leading to ILS.
Methods
Mutation screening
This study was performed in compliance with the guidelines of Asahikawa Medical University. After obtaining written informed consent from her parents, genomic DNA was extracted from the peripheral blood leukocytes of both the patient and her parents, and was used for variant screening. Appropriate primers were used to yield DNA fragments spanning the entire coding region and intron–exon boundaries of PAFAH1B1 [5]. The PCR fragments were analyzed by automated sequencing. Gene dosage analysis was performed using the SALSA MLPA Kit P061-B1 Lissencephaly (MRC-Holland, Amsterdam, The Netherlands). To define the boundary of the duplicated region, array CGH analysis was performed using a high-resolution 1 M array (Agilent Technologies Inc.; Santa Clara, CA).
Long-range PCR amplification and DNA sequencing analysis
Long-range PCR was performed using TaKaRa LA Taq Polymerase (TaKaRa Bio Inc.; Otsu, Japan). Primers were designed for amplification and sequencing of the duplication junction within PAFAH1B1 including a forward primer in intron 2 (intron2F; 5′-GTGTGGAAGACACTTAGTGGC-3′) and a reverse primer in intron 1 (intron1R; 5′-GGCCTACATCCTGACTTGAC-3′). To verify the DNA quality for PCR amplification, the following primers for amplification of PAFAH1B1 exon 2 were also used: a forward primer in intron 1 (intron1F; 5′- TGTGGAAGACACTTAGTGGCA-3′) and a reverse primer in intron 2 (intron2R; 5′- AAGAGACCTCCCAAAGCTGTA-3′), which generated a 269-bp product [5]. The obtained PCR products were directly sequenced on an ABI 3130 Genetic Analyzer (Applied Biosystems; Foster City, CA).
RNA isolation and RT-PCR
Total RNA was extracted from the peripheral blood leukocytes using the PAXgene Blood RNA Kit (Qiagen GmbH; Hilden, Germany). The RT step was performed using the SuperScript First-Strand Synthesis System (Invitrogen Corporation; Carlsbad, CA) to generate cDNA by using 1 μg of total RNA in a 20-μl reaction. The following primers were used: for PAFAH1B1, 5′-GAGTGAAGGACGGAAGAGGC-3′ and 5′-GAATATGCCTCTTCATAGCC-3′, generating a 314-bp product; for YWHAE, 5′-ACGACGAAATGGTGGAGT-3′ and 5′-AGCTGCTGGAATGAGGTG-3′, generating a 278-bp product; and for GAPDH, 5′-CCAGCCGAGCCACATCGCTC-3′ and 5′-ATGAGCCCCAGCCTTCTCCAT-3′, generating a 360-bp product. The PCR products were visualized by ethidium bromide staining, following electrophoresis on 2 % agarose gels. The optical densities of the bands were quantified using an image analysis system and ImageJ software (National Institutes of Health; Bethesda, MD). Selected RT-PCR products were subsequently cloned into the pCR4-TOPO vector (Invitrogen Corporation; Carlsbad, CA) for sequencing.
Consent
Written informed consent was obtained from the parents of the patients for publication of this case report. A copy of the written consent is avaible by the Editor-in –chief of this journal.
Acknowledgements
We sincerely thank the members of the family described here, whose help and participation made this work possible.
Abbreviations
- ACTH
adrenocorticotropic hormone
- CGH
comparative genomic hybridization
- CNV
copy number variation
- DCX
doublecortin
- EEG
electroencephalography
- FISH
fluorescence in situ hybridization
- ILS
isolated lissencephaly sequence
- MDS
Miller–Dieker syndrome
- MLPA
multiplex ligation-dependent probe amplification
- MRI
magnetic resonance imaging
- PAFAH1B1
platelet-activating factor acetylhydrolase 1b, regulatory subunit 1
- PCR
polymerase chain reaction
- RT
reverse transcription
- TUBA1A
tubulin alpha-1a
- UTR
untranslated region
- YWHAE
tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, epsilon
Footnotes
Competing interests
None of the authors declare any conflict of interest related to this study.
Authors’ contributions
ST carried out most of the molecular genetic studies and drafted the manuscript. RT, SO, AO and NS were responsible for the medical treatment of the patient and contributed to the clinical description. HA helped designing the study and drafting the final version of the manuscript. All authors read and approved the final manuscript.
References
- 1.Dobyns WB, Elias ER, Newlin AC, Pagon RA, Ledbetter DH. Causal heterogeneity in isolated lissencephaly. Neurology. 1992;42:1375–88. doi: 10.1212/WNL.42.7.1375. [DOI] [PubMed] [Google Scholar]
- 2.Lo Nigro C, Chong CS, Smith AC, Dobyns WB, Carrozzo R, Ledbetter DH. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet. 1997;6:157–64. doi: 10.1093/hmg/6.2.157. [DOI] [PubMed] [Google Scholar]
- 3.Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/S0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 4.Dobyns WB, Truwit CL, Ross ME, Matsumoto N, Pilz DT, Ledbetter DH, et al. Differences in the gyral pattern distinguish chromosome 17-linked and X-linked lissencephaly. Neurology. 1999;53:270–7. doi: 10.1212/WNL.53.2.270. [DOI] [PubMed] [Google Scholar]
- 5.Pilz DT, Matsumoto N, Minnerath S, Mills P, Gleeson JG, Allen KM, et al. LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Hum Mol Genet. 1998;7:2029–37. doi: 10.1093/hmg/7.13.2029. [DOI] [PubMed] [Google Scholar]
- 6.Cardoso C, Leventer RJ, Ward HL, Toyo-Oka K, Chung J, Gross A, et al. Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am J Hum Genet. 2003;72:918–30. doi: 10.1086/374320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vallee RB, Tsai JW. The cellular roles of the lissencephaly gene LIS1, and what they tell us about brain development. Genes Dev. 2006;20:1384–93. doi: 10.1101/gad.1417206. [DOI] [PubMed] [Google Scholar]
- 8.Mei D, Lewis R, Parrini E, Lazarou LP, Marini C, Pilz DT, et al. High frequency of genomic deletions--and a duplication--in the LIS1 gene in lissencephaly: implications for molecular diagnosis. J Med Genet. 2008;45:355–61. doi: 10.1136/jmg.2007.056507. [DOI] [PubMed] [Google Scholar]
- 9.Haverfield EV, Whited AJ, Petras KS, Dobyns WB, Das S. Intragenic deletions and duplications of the LIS1 and DCX genes: a major disease-causing mechanism in lissencephaly and subcortical band heterotopia. Eur J Hum Genet. 2009;17:911–8. doi: 10.1038/ejhg.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vissers LE, Bhatt SS, Janssen IM, Xia Z, Lalani SR, Pfundt R, et al. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum Mol Genet. 2009;18:3579–93. doi: 10.1093/hmg/ddp306. [DOI] [PubMed] [Google Scholar]
- 11.Poirier K, Keays DA, Francis F, Saillour Y, Bahi N, Manouvrier S, et al. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A) Hum Mutat. 2007;28:1055–64. doi: 10.1002/humu.20572. [DOI] [PubMed] [Google Scholar]
- 12.Kurahashi H, Sakamoto M, Ono J, Honda A, Okada S, Nakamura Y. Molecular cloning of the chromosomal breakpoint in the LIS1 gene of a patient with isolated lissencephaly and balanced t(8;17) Hum Genet. 1998;103:189–92. doi: 10.1007/s004390050805. [DOI] [PubMed] [Google Scholar]