

Abstract
Emerging evidence suggests a role for sphingosine-1-phosphate (S1P) in various aspects of rheumatoid arthritis (RA) pathogenesis. In this study we compared the effect of chemical hypoxia induced by cobalt chloride (CoCl2) on the expression of S1P metabolic enzymes and cytokine/chemokine secretion in normal fibroblast-like synoviocytes (FLS) and RAFLS. RAFLS incubated with CoCl2, but not S1P, produced less IL-8 and MCP-1 than normal FLS. Furthermore, incubation with the S1P2 and S1P3 receptor antagonists, JTE-013 and CAY10444, reduced CoCl2-mediated chemokine production in normal FLS but not in RAFLS. RAFLS showed lower levels of intracellular S1P and enhanced mRNA expression of S1P phosphatase 1 (SGPP1) and S1P lyase (SPL), the enzymes that are involved in intracellular S1P degradation, when compared to normal FLS. Incubation with CoCl2 decreased SGPP1 mRNA and protein and SPL mRNA as well. Inhibition of SPL enhanced CoCl2-mediated cytokine/chemokine release and restored autocrine activation of S1P2 and S1P3 receptors in RAFLS. The results suggest that the sphingolipid pathway regulating the intracellular levels of S1P is dysregulated in RAFLS and has a significant impact on cell autocrine activation by S1P. Altered sphingolipid metabolism in FLS from patients with advanced RA raises the issue of synovial cell burnout due to chronic inflammation.
1. Introduction
Rheumatoid arthritis (RA) is a chronic systemic disorder that causes destruction of joints through inflammation and proliferation of the synovial membrane [1, 2]. In RA, the synovial tissue lining the joints becomes inflamed. In comparison with the normal synovial membrane, which is normally 1-2 cell layers thick, RA synovial tissue is hypertrophic and invaded by an excess of various leukocytes including neutrophils, T cells, macrophages, and monocytes [3]. This recruitment of leukocytes is likely to be mediated by selective chemotactic factors, such as interleukin-8 (IL-8) that recruits neutrophils and T cells, and monocyte chemotactic protein-1 (MCP-1) that recruits monocytes, into the synovium [4, 5]. A role for IL-8 [6, 7] and MCP-1 [8, 9] in these processes has been highlighted. The synthesis of chemokines in RA may be dependent, at least in part, on the production of inflammatory cytokines, such as IL-1β and tumor necrosis factor-α (TNF-α) [4], by the hypertrophic synovium and activated leukocytes. The complex cascade of production of chemokines, cytokines, and tissue-remodelling enzymes associated with leukocyte recruitment plays a role in synovial cell proliferation and joint erosion in RA [1, 2, 10]. Eventually, the thickened synovial membrane decreases capillary density and the oxygen tension in the joint [11–13]. Severe reduction of mean oxygen pressure in the RA synovium compared to that of healthy joints correlates with severity of inflammation [14–16]. The hypoxic RA joint environment in turn affects a host of genes involved in angiogenesis, apoptosis, cellular metabolism, matrix degradation, and inflammation [17]. Hypoxia drives vascular endothelial growth factor (VEGF) expression leading to angiogenesis [18–20]. The expression of cyclooxygenase-2 (COX-2) [21], matrix metalloproteinases (MMPs) [22], stromal cell-derived factor 1 [23], IL-6 and IL-8 [22, 24], and migration [25, 26] and proliferation of synovial fibroblasts as well [27], are exacerbated in response to hypoxia.
Sphingosine-1-phosphate (S1P) is a bioactive sphingolipid implicated in various pathological processes through binding to and activation of five G protein-coupled receptors designated as S1P1–5 [28]. Intracellular S1P is transported outside cells and gains access to cognate receptors for autocrine or paracrine signalling [28, 29]. The steady state level of intracellular S1P is regulated through synthesis by two sphingosine kinases (SphK1 and SphK2) and degradation either via dephosphorylation by S1P phosphatases (SGPP1 and SGPP2) or irreversible cleavage by S1P lyase (SPL) [30]. Moreover, S1P exported outside cells is dephosphorylated back to sphingosine by lipid phosphate phosphatases (LPPs), thereby attenuating its effects on the activation of surface receptors [31]. Alteration in the enzymes involved in S1P synthesis and catabolism may mediate many pathological states including arthritis (reviewed in [28, 32]).
Fibroblast-like synoviocytes from RA patients (RAFLS) express S1P1, S1P2, and S1P3 receptors [33]. RAFLS stimulation with S1P promotes the synthesis of cytokines/chemokines, COX-2 expression and release of prostaglandin E2 (PGE2), and cell migration, proliferation, and survival as well [33, 34]. SphK activation and high S1P levels have been reported in the synovium and synovial fluids of patients with RA [34–36]. Studies suggest a role for S1P in the pathophysiology of RA since SphK1 deficiency and blockade of S1P1 receptors attenuate collagen-induced arthritis in mice [37, 38]. Though SphKs can be activated by TNF-α and IL-1β to generate S1P, new evidence suggests a potential link between S1P and hypoxia in cancer and cardiovascular diseases [39, 40]. In this study we evaluated the impact of chemical hypoxia induced by CoCl2 on chemokine synthesis by normal FLS and RAFLS. We report that the blockade of S1P2 or S1P3 receptors attenuates CoCl2-mediated IL-8 and MCP-1 secretion in normal FLS but not in RAFLS. Furthermore, we provide evidence that low levels of intracellular S1P in RAFLS attenuate the S1P2 and S1P3 receptor-dependent synthesis of chemokines under conditions of chemical hypoxia.
2. Materials and Methods
2.1. Reagents
Cobalt chloride (CoCl2) was from Sigma Aldrich (Oakville, ON, Canada). S1P was purchased from Biomol (Plymouth Meeting, PA, USA). Human IL-8 and MCP-1 ELISA (Enzyme-Linked Immunosorbent Assay) kits were purchased from BioSource International Inc. (Camarillo, CA, USA) and R&D Systems (Minneapolis, MN, USA), respectively. The S1P2 and S1P3 receptor antagonists (JTE-013 and CAY10444) were from Cayman Chemical (Ann Arbor, MI, USA). The S1P assay kit was from Echelon Biosciences (Salt Lake City, UT, USA). SYBR Green JumpStart Ready Mix kits were obtained from Sigma (Oakville, ON, Canada). TRIzol reagent and Superscript II were purchased from Life Technologies (Burlington, ON, Canada). Anti-SGPP1 and SPL antibodies were from Novus Biologicals (Oakville, ON, Canada) and R&D Systems (Minneapolis, MN, USA), respectively. Anti-PI3 kinase p85 (06-195) was purchased from Upstate Biotechnology Associates (Billerica, MA, USA). The Proteome Profiler Human Cytokine Array (panel A) was bought from R&D Systems (Minneapolis, MN, USA). Cell culture reagents were from Wisent Inc. (St-Bruno, QC, Canada).
2.2. Synthesis of SPL Inhibitor
Starting chemicals and solvents were purchased from Sigma Aldrich (Oakville, ON, Canada) and Alfa Aesar (Ward Hill, MA, USA). A Biotage initiator system was used for microwave heating. Nuclear magnetic resonance (NMR) spectra were collected on a Bruker Avance III 400 MHz spectrometer with chemical shifts referenced to residual solvent peaks as secondary reference for 1H and 13C spectra. Crude products were purified using a Sg100c (Teledyne Isco) flash chromatographic instrument.
Compounds SM4 (SPL inhibitor) and SM3 (the inactive enantiomer) (Figure 1) were prepared as previously described [41] and as shown in Scheme 1. Briefly, the substitution of the chlorine of the commercially available 1-benzyl-4-chlorophthalazine (1) with (R)-methylpiperazine or (S)-methylpiperazine followed by a second substitution of the chlorine of 6-chloronicotinonitrile with compound 2 or 3 gives us the desired compounds SM4 and SM3. The 1H NMR of compounds 2, 3, SM4, and SM3 were identical to those reported previously [41].
Figure 1.
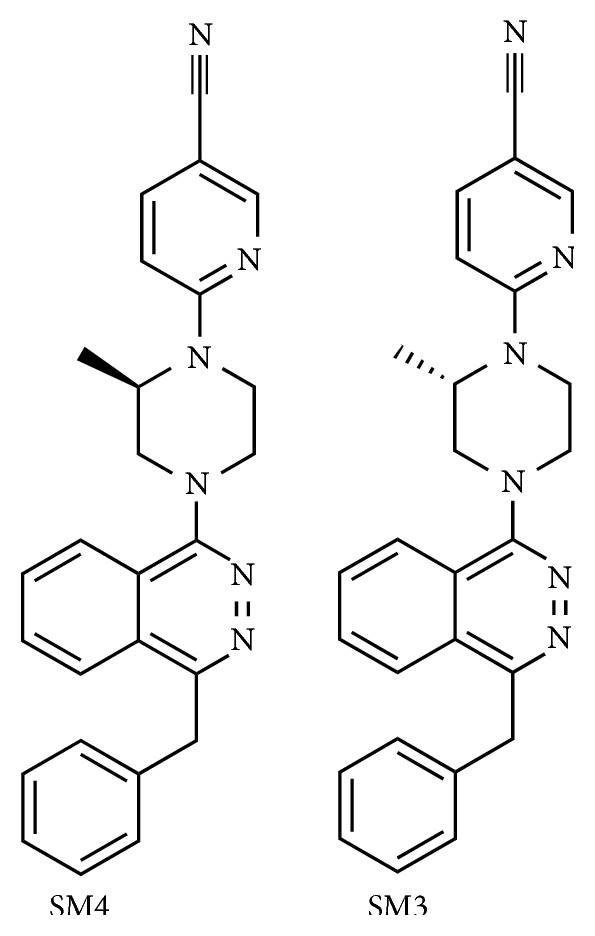
Structures of SM4 and SM3.
Scheme 1.

Reagents and conditions: (i) (2): Na2CO3, (R)-methylpiperazine, dioxane, 100°C, 48 h, 91%; (3): Na2CO3, (S)-methylpiperazine, dioxane, 100°C, 48 h, 88%; (ii): Na2CO3, 6-chloronicotinonitrile, DMF/dioxane, 180°C, microwave (SM4: 11%, SM3: 16%).
2.3. Cell Treatment and Viability
Human primary FLS were isolated from articular synovia of donors with RA (RAFLS) or without history of arthritis (normal FLS). Patients from whom synovial specimens were obtained were diagnosed based on the criteria developed by the American College of Rheumatology Diagnostic Subcommittee for RA [42] and underwent arthroplasty. FLS were isolated by sequential enzymatic digestion as described previously [43]. Briefly, FLS were released by sequential enzymatic digestion with 1 mg/mL pronase for 1 h, followed by 6 h with 2 mg/mL collagenase at 37°C in DMEM supplemented with 10% FBS, 1% sodium pyruvate, 100 U/mL penicillin, and 100 mg/mL streptomycin. Released cells were incubated for 1 h at 37°C in tissue culture flasks allowing the adherence of nonfibroblastic cells possibly present in the synovial preparation. The cells were cultured in DMEM supplemented with 10% FBS and antibiotics at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Semiconfluent cells were starved with serum-free medium for 24 h before treatment. At the moment of cell treatment, the culture medium was replaced with fresh serum-free medium containing various concentrations of the tested compounds as indicated below. Cells were used between passages 3 and 9. Propidium iodide (PI) was used to evaluate the viability of RAFLS by flow cytometry. Cells were detached using Accutase cell detachment solution and incubated with PI (5 mg/mL). PI negative RAFLS were considered viable.
2.4. IL-8 and MCP-1 ELISA
FLS (5 × 104 cells/well) were plated in 24-well plates and serum starved for 24 h prior to stimulation with 200 μM CoCl2 or 5 μM S1P for an additional 24 h. Where indicated, cells were pretreated for 30 min with 5 μM of the selective S1P2 receptor antagonist JTE-013 and/or selective S1P3 receptor antagonist CAY10444, prior to stimulation with CoCl2 or S1P. To evaluate the effect of SPL inhibition on CoCl2-mediated chemokine secretion, cells were treated with the SPL inhibitor SM4 (or the inactive enantiomer SM3) for 24 h in the absence or the presence of CoCl2 and/or sphingosine. Cell culture supernatants were collected and stored at −80°C until the ELISAs were performed. IL-8 and MCP-1 in all samples were monitored in triplicate, according to the manufacturer's protocol. Optical densities were determined using a SoftMaxPro40 plate reader at 450 nm. The results were compared with a standard curve that was generated using known concentrations (pg/mL) of the chemokines. The detection limit of IL-8 and MCP-1 ELISA was 12.5 pg/mL and 15.625 pg/mL, respectively. Data are expressed either as pg/mL or as the percentage of chemokines secreted relative to the appropriate controls.
2.5. Quantitative Real-Time PCR
FLS (5 × 105 cells) were plated in 6-well plates and serum starved for 24 h prior to stimulation with or without 200 μM CoCl2 in serum-free medium for various times. Total cellular RNA was extracted using TRIzol reagent according to the manufacturer's instructions. RNA (1 μg) was reverse-transcribed using random priming and the Superscript II Reverse Transcriptase system. Real-time PCR was performed to assess the expression of SGPP1, SGPP2, and SPL and their regulation by CoCl2. The following sets of primers were used: SGPP1 forward (5′-GCCGCTGGCAGTACCCT-3′) and reverse (5′-AATAGAGTGCATTCCCATGTAAATTCT-3′); SGPP2 forward (5′-TTCAGAACATCCCACCACTCACCA-3′) and reverse (5′-TTCCTGGTGACCACCTTGAACCAT-3′); and SPL forward (5′-GCCAGAGAGTTTATGGTCAAGGTT-3′) and reverse (5′-CAACTTGTCTTGAATCTTACGACCAA-3′). The ribosomal protein RPLP0 mRNA was used as an internal PCR control. RPLP0 primer sequences were as follows: forward (5′-GTTGTAGATGCTGCC-ATTG-3′) and reverse (5′-CCATGTGAAGTCACTGTGC-3′). Amplicon expression in each sample was normalized to its RPLP0 content. The thermal cycling conditions were as follows: 95°C (initial denaturation, 3 min) followed by 40 cycles of 95°C (denaturation, 15 sec), 54°C (annealing, 20 sec), and 72°C (extension, 20 sec).
2.6. Western Blot
Cells were exposed to 200 μM CoCl2 for various times (0–48 h) and lysed in boiling sample buffer [50 mM Tris/HCL (pH 6.8), 10% (v/v) glycerol, 50 mM DTT, and 4% (v/v) SDS] for 7–10 min. Equal amounts of protein were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred to methanol-soaked Immobilon PVDF membranes (Millipore Corporation, Bedford, MA, USA). Primary antibody incubation was performed either overnight at 4°C (anti-SGPPL, SPL) or 1 h at 37°C (anti-PI3 kinase p85). The membranes were then washed three times and incubated with appropriate horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 h. Membranes were washed three times and antibody-antigen complexes were revealed using Western Lightening ECL+ according to the manufacturer's instructions (Perkin Elmer Life Sciences, Woodbridge, ON, Canada).
2.7. S1P ELISA
FLS from 2 normal and 2 RA donors were cultured up to 80–85% confluence in 75 cm2 flasks and serum starved for 24 h. Cells were lysed in 400 μL of lysis buffer provided with the S1P ELISA kit. Protein concentration was measured by the BCA method and S1P in cell lysates (1 : 10 in delipidated human serum) was monitored according to the manufacturer's instructions.
2.8. Cytokine/Chemokine Profiling Analysis
RAFLS were treated with the SPL inhibitor SM4 for 24 h in the absence/presence of CoCl2 and sphingosine. Cell culture supernatants were collected and stored at −80°C until the Proteome Profiler Human Cytokine Array (panel A) was performed.
2.9. Statistical Analysis
Unless otherwise stated, experiments were performed three times for each donor and results presented are expressed as mean ± SE or as representative studies. All statistical analyses were performed using Prism 4.0 software. Statistical significance of the difference between samples of two different treatments was determined by t-test (two-tailed p value). For multiple comparisons, statistical significance was determined by one-way ANOVA, Dunnett's multiple comparison test. p values less than 0.05 were considered statistically significant.
3. Results
3.1. Chemokine Secretion by Normal FLS and RAFLS in Response to Hypoxic Stress
To mimic hypoxia, FLS were incubated with CoCl2, a chemical inducer of hypoxia-inducible factor-1 (HIF-1) [44]. The effect of chemical hypoxia on chemokine synthesis was assessed using ELISA assays and CoCl2-dependent secretion of IL-8 and MCP-1 by normal FLS and RAFLS was compared (Figure 2). Small amounts of IL-8 (<3 pg/mL) (Figure 2(a)) and MCP-1 (<35 pg/mL) (Figure 2(b)) were produced by both normal FLS and RAFLS cultured under normoxic conditions. When incubated with CoCl2, normal FLS released significantly larger amounts of IL-8 (644.3 ± 125.9 pg/mL) and MCP-1 (1092 ± 138.6) than RAFLS with similar passage number (125.7 ± 26.5 pg/mL for IL-8 and 195.3 ± 31.9 for MCP-1) (p < 0.001). In both control FLS and RAFLS there was a similar trend of decreased synthesis of IL-8 and MCP-1 in response to CoCl2 with increased number of cell passages (data not show).
Figure 2.

Effect of CoCl2 stimulation on IL-8 and MCP-1 secretion in normal FLS and RAFLS. Human primary FLS from normal (n = 4) and RA (n = 4) donors were incubated with 200 μM CoCl2. The amounts of IL-8 (a) and MCP-1 (b) released in the supernatants were monitored 24 h after stimulation. The data are the means ± SE from four experiments (4 different donors) performed in triplicate (3 independent experiments). For statistical comparative analyses, we compared RA to normal FLS treated with CoCl2. ∗∗∗ p < 0.001.
3.2. S1P Receptor(s) Dependency of Chemokine Secretion in Normal FLS and RAFLS
S1P regulates a variety of cellular processes through binding to G protein-coupled receptors [45]. We previously reported a role for S1P2 and S1P3 in S1P-mediated IL-8 secretion in RAFLS [33]. As expected, the addition of S1P to normal FLS and RAFLS stimulated the secretion of IL-8 and MCP-1. The amounts of IL-8 and MCP-1 released by normal FLS and RAFLS in response to S1P were not statistically different (149.0 ± 28.62 versus 126.9 ± 14.3 pg/mL for IL-8 (p = 0.47) and 800.3 ± 116.4 pg/mL versus 546.5 ± 69.42 pg/mL for MCP-1 (p = 0.10)). Under these conditions the S1P3 antagonist CAY10444 and the S1P2 antagonist JTE-013 significantly decreased S1P-induced IL-8 by 51.3 ± 5.0% (p < 0.01) and 80.1 ± 5.4% (p < 0.01) in normal FLS and by 63.9 ± 7.8% (p < 0.01) and 93.3 ± 0.6% (p < 0.01) in RAFLS, respectively (Figure 3(a)). CAY10444 and JTE-013 also reduced S1P-mediated MCP-1 secretion by 46.7 ± 8.9% (p < 0.001) and 80.3 ± 2.7% (p < 0.001) in normal FLS and that of RAFLS by 46.4 ± 4.5% (p < 0.001) and 89.6 ± 1.6% (p < 0.001), respectively (Figure 3(b)). Similarly, the incubation in normal FLS with CAY10444 and JTE-013 in combination with CoCl2 reduced IL-8 secretion by 59.0 ± 6.8% (p < 0.001) and 22.0 ± 7.5% (p < 0.01) and that of MCP-1 by 77.6 ± 4.2% (p < 0.001) and 66.4 ± 5.0% (p < 0.001), respectively (Figures 3(c) and 3(d)). In contrast, the production of chemokines by RAFLS incubated with CoCl2 was not inhibited by the S1P3 or the S1P2 receptor antagonist (Figures 3(c) and 3(d)). The percentage of PI positive cells treated with 200 μM CoCl2 together with 5 μM CAY10444 and 5 μM JTE-013 for 24 h was identical to that of untreated cells (1.25 ± 0.15% versus 1.4 ± 0.3% for normal FLS treated with CoCl2/CAY10444 versus untreated, 1.15 ± 0.15% versus 1.4 ± 0.3% for normal FLS treated with CoCl2/JTE-013 versus untreated; 1.2 ± 0% versus 1.0 ± 0.1% for RAFLS treated with CoCl2/CAY10444 versus untreated, and 1.15 ± 0.25% versus 1.0 ± 0.1% for RAFLS treated with CoCl2/JTE-013 versus untreated), indicating that inhibition of chemokine synthesis was not mediated by a cytotoxic effect of these compounds.
Figure 3.

Differential involvement of S1P receptors in S1P- and CoCl2-mediated chemokine secretion by normal FLS and RAFLS. Human primary FLS from normal (n = 4) and RA (n = 4) donors were incubated with 5 μM S1P (a, b) or 200 μM CoCl2 (c, d). Where indicated, cells were pretreated with S1P3 antagonist CAY10444 (5 μM) or S1P2 antagonist JTE-013 (5 μM) for 30 min before stimulation with S1P or CoCl2. The amounts of chemokines released in the supernatants were monitored after 24 h. Data are expressed as percentage of chemokine production induced by S1P (a, b) or CoCl2 (c, d). The data are the means ± SE from four experiments (4 different donors) performed in triplicate (3 independent experiments). For statistical comparative analyses, the samples stimulated with S1P (a, b) or CoCl2 (c, d) were compared to those stimulated in the presence of CAY10444 or JTE-013, respectively. ∗∗ p < 0.01; ∗∗∗ p < 0.001.
3.3. Intracellular Levels of S1P in Normal FLS and RAFLS
The response of normal FLS and RAFLS to exogenously added S1P and inhibition of chemokine secretion by the S1P3 and S1P2 receptor antagonists provide evidence for functional S1P receptors in both types of FLS. On the other hand, inhibition of CoCl2-dependent chemokine synthesis by the S1P antagonists in normal FLS but not in RAFLS points toward alteration of an autocrine positive feedback loop driven by S1P. This could be due to impaired steady levels of intracellular S1P and/or export outside cells. To gain insight into the possible mechanisms we monitored the intracellular levels of S1P in normal FLS and in RAFLS. As shown in Table 1 the basal level of intracellular S1P was more elevated in normal FLS as compared to RAFLS.
Table 1.
S1P content in normal FLS and RAFLS.
| S1P content (pmol/mg of protein) | |
|---|---|
| Normal FLS | |
| Donor #1 (S3618) | 64.5 ± 1.5 |
| Donor #2 (S3739) | 273.0 ± 21 |
| RAFLS | |
| Donor #1 (37158A1-S) | 19.5 ± 1.5 |
| Donor #2 (87546A1-S) | 22.5 ± 1.5 |
Cell lysates from human primary FLS of normal (n = 2) and RA (n = 2) donors were prepared. S1P content in cell lysates (50 μg protein) was measured using the S1P assay kit from Echelon Inc. according to the manufacturer's instruction.
3.4. Regulation of the Expression of the S1P Degradation Enzymes by CoCl2 in Normal FLS and RAFLS
Decreased steady state levels of intracellular S1P in RAFLS could be due to altered production of S1P by SphKs, increased degradation by S1P phosphatases (SGPP1 and SGPP2) or S1P lyase (SPL), and/or a combination of the two mechanisms. In this study we focussed on the impact of CoCl2 on S1P phosphatases and SPL gene/protein expression in normal FLS and in RAFLS. Quantitative real-time PCR (qPCR) analyses highlighted the expression of SGPP1 and SPL mRNA in cells (Figure 4(a)). SGPP2 mRNA was not detected with the primers we designed for this study (data not shown). As shown in Figure 4(a), SGPP1 and SPL mRNA were ~1.5- and 1.53-fold more abundant in RAFLS than in normal FLS (p < 0.05). Moreover, incubation with CoCl2 decreased SGPP1 mRNA levels by 34.7 ± 2.5% (p < 0.001) and 64.8 ± 6.2% (p < 0.05) and those of SPL mRNA by 45.9 ± 3.0% (p < 0.001) and 67.7 ± 6.9% (p < 0.01) in normal FLS and RAFLS, respectively (Figure 4(a)). Decreased expression of SGPP1 was confirmed at the protein level in RAFLS with a 48.8 ± 13.1% decrease in SGPP1 protein (p < 0.05) after treatment with CoCl2 for 48 h (Figure 4(b)). SPL protein levels in RALFS were not significantly reduced by CoCl2 as estimated by immunoblotting (Figure 4(b)).
Figure 4.

Differential expression of SGPP1 and SPL in normal FLS and RAFLS. Human primary FLS from normal (n = 4) and RA (n = 4) donors were incubated with or without 200 μM CoCl2 for 24 h. Total RNA was extracted for quantitative PCR analyses and RPLP0 was used as an internal control and data normalized to that of normal FLS (a). The data are the means ± SE from four experiments (4 different donors) performed in triplicate (3 independent experiments). For statistical analyses, we compared the cells stimulated with CoCl2 to those without CoCl2, or normal FLS to RAFLS. ∗ p < 0.05; ∗∗ p < 0.01; ∗∗∗ p < 0.001. Human primary FLS from RA patients (n = 3) were incubated with 200 μM CoCl2 for up to 48 h (b). Proteins from whole cell lysates were prepared for Western blot. Total PI3-kinase p85 subunit was used as a control for protein loading. Data presented are from a representative blot (upper panel) or the means ± SE from three experiments (lower panel). For statistical comparative analyses, the samples stimulated with CoCl2 at 0 h were compared to those treated for indicated times. ∗ p < 0.05.
3.5. Effect of SPL on CoCl2-Mediated Chemokine Secretion by Normal FLS and RAFLS
To determine whether the levels of intracellular S1P in FLS may impact its transport outside cells and access to its cognate receptors for autocrine signalling we incubated the cells with CoCl2 in the presence or absence of a SPL inhibitor [46, 47]. When normal FLS and RAFLS were incubated with CoCl2 in combination with increasing concentrations of the SPL inhibitor SM4 there was a trend towards increased secretion of IL-8 and MCP-1 (Figure 5 and data not shown). However, even with 3 μM SM4, the highest concentration tested, the increase in chemokine synthesis was not significant compared to cells treated with CoCl2 alone (data not shown). Since the addition of sphingosine to cell line or primary cell cultures has been shown to provide a source of intracellular S1P that is susceptible to degradation by SPL [46, 47], we evaluated the impact of exogenously added sphingosine in combination with the SPL inhibitor on CoCl2-mediated chemokine synthesis. Figure 5 shows that the inhibition of SPL in the presence of sphingosine significantly increased CoCl2-induced chemokine secretion in RAFLS (Figures 5(b) and 5(d)) and in normal FLS as well (Figures 5(a) and 5(c)). In RAFLS SM4 increased the secretion of IL-8 and MCP-1 by 232 ± 23.8% (p < 0.001) and 158.7 ± 10.7% (p < 0.05), respectively, while in normal FLS SM4 increased IL-8 and MCP-1 secretion by 243.3 ± 73.4% (p < 0.05) and 368.5 ± 109.7% (p < 0.01), respectively. No significant increase in chemokine synthesis was observed when cells were incubated with CoCl2 in the presence of sphingosine without the SPL inhibitor, with the SPL inhibitor but without sphingosine, or with the inactive enantiomer SM3. The Proteome Profiler Antibody Array confirmed in RAFLS that inhibition of SPL in combination with sphingosine increases CoCl2-mediated IL-8 secretion and possibly that of other cytokines such as IL-6 and IL-23 (Figure 5(e)). When RAFLS were treated with the SPL inhibitor in the presence of sphingosine (Figure 5(f)), CoCl2-mediated secretion of IL-8 and MCP-1 becomes sensitive to inhibition by the S1P3 receptor antagonist CAY10444 (46.9 ± 10.3% and 55.3 ± 4.0% decrease, p < 0.01, resp.) and the S1P2 receptor antagonist JTE-013 (45.9 ± 15.9% and 23.5 ± 7.0% decrease, p < 0.01, resp.).
Figure 5.

Impact of SPL inhibition on CoCl2-mediated chemokine/cytokine secretion in normal FLS and RAFLS. Human primary FLS from normal (a, c) and RA (b, d, e, f) donors were incubated with 200 μM CoCl2 in the presence of SPL inhibitor SM4 (3 μM) or the inactive analog SM3 (3 μM) and sphingosine (1 μM) for 24 h. Where indicated, cells were pretreated with S1P3 antagonist CAY10444 (5 μM) or S1P2 antagonist JTE-013 (5 μM) for 30 min before stimulation with CoCl2 in combination with sphingosine (Sph), SM4, or SM3. The data are the means ± SE from three experiments. For statistical comparative analyses, chemokine levels in the samples stimulated with CoCl2 were compared to that of other samples (a–d) or chemokines produced by cells stimulated with CoCl2 in combination with Sph and SM4 were compared to those produced by cells incubated with the S1P receptor antagonists prior to cell stimulation (f). ∗ p < 0.05; ∗∗ p < 0.01; ∗∗∗ p < 0.001. Cytokine/chemokine secretion in RAFLS supernatants was analyzed using Proteome Profiler Human Cytokine Array panel A (e). Circled pairs of duplicate spots represent one cytokine/chemokine.
4. Discussion
FLS are key effector cells in RA. They spread arthritis to unaffected joints [48] and their altered phenotypes in RA have been associated with changes in signalling cascades, apoptotic responses, and the expression of adhesion molecules as well as matrix-degrading enzymes [49, 50]. The cell microenvironment plays an essential role in determining cell phenotype and phenotypic and metabolic characterization of those changes will further our understanding of the pathogenesis of RA. Herein, we report novel characteristics of RAFLS that distinguish these cells from their normal counterparts: (1) RAFLS are less prone to release IL-8 and MCP-1 in response to the hypoxia mimetic CoCl2; (2) CoCl2-mediated chemokine production is, at least in part, due to autocrine activation of S1P receptors in control FLS but not in RAFLS; (3) expression of SGPP1 and SPL mRNA is elevated whereas intracellular levels of S1P are reduced in RAFLS when compared to normal FLS; (4) whereas CoCl2 reduces SGPP1 mRNA and protein expression, the combination of the hypoxic-like stress, sphingosine, and inhibition of SPL is required to enhance chemokine/cytokine synthesis and to restore a positive autocrine feedback loop of chemokine synthesis depending on S1P receptor activation in RAFLS. The data suggest that sphingolipid metabolism is altered in RAFLS collected from patients with advanced RA.
Hypoxia was reported to potentiate the expression of inflammatory cytokines, MMPs, and VEGF in RAFLS stimulated with TLR ligands [51]. Moreover, hypoxia has been shown to induce the expression of IL-8 mRNA in RAFLS [24]. In agreement with those findings we report that the hypoxia mimetic agent CoCl2 stimulated IL-8 and MCP-1 production in normal FLS and RAFLS. Surprisingly, the amounts of IL-8 and MCP-1 released by RAFLS incubated with CoCl2 were less than those produced by normal FLS, indicative of altered molecular pathways regulating chemokine synthesis in RAFLS. RAFLS phenotypic changes are possibility related to genetic/epigenetic determinants and genetic mutation due to chronic exposure to a hypoxic inflammatory environment [52]. Indeed, the expression of many genes involved in immune and inflammatory function is differently regulated by hypoxia in normal FLS and RAFLS [53]. The proinflammatory chemokines/cytokines MCP-2, MIP-2α, MIP-2β, and IL-12A for instance are downregulated whereas the anti-inflammatory mediators CD300a and AMPD3 are upregulated by hypoxia in RAFLS [53].
Upregulation of SphK1 expression and activation by hypoxia has been linked to increases in intracellular and extracellular S1P levels [54]. Previous studies have highlighted the expression of S1P1, S1P2, and S1P3 receptors in RAFLS [33, 34]. High expression of S1PR1 in RA synovial tissue was observed in the synovial lining, vascular endothelial cells, and mononuclear cells when compared to osteoarthritis and normal synovial tissues [34]. In vitro S1P induces RAFLS migration, expression of cytokines/chemokines and COX-2, prostaglandin synthesis, and cell proliferation and survival [33, 34]. S1P receptors expressed by RAFLS have redundant functions. In a wound-closing assay S1P induced RAFLS migration through S1P1 and S1P3 receptors [33]. On the other hand, S1P stimulated the secretion of numerous cytokines/chemokines (IL-8, IL-6, MCP-1, and RANTES) through S1P2 and S1P3 receptors. In the present study we provide evidence that the mechanism by which CoCl2 induces the secretion of chemokines is, at least in part, through autocrine activation of S1P2 and S1P3 receptors in normal FLS. Although RAFLS express functional S1P2 and S1P3 receptors, CoCl2-mediated chemokine synthesis was not reduced by S1P receptor antagonists. This was related to low levels of intracellular S1P in RAFLS since incubation of cells with an inhibitor of SPL and sphingosine, a condition that has been shown to increase intracellular amounts of S1P and its release by various cells [46, 47], restores autocrine signalling through S1P2 and S1P3 receptors in RAFLS stimulated with CoCl2.
S1P synthesis requires the concerted action of ceramidase and sphingosine kinases and once formed, S1P is either metabolized to hexadecenal and ethanolamine phosphate by SPL or recycled to sphingosine by S1P phosphatases [30]. Upregulation of SGPP2 has been detected in samples of skin lesions from patients with psoriasis, a chronic inflammatory skin disease [55]. Other studies investigating sphingolipid metabolism have shown that oxygen deprivation in microendothelial cells resulted in reduced SPL activity [56] and that adipocytes respond to hypoxia by downregulating SPL expression [57]. In this study we provide evidence for increased expression of SGPP1 and SPL mRNA in RAFLS, suggesting that the lower level of intracellular S1P in these cells is possibly driven by a hypercatabolic state. Targeting S1P1 receptor with a selective antagonist [38] or with the sphingosine analogue FTY720 [58, 59] and pharmacological inhibition of SPL in mice [60], all decreased the development of collagen-induced arthritis (CIA). The anti-inflammatory properties of these compounds are associated with abnormal B and T cell maturation and lymphocyte egress from lymphoid organs due to local S1P gradient breakdown or S1P1 receptor degradation [38, 61, 62]. Whereas inhibition of SPL may have a beneficial effect through targeting lymphocyte trafficking from lymphoid organs, we suggest that inhibition of SPL may have adverse inflammatory effects by increasing the steady state levels of intracellular S1P, S1P export, and synthesis of proinflammatory chemokines/cytokines through autocrine/paracrine activation of S1P2 and S1P3 receptors. Allende et al. recently reported that SPL deficiency in mice promotes an inflammatory response [63].
A few studies have evaluated S1P levels and S1P metabolizing enzymes in RA synovial biopsy. For example, expression of SphK2 and elevated levels of S1P were detected in the synovium and synovial fluids of RA patients [34–36]. Animal models have been used to evaluate the role of S1P in inflammatory arthritis. In the CIA model, administration of a nonspecific inhibitor of SphKs or of a siRNA to silence SphK1 markedly suppressed cartilage and bone erosion, synovial hyperplasia, and leukocyte infiltration into the joint compartments [36]. While SphK1 activity is proinflammatory, SphK2 has an opposite role since the silencing of this enzyme in mice promotes CIA-mediated synovitis [64]. However, depending on the animal models of arthritis employed, studies with KO mice have produced conflicting information. Whereas SphK1 deficiency has been reported to reduce synovial inflammation and bone erosions in human TNF-α transgenic mice, which spontaneously develop inflammatory arthritis [37], SphK2 deficiency has no impact on disease severity and progression [65]. Our preliminary data suggest that CoCl2 induces SphK1 expression in normal FLS whereas SphK1 seems to be less prone to upregulation by CoCl2 in RAFLS (data not shown). Further characterization is underway to determine whether altered expression and/or activation of Sphks contribute to reduced steady state levels of intracellular S1P in RAFLS.
In summary, the results of this study suggest that the sphingolipid metabolism involved in the production and/or release of S1P under hypoxic-like conditions is altered in RAFLS. Decreased steady state levels of intracellular S1P in RAFLS were associated with reduced production of chemokine/cytokine and autocrine activation of S1P2 and S1P3 receptors in response to chemical hypoxia. Our data provide new insights into the mechanisms that may regulate inflammation and possibly joint destruction in advanced cases of RA.
Acknowledgments
The authors thank Ms. Lynn Davis for her editorial assistance. This project was supported by a research grant from the Arthritis Society of Canada (RG10/011). Mohamed Touaibia acknowledges the contribution of the Canadian Foundation for Innovation (CFI), the New Brunswick Innovation Foundation (NBIF), and Université de Moncton (NB, Canada) for the acquisition of the NMR instrument.
Conflict of Interests
The authors have declared no conflict of interests.
References
- 1.Firestein G. S. Evolving concepts of rheumatoid arthritis. Nature. 2003;423(6937):356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 2.Müller-Ladner U., Pap T., Gay R. E., Neidhart M., Gay S. Mechanisms of disease: the molecular and cellular basis of joint destruction in rheumatoid arthritis. Nature Clinical Practice Rheumatology. 2005;1(2):102–110. doi: 10.1038/ncprheum0047. [DOI] [PubMed] [Google Scholar]
- 3.Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nature Reviews Immunology. 2002;2(5):364–371. doi: 10.1038/nri802. [DOI] [PubMed] [Google Scholar]
- 4.Badolato R., Oppenheim J. J. Role of cytokines, acute-phase proteins, and chemokines in the progression of rheumatoid arthritis. Seminars in Arthritis and Rheumatism. 1996;26(2):526–538. doi: 10.1016/s0049-0172(96)80041-2. [DOI] [PubMed] [Google Scholar]
- 5.Hosaka S., Akahoshi T., Wada C., Kondo H. Expression of the chemokine superfamily in rheumatoid arthritis. Clinical & Experimental Immunology. 1994;97(3):451–457. doi: 10.1111/j.1365-2249.1994.tb06109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Endo H., Akahoshi T., Takagishi K., Kashiwazaki S., Matsushima K. Elevation of interleukin-8 (IL-8) levels in joint fluids of patients with rheumatoid arthritis and the induction by IL-8 of leukocyte infiltration and synovitis in rabbit joints. Lymphokine and Cytokine Research. 1991;10(4):245–252. [PubMed] [Google Scholar]
- 7.Nishiura H., Tanaka J., Takeya M., Tsukano M., Kambara T., Imamura T. IL-8/NAP-1 is the major T-cell chemoattractant in synovial tissues of rheumatoid arthritis. Clinical Immunology and Immunopathology. 1996;80(2):179–184. doi: 10.1006/clin.1996.0112. [DOI] [PubMed] [Google Scholar]
- 8.Akahoshi T., Wada C., Endo H., et al. Expression of monocyte chemotactic and activating factor in rheumatoid arthritis. Regulation of its production in synovial cells by interleukin-1 and tumor necrosis factor. Arthritis and Rheumatism. 1993;36(6):762–771. doi: 10.1002/art.1780360605. [DOI] [PubMed] [Google Scholar]
- 9.Koch A. E., Kunkel S. L., Harlow L. A., et al. Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. The Journal of Clinical Investigation. 1992;90(3):772–779. doi: 10.1172/jci115950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McInnes I. B., Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nature Reviews Immunology. 2007;7(6):429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 11.Blake D. R., Winyard P. G., Marok R. The contribution of hypoxia-reperfusion injury to inflammatory synovitis: the influence of reactive oxygen intermediates on the transcriptional control of inflammation. Annals of the New York Academy of Sciences. 1994;723:308–317. doi: 10.1111/j.1749-6632.1994.tb36736.x. [DOI] [PubMed] [Google Scholar]
- 12.Edmonds S. E., Blake D. R., Morris C. J., Winyard P. G. An imaginative approach to synovitis—the role of hypoxic reperfusion damage in arthritis. The Journal of Rheumatology. Supplement. 1993;37:26–31. [PubMed] [Google Scholar]
- 13.Edmonds S. E., Ellis G., Gaffney K., Archer J., Blake D. R. Hypoxia and the rheumatoid joint: immunological and therapeutic implications. Scandinavian Journal of Rheumatology, Supplement. 1995;101:163–168. doi: 10.3109/03009749509100921. [DOI] [PubMed] [Google Scholar]
- 14.Falchuk K. H., Goetzl E. J., Kulka J. P. Respiratory gases of synovial fluids. an approach to synovial tissue circulatory-metabolic imbalance in rheumatoid arthritis. The American Journal of Medicine. 1970;49(2):223–231. doi: 10.1016/s0002-9343(70)80078-x. [DOI] [PubMed] [Google Scholar]
- 15.Richman A. I., Su E. Y., Ho G., Jr. Reciprocal relationship of synovial fluid volume and oxygen tension. Arthritis and Rheumatism. 1981;24(5):701–705. doi: 10.1002/art.1780240512. [DOI] [PubMed] [Google Scholar]
- 16.Treuhaft P. S., MCCarty D. J. Synovial fluid pH, lactate, oxygen and carbon dioxide partial pressure in various joint diseases. Arthritis and Rheumatism. 1971;14(4):475–484. doi: 10.1002/art.1780140407. [DOI] [PubMed] [Google Scholar]
- 17.Konisti S., Kiriakidis S., Paleolog E. M. Hypoxia—a key regulator of angiogenesis and inflammation in rheumatoid arthritis. Nature Reviews Rheumatology. 2012;8(3):153–162. doi: 10.1038/nrrheum.2011.205. [DOI] [PubMed] [Google Scholar]
- 18.Koch A. E., Harlow L. A., Haines G. K., et al. Vascular endothelial growth factor. A cytokine modulating endothelial function in rheumatoid arthritis. The Journal of Immunology. 1994;152(8):4149–4156. [PubMed] [Google Scholar]
- 19.Paleolog E. M., Young S., Stark A. C., McCloskey R. V., Feldmann M., Maini R. N. Modulation of angiogenic vascular endothelial growth factor by tumor necrosis factor α and interleukin-1 in rheumatoid arthritis. Arthritis and Rheumatism. 1998;41(7):1258–1265. doi: 10.1002/1529-0131(199807)41:7lt;1258::aid-art1762;3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 20.Ke J., Liu Y., Long X., et al. Up-regulation of vascular endothelial growth factor in synovial fibroblasts from human temporomandibular joint by hypoxia. Journal of Oral Pathology and Medicine. 2007;36(5):290–296. doi: 10.1111/j.1600-0714.2007.00532.x. [DOI] [PubMed] [Google Scholar]
- 21.Demasi M., Cleland L. G., Cook-Johnson R. J., James M. J. Effects of hypoxia on the expression and activity of cyclooxygenase 2 in fibroblast-like synoviocytes: Interactions with monocyte-derived soluble mediators. Arthritis and Rheumatism. 2004;50(8):2441–2449. doi: 10.1002/art.20429. [DOI] [PubMed] [Google Scholar]
- 22.Cha H.-S., Ahn K.-S., Jeon C. H., Kim J., Song Y. W., Koh E.-M. Influence of hypoxia on the expression of matrix metalloproteinase-1, -3 and tissue inhibitor of metalloproteinase-1 in-rheumatoid synovial fibroblasts. Clinical and Experimental Rheumatology. 2003;21(5):593–598. [PubMed] [Google Scholar]
- 23.Hitchon C., Wong K., Ma G., Reed J., Lyttle D., El-Gabalawy H. Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis and Rheumatism. 2002;46(10):2587–2597. doi: 10.1002/art.10520. [DOI] [PubMed] [Google Scholar]
- 24.Ahn J. K., Koh E.-M., Cha H.-S., et al. Role of hypoxia-inducible factor-1α in hypoxia-induced expressions of IL-8, MMP-1 and MMP-3 in rheumatoid fibroblast-like synoviocytes. Rheumatology. 2008;47(6):834–839. doi: 10.1093/rheumatology/ken086. [DOI] [PubMed] [Google Scholar]
- 25.Akhavani M. A., Madden L., Buysschaert I., Sivakumar B., Kang N., Paleolog E. M. Hypoxia upregulates angiogenesis and synovial cell migration in rheumatoid arthritis. Arthritis Research and Therapy. 2009;11(3, article R64) doi: 10.1186/ar2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ng C. T., Biniecka M., Kennedy A., et al. Synovial tissue hypoxia and inflammation in vivo. Annals of the Rheumatic Diseases. 2010;69(7):1389–1395. doi: 10.1136/ard.2009.119776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nonomura Y., Mizoguchi F., Suzuki A., et al. Hypoxia-induced abrogation of contact-dependent inhibition of rheumatoid arthritis synovial fibroblast proliferation. Journal of Rheumatology. 2009;36(4):698–705. doi: 10.3899/jrheum.080188. [DOI] [PubMed] [Google Scholar]
- 28.Proia R. L., Hla T. Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. The Journal of Clinical Investigation. 2015;125(4):1379–1387. doi: 10.1172/jci76369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spiegel S., Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nature Reviews Immunology. 2011;11(6):403–415. doi: 10.1038/nri2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saba J. D., Hla T. Point-counterpoint of sphingosine 1-phosphate metabolism. Circulation Research. 2004;94(6):724–734. doi: 10.1161/01.res.0000122383.60368.24. [DOI] [PubMed] [Google Scholar]
- 31.Tang X., Benesch M. G., Brindley D. N. Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. Journal of Lipid Research. 2015 doi: 10.1194/jlr.r058362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Snider A. J. Sphingosine kinase and sphingosine-1-phosphate: regulators in autoimmune and inflammatory disease. International Journal of Clinical Rheumatology. 2013;8(4):453–463. doi: 10.2217/ijr.13.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao C., Fernandes M. J., Turgeon M., et al. Specific and overlapping sphingosine-1-phosphate receptor functions in human synoviocytes: impact of TNF-alpha. Journal of Lipid Research. 2008;49(11):2323–2337. doi: 10.1194/jlr.m800143-jlr200. [DOI] [PubMed] [Google Scholar]
- 34.Kitano M., Hla T., Sekiguchi M., et al. Sphingosine 1-phosphate/sphingosine 1-phosphate receptor 1 signaling in rheumatoid synovium: regulation of synovial proliferation and inflammatory gene expression. Arthritis and Rheumatism. 2006;54(3):742–753. doi: 10.1002/art.21668. [DOI] [PubMed] [Google Scholar]
- 35.Kamada K., Arita N., Tsubaki T., et al. Expression of sphingosine kinase 2 in synovial fibroblasts of rheumatoid arthritis contributing to apoptosis by a sphingosine analogue, FTY720. Pathology International. 2009;59(6):382–389. doi: 10.1111/j.1440-1827.2009.02381.x. [DOI] [PubMed] [Google Scholar]
- 36.Lai W.-Q., Irwan A. W., Goh H. H., et al. Anti-inflammatory effects of sphingosine kinase modulation in inflammatory arthritis. Journal of Immunology. 2008;181(11):8010–8017. doi: 10.4049/jimmunol.181.11.8010. [DOI] [PubMed] [Google Scholar]
- 37.Baker D. A., Barth J., Chang R., Obeid L. M., Gilkeson G. S. Genetic sphingosine kinase 1 deficiency significantly decreases synovial inflammation and joint erosions in murine TNF-α-induced arthritis. The Journal of Immunology. 2010;185(4):2570–2579. doi: 10.4049/jimmunol.1000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fujii Y., Hirayama T., Ohtake H., et al. Amelioration of collagen-induced arthritis by a novel S1P 1 antagonist with immunomodulatory activities. Journal of Immunology. 2012;188(1):206–215. doi: 10.4049/jimmunol.1101537. [DOI] [PubMed] [Google Scholar]
- 39.Michaud M. D., Robitaille G. A., Gratton J.-P., Richard D. E. Sphingosine-1-phosphate: a novel nonhypoxic activator of hypoxia-inducible factor-1 in vascular cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29(6):902–908. doi: 10.1161/atvbaha.109.185280. [DOI] [PubMed] [Google Scholar]
- 40.Ader I., Malavaud B., Cuvillier O. When the sphingosine kinase 1/sphingosine 1-phosphate pathway meets hypoxia signaling: new targets for cancer therapy. Cancer Research. 2009;69(9):3723–3726. doi: 10.1158/0008-5472.can-09-0389. [DOI] [PubMed] [Google Scholar]
- 41.Miller-Moslin K., Peukert S., Jain R. K., et al. 1-Amino-4-benzylphthalazines as orally bioavailable smoothened antagonists with antitumor activity. Journal of Medicinal Chemistry. 2009;52(13):3954–3968. doi: 10.1021/jm900309j. [DOI] [PubMed] [Google Scholar]
- 42.Arnett F. C., Edworthy S. M., Bloch D. A., et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis and Rheumatism. 1988;31(3):315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 43.Mancini A., Jovanovic D. V., He Q. W., Di Battista J. A. Site-specific proteolysis of cyclooxygenase-2: a putative step in inflammatory prostaglandin E(2) biosynthesis. Journal of Cellular Biochemistry. 2007;101(2):425–441. doi: 10.1002/jcb.21191. [DOI] [PubMed] [Google Scholar]
- 44.Yuan Y., Hilliard G., Ferguson T., Millhorn D. E. Cobalt inhibits the interaction between hypoxia-inducible factor-α and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-α . The Journal of Biological Chemistry. 2003;278(18):15911–15916. doi: 10.1074/jbc.m300463200. [DOI] [PubMed] [Google Scholar]
- 45.Kluk M. J., Hla T. Signaling of sphingosine-1-phosphate via the S1P/EDG-family of G-protein-coupled receptors. Biochimica et Biophysica Acta. 2002;1582(1–3):72–80. doi: 10.1016/s1388-1981(02)00139-7. [DOI] [PubMed] [Google Scholar]
- 46.Billich A., Beerli C., Bergmann R., Bruns C., Loetscher E. Cellular assay for the characterization of sphingosine-1-phosphate lyase inhibitors. Analytical Biochemistry. 2013;434(2):247–253. doi: 10.1016/j.ab.2012.11.026. [DOI] [PubMed] [Google Scholar]
- 47.Loetscher E., Schneider K., Beerli C., Billich A. Assay to measure the secretion of sphingosine-1-phosphate from cells induced by S1P lyase inhibitors. Biochemical and Biophysical Research Communications. 2013;433(3):345–348. doi: 10.1016/j.bbrc.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 48.Lefèvre S., Knedla A., Tennie C., et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nature Medicine. 2009;15(12):1414–1420. doi: 10.1038/nm.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huber L. C., Distler O., Tarner I., Gay R. E., Gay S., Pap T. Synovial fibroblasts: key players in rheumatoid arthritis. Rheumatology. 2006;45(6):669–675. doi: 10.1093/rheumatology/kel065. [DOI] [PubMed] [Google Scholar]
- 50.Bartok B., Firestein G. S. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunological Reviews. 2010;233(1):233–255. doi: 10.1111/j.0105-2896.2009.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu F., Mu R., Zhu J., et al. Hypoxia and hypoxia-inducible factor-1α provoke toll-like receptor signalling-induced inflammation in rheumatoid arthritis. Annals of the Rheumatic Diseases. 2014;73(5):928–936. doi: 10.1136/annrheumdis-2012-202444. [DOI] [PubMed] [Google Scholar]
- 52.Bottini N., Firestein G. S. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nature Reviews Rheumatology. 2013;9(1):24–33. doi: 10.1038/nrrheum.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Del Rey M. J., Izquierdo E., Usategui A., et al. The transcriptional response of normal and rheumatoid arthritis synovial fibroblasts to hypoxia. Arthritis Care and Research. 2010;62(12):3584–3594. doi: 10.1002/art.27750. [DOI] [PubMed] [Google Scholar]
- 54.Anelli V., Gault C. R., Cheng A. B., Obeid L. M. Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells. Role of hypoxia-inducible factors 1 and 2. The Journal of Biological Chemistry. 2008;283(6):3365–3375. doi: 10.1074/jbc.m708241200. [DOI] [PubMed] [Google Scholar]
- 55.Mechtcheriakova D., Wlachos A., Sobanov J., et al. Sphingosine 1-phosphate phosphatase 2 is induced during inflammatory responses. Cellular Signalling. 2007;19(4):748–760. doi: 10.1016/j.cellsig.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 56.Testai F. D., Kilkus J. P., Berdyshev E., Gorshkova I., Natarajan V., Dawson G. Multiple sphingolipid abnormalities following cerebral microendothelial hypoxia. Journal of Neurochemistry. 2014;131(4):530–540. doi: 10.1111/jnc.12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ito S., Iwaki S., Koike K., et al. Increased plasma sphingosine-1-phosphate in obese individuals and its capacity to increase the expression of plasminogen activator inhibitor-1 in adipocytes. Coronary Artery Disease. 2013;24(8):642–650. doi: 10.1097/mca.0000000000000033. [DOI] [PubMed] [Google Scholar]
- 58.Matsuura M., Imayoshi T., Okumoto T. Effect of FTY720, a novel immunosuppressant, on adjuvant- and collagen-induced arthritis in rats. International Journal of Immunopharmacology. 2000;22(4):323–331. doi: 10.1016/s0192-0561(99)00088-0. [DOI] [PubMed] [Google Scholar]
- 59.Wang F., Tan W., Guo D., He S. Reduction of CD4 positive T cells and improvement of pathological changes of collagen-induced arthritis by FTY720. European Journal of Pharmacology. 2007;573(1–3):230–240. doi: 10.1016/j.ejphar.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 60.Bagdanoff J. T., Donoviel M. S., Nouraldeen A., et al. Inhibition of sphingosine 1-phosphate lyase for the treatment of rheumatoid arthritis: discovery of (E)-1-(4-((1R,2 S,3R)-1,2,3,4-tetrahydroxybutyl)-1H -imidazol-2-yl)ethanone oxime (LX2931) and (1R,2 S,3R)-1-(2-(Isoxazol-3-yl)-1H -imidazol-4-yl)butane-1,2,3,4-tetraol (LX2932) Journal of Medicinal Chemistry. 2010;53(24):8650–8662. doi: 10.1021/jm101183p. [DOI] [PubMed] [Google Scholar]
- 61.Matloubian M., Lo C. G., Cinamon G., et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 62.Schwab S. R., Pereira J. P., Matloubian M., Xu Y., Huang Y., Cyster J. G. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309(5741):1735–1739. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- 63.Allende M. L., Bektas M., Lee B. G., et al. Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. The Journal of Biological Chemistry. 2011;286(9):7348–7358. doi: 10.1074/jbc.m110.171819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lai W.-Q., Irwan A. W., Goh H. H., Melendez A. J., McInnes I. B., Leung B. P. Distinct roles of sphingosine kinase 1 and 2 in murine collagen-induced arthritis. Journal of Immunology. 2009;183(3):2097–2103. doi: 10.4049/jimmunol.0804376. [DOI] [PubMed] [Google Scholar]
- 65.Baker D. A., Eudaly J., Smith C. D., Obeid L. M., Gilkeson G. S. Impact of sphingosine kinase 2 deficiency on the development of TNF-alpha-induced inflammatory arthritis. Rheumatology International. 2013;33(10):2677–2681. doi: 10.1007/s00296-012-2493-2. [DOI] [PMC free article] [PubMed] [Google Scholar]