

Summary
Human induced pluripotent stem cells (iPSCs) and genome editing provide a precise way to generate gene-corrected cells for disease modeling and cell therapies. Human iPSCs generated from sickle cell disease (SCD) patients have a homozygous missense point mutation in the HBB gene encoding adult β-globin proteins, and are used as a model system to improve strategies of human gene therapy. We demonstrate that the CRISPR/Cas9 system designer nuclease is much more efficient in stimulating gene targeting of the endogenous HBB locus near the SCD point mutation in human iPSCs than ZFNs and TALENs. Using a specific guide RNA and Cas9, we readily corrected one allele of the SCD HBB gene in human iPSCs by homologous recombination with a donor DNA template containing the wild-type HBB DNA and a selection cassette that was subsequently removed to avoid possible interference of HBB transcription and translation. We chose targeted iPSC clones that have one corrected and one disrupted SCD allele for erythroid differentiation assays, using an improved xeno-free and feeder-free culture condition we recently established. Erythrocytes from either the corrected or its parental (uncorrected) iPSC line were generated with similar efficiencies. Currently ~6%-10% of these differentiated erythrocytes indeed lacked nuclei, characteristic of further matured erythrocytes called reticulocytes. We also detected the 16-kD β-globin protein expressed from the corrected HBB allele in the erythrocytes differentiated from genome-edited iPSCs. Our results represent a significant step towards the clinical applications of genome editing using patient-derived iPSCs to generate disease-free cells for cell and gene therapies.
Keywords: Human iPSCs, genome editing, erythroid cells, globin switching
Introduction
Patient-derived human induced pluripotent stem cells (iPSCs), which can be expanded unlimitedly in culture while maintaining pluripotency, provide advantages for selecting and expanding rare cells whose genome is precisely edited or engineered by homologous recombination (HR). HR as well as another form of DNA repair, non-homologous end joining (NHEJ) independent of a DNA template, are the two major mechanisms for a mammalian cell to repair a DNA double strand break (DSB). In the past decade, scientists developed tools to induce a specific DSB that triggers DNA repair machinery and greatly stimulate either NHEJ or HR at a predetermined DNA sequence in human and other mammalian cells [1–5]. NHEJ does not need a template for DNA repair but rather simply and promiscuously joins the DNA ends together in an error-prone process, often resulting in small deletions or insertions (indels) at the rejoining junction. In contrast, HR requires a DNA template to replace the DNA sequence surrounding the DSB site. HR-mediated precise gene targeting is much less efficient than NHEJ even in the presence of exogenous donor DNA template [1, 2]. To increase the efficiency of either form of gene targeting, designer DNA endonucleases such as zinc finger nucleases (ZFNs) have been engineered and used in various cell types such as human iPSCs and embryonic stem cells (ESCs) [1–7]. In addition to making targeted insertion, ZFNs were also used to correct a point mutation in human iPSCs derived from adult patients of sickle cell disease (SCD), which harbor the homozygous βs mutation in the HBB gene [8, 9]. This was achieved by HR-mediated gene targeting with a donor DNA that provides a correct HBB DNA sequence and an expression cassette rendering drug selection of rare clones after HR-mediated DNA replacement resulting in a targeted insertion. However, the reported efficiencies of gene targeting in these two early studies were low even among the vastly enriched clones after drug-selection: 1.3% and 9.8% of selected clones, respectively [8, 9]. This was not totally surprising, because the HBB gene is not expressed in human ESCs or iPSCs, and it is challenging to find a pair of ZFNs that efficiently and specifically cut the HBB gene without affecting the nearby and highly homologous genes HBD, HBE or HBG (the later two encoding embryonic and fetal forms of hemoglobin β subunits). Importantly, the two studies did not show the production of the corrected HBB protein, after hematopoietic differentiation of the gene-corrected iPSCs [8, 9]. The lack of evidence in producing HBB protein after iPSC gene correction and differentiation could be due to: 1) defects in the donor vector that is unable to express the corrected gene from the endogenous regulatory element after genome targeting; 2) defects in the iPSC lines in these earlier studies that were derived from adult fibroblasts by vectors inserting and disputing the native human genome; 3) lack of an efficient differentiation method to generate mature erythrocytes that produce the adult form of beta-globin encoded by the HBB gene; or 4) combinations of the above.
Recently, several groups used TALENs and CRISPR/Cas9 to develop better designer nucleases that target the HBB gene near the SCD point mutation (nt. 69A>T in exon 1). Although these studies reported greater efficiencies measuring both NHEJ and HR rates in human iPSCs and simpler cell models such as cancer cell lines [10–13], the specificity as well as efficiencies of these TALENs and CRIPSR/Cas9 nucleases remained to be fully analyzed. An ultimate test would be to efficiently and specifically target the SCD mutation in high quality iPSCs (such as those free of vector insertion), differentiate them into erythrocytes expressing a high-level of HBB mRNA, and demonstrate the production of HBB-encoded protein from the corrected allele. In this report, we demonstrated that we achieved these objectives, and established a method that is applicable to other human diseases such as β-thalassemia.
Methods
Approvals of Using Human iPSCs and Primary Cells from Anonymous Donors
The experiments using human iPSCs were approved by the Institutional Stem Cell Research Oversight (ISCRO) committee in the Johns Hopkins University. Use of anonymous human samples for laboratory research was approved by the Institutional Review Board of the Johns Hopkins University.
Human iPSC maintenance and expansion
Human iPSC lines BC1 and TNC1, derived from human adult hematopoietic cells by transient expression of one or two episomal vectors [14, 15], were used in this study. While TNC1 was derived from cultured erythroblasts after selective expansion of a SCD patient’s peripheral blood (PB) mononuclear cells (MNCs), BC1 was derived from expanded hematopoietic progenitors in bone marrow (BM) of an adult healthy donor [14, 15]. They lack overt genomic alteration or V(D)J rearrangement associated with lymphocytes [14, 15]. Although they were initially derived and expanded on mouse embryonic fibroblasts, these human iPSCs (around passages 20–25) were subsequently cultured under a feeder-free and xeno-free condition with a simplified culture medium E8 as described before [16–18].
Designing CRISPR/Cas9 vectors for targeting the endogenous HBB locus
We used the CRIPSR/Cas9 system previously described [18, 25], for which an expression vector encoding humanized (h) Cas9 protein was obtained from Addgene.org (Plasmid #41815). 455-bp guide RNA (gRNA) expression cassettes including a 20-bp target-specific sequence targeting the human HBB gene in the first exon were synthesized (Integrated DNA Technology), and cloned into Zero-blunt TOPO vector (Life Technologies). Alternatively, two 60-bp oligonucleotides that are complementary at the 20-bp target sequence were used as a template encoding a guide RNA, and cloned by Gibson assembly into a guide RNA vector (Addgene plasmid #41824) linearized with AflII [25]. The tests of CRIPSR gRNA and Cas9 in human 293T and iPSCs, including Miseq analyses, were described in details in the supplemental materials as recently published [18].
Gene targeting in human iPSCs
Gene targeting in various human iPSC lines including TNC1 was conducted as described [18]. Briefly, two million iPSCs were re-suspended in 100 μl P3 Primary Cell Solution (for 4D Nucleofector, Lonza) and mixed with 10 μg DNA. To target the SCD mutation in TNC1 iPSCs, we used the donor vector (briefed as MGH) that was made previously by MGH investigators and a gift from Dr. J. Keith Joung [9]. The MGH donor vector provides a wild-type HBB DNA template and a PGK-puromycin expression cassette to be inserted into the intron 1 for HR-mediated replacement [9]. In addition to 5 μg MGH donor vector, the mixture also contained 2.5 μg hCas9 plasmid, and 2.5 μg of guide RNA, or 2.5 μg each of the two plasmids expressing of ZFN or TALEN pairs. The nucleofected iPSCs were plated onto Matrigel- or vitronectin-coated 6-well plates immediately following nucleofection. Ten μM ROCK Inhibitor Y-27632 was added in the E8 medium for the first 24-hour. Starting at 72–96 hours post nucleofection, puromycin (0.5 μg/ml) was added to the medium for the selection of targeted events. After puromycin selection, colonies were manually picked from different wells and expanded. Genomic DNAs were isolated from the expanded clones and analyzed for targeted integration [18]. To excise loxP flanked PGK-puromycin selection cassette, iPSCs were transfected with a pCAG-Cre-IRES2-GFP plasmid ([19], plasmid # 26646 in Addgene.org) and plated in low density in 6-well plates for clonal selection. Individual clones were picked, expanded, screened for excision by genomic PCR that amplifies the DNA fragment with shortened insertion in intron 1.
Hematopoietic and erythroblast differentiation of human iPSCs
Hematopoietic and erythroblast differentiation of BC1, TNC1 and targeted iPSCs were done as described previously [17, 20].
Erythroid terminal maturation of erythroblasts generated from iPSCs
We counted the erythroblast cell number at day 10 of erythroblast differentiation and expansion culture and washed erythroblasts twice with PBS after we confirmed the normal erythroblast phenotype by flow cytometry. We then seeded erythroblasts in erythroid terminal maturation medium (TM) at 1 million per ml at 37°C in a humid atmosphere of 5% CO2 in air. The erythroid TM medium is made by mixing with IMDM (Invitrogen) plus 3 U/ml Epo (R&D Systems), 500 μg/ml human holo-transferrin (R&D systems), 4 U/ml heparin (Sigma), 10 μg/ml insulin (Sigma), and 10% pooled normal adult human peripheral blood plasma (innovative-research, Cat# IPLA-N). Fresh medium with cytokines was added every 2 to 3 days. The cell density is maintained below 4 million per ml. The maturating cells were taken for analysis at different days up to ten days from the beginning of terminal maturation.
Flow cytometry of differentiated hematopoietic cells
For flow cytometry analysis of HSPCs and erythroblasts differentiated from human iPSCs, we harvested ~200,000 cells, washed with PBS, and re-suspended the cell pellet in PBS+1%FBS for antibody staining. We used CD34-APC (BD Biosciences), CD45-AF488 or CD45-APC (Invitrogen), CD235a-FITC (Invitrogen), Band3-FITC or Band3-APC (kindly provided by Dr. Xiuli An at NYBC), CD49d-FITC or CD49d-PE (Miltenyi Biotec, San Diego, CA) to characterize expanded and differentiated cells. Dead cells were excluded from analysis by positive staining 7-AAD (Viaprobe, BD Biosciences).
For flow cytometric analysis of erythroid terminal maturation of erythroblasts, we harvested suspension cells from terminal maturation culture at different time points of maturation, washed with PBS, and re-suspended in PBS+1%FBS and stained with appropriate combinations of CD235a-FITC, Band3-FITC or APC, CD45-APC and CD49d-FITC (BD BioSciences and Life Technologies), and DRAQ5, a cell permeable dye for DNA staining (Cell Signaling Technology, Danvers, MA). Intracellular hemoglobin staining was done as described before [21].
Morphological analysis of cells spun on slides
Cytospin and histology staining were done as described before [21].
RNA extraction and quantitative RT-PCR to measure gene expression
RT-PCR analysis of globin gene expression was done as described before [21]. All experiments were performed in triplicate and a non-template control (lacking cDNA template) was included in each assay. Relative gene expression was normalized to the housekeeping gene GAPDH.
Western Blot of HBB protein production
Western blot for analyzing HBB protein level in differentiated cells is carried out using a monoclonal antibody against human HBB (Santa Cruz Biotechnology, sc-21757) and GAPDH (Cell Signaling Technology, Cat. 5174), according to the manufacturer’s instructions. We used the protein extract from the same number of cells at different stages of differentiation to estimate HBB content in differentiated cells.
O2 binding capacity and affinity
O2 equilibrium curves of day 8 erythrocytes after terminal maturation (TM8) differentiated from iPSCs or fresh human CB as a control were determined using a Hemox-Analyzer (TCS Scientific, New Hope, PA) in accordance to manufacturer’s recommendations as previously described [21].
Results
Generation of enucleated erythrocytes from human iPSC lines derived from blood
Recently we developed a method to generate human iPSC lines that are free of vector insertion in the genome from hematopoietic cells in cord blood (CB), adult PB or BM [14, 15]. For example, we used PB MNCs from an adult SCD patient and established a proliferative erythroblast culture that was reprogrammed to iPSCs by one to two plasmids [14]. One of such established iPSC lines, TNC1, contains the homozygous SCD mutation (nt. 69A>T) in the HBB gene [14]. After extensive expansion under a feeder-free culture condition such as one with the E8 culture medium on vitronectin as substrates, the TNC1 and BC1 iPSCs maintain pluripotency and are karyotypically stable [17]. We also analyzed the hematopoietic and erythrocytic differentiation potential of blood-derived iPSCs by first generating definitive CD34+CD45+ hematopoietic progenitor cells (HPCs) followed by erythrocyte production ex vivo. In the first step, we used an improved method of forming individual embryoid bodies (EBs) followed by hematopoietic differentiation (Fig. 1A), under a feeder-free and serum-free condition previously described [17]. Many hematopoietic cells released from EBs to culture medium by day 11–14 (Fig. 1B). We found that 55–65% of the cells harvested from culture suspension co-expressed CD45 and CD34, characteristic phenotypes of definitive HPCs (Fig. 1C). Compared to the input number of iPSCs, this was >3-fold expansion (Fig 1D).
Figure 1. A feeder- and xeno-free culture condition for the hematopoietic differentiation and terminal maturation of human iPSCs.
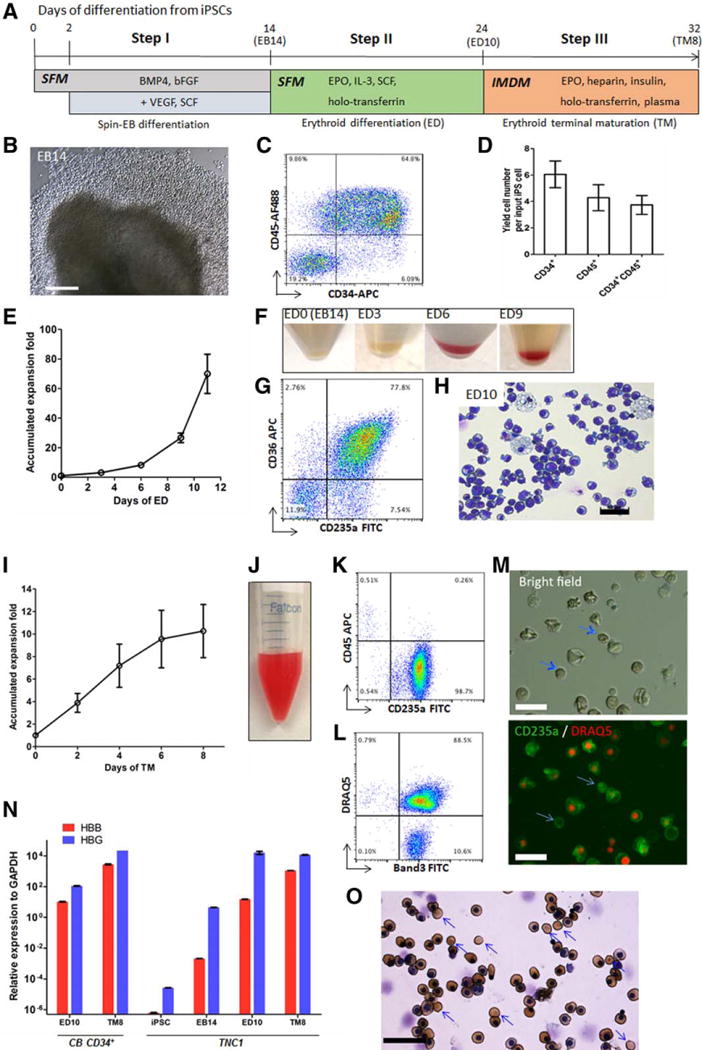
(A) A three-step differentiation strategy from iPSCs to hemoglobinized and enucleated erythrocytes, including spin-EB mediated hematopoietic differentiation (EB), erythroid differentiation (ED), and erythroid terminal maturation (TM). (B–D) Characterization of EB day 14 cells of TNC1 iPSC cultures. (B) Phase-contrast image (10×) of an EB and surrounding hematopoietic cells. Scale bar: 200 μm. (C) Representative flow cytometric analysis of suspension cells harvested EB14 cells that co-express CD34 and CD45. The gates in all flow plots were set based on corresponding fluorescent isotype controls. (D) The yield of CD34+, CD45+, and CD34+CD45+ cells derived from one input iPSC. Means ± s.d., n = 4. (E–H) Erythroid differentiation (ED) step resulting in erythroid commitment, and erythroblast formation and expansion. (E) The accumulated expansion of cell numbers during the ED step. Means ± s.d., n = 3. (F) Gradual color changes of cell pellets along the ED step. (G) A representative flow cytometric plot indicates that the majority of the ED10 cells express immature erythrocyte (erythroblast) markers CD235a and CD36. (H) Wright-Giemsa staining of ED10 cells. Scale bar: 50 μm. (I-O) Erythroid terminal maturation (TM) of TNC1 iPSC derivatives. (I) Cell expansion during the TM step. Means ± s.d., n = 3. (J) Suspension of 90 million TM day 8 (TM8) cells in 1 ml PBS. (K, L) Representative flow cytometric plots of TM8 cells, with >98% cells co-express CD235a and Band3, and ~10% enucleation marked by negative staining of DRAQ5. (M) Phase-contrast and fluorescent images of TM8 cells. Blue arrows indicate enucleated erythrocytes. Scale bars: 30 μm. (N) Quantitative PCR analysis of gene expression of HBB (adult) and HBG (fetal) at different differentiation stages of TNC1 cells, comparing to the cultures of CB CD34+ cells. Means ± s.d., n = 3. (O) benzidine (brown color) for hemoglobin and Wright-Giemsa (nuclear DNA) staining of TM8 cells. 40× image. Scale bar: 50 μm. Blue arrows indicate enucleated erythrocytes lacking nucleus.
Next, the harvested cells from culture suspension at day 14 after EB formation or the purified CD45+CD34+ cells were expanded in erythroid differentiation (ED) culture in the presence of SCF, EPO, IL-3 and holo-transferrin (Step II in Fig. 1A). After 10 days, the total cells expanded ~40-fold (Fig. 1E), turned reddish gradually (Fig. 1F), expressed erythroblast markers such as CD36 and CD235a (Glycophorin A) and CD45 (not shown) at day 9–10 (Fig. 1G), and displayed erythroblast morphology (Fig. 1H). The erythroblast-like cells were allowed to terminally differentiate in culture with EPO, heparin, insulin and a high concentration of holo-transferrin (Step III in Fig. 1A). We also found that adding human plasma, which enhanced erythrocyte enucleation of erythroblasts derived from primary CD34+ cells [22], also stimulated terminal differentiation of human iPSC-derived erythroblasts as reported recently [23, 24]. Upon terminal differentiation, the cells expanded for another 10-fold (>3 cell divisions) after 7–8 days before ceasing cell proliferation. The differentiated cells at this stage turned reddish, lost CD45 expression in the majority of cells (>98%), continued CD235a expression, and gained the expression of a maturation marker Band3 in both nucleated and enucleated (~10.6%) cells (Fig. 1I to 1L). In addition to the Band3+ and CD235a+ erythrocytes (>98%), we consistently observed a small population (0.2% to 1.6%) of nucleated cells in the harvested suspension cells after 6–8 days of terminal differentiation that express CD45, but lack CD235a and Band 3 expression (Fig. 1K). Morphological staining suggests that these residual nucleated CD45+ cells resemble monocytes or macrophages (data not shown). Using quantitative RT-PCR analysis, we observed that the HBB gene expression in erythroblasts (ED10) and erythrocytes (TM8) from either TNC1 iPSCs or CB CD34+ cells under the same improved culture condition, while the HBG gene expression is ~10–fold than HBB (Fig. 1N). We further confirmed that enucleated cells expressed hemoglobin as stained by benzidine (Fig. 1O). Similar data were obtained using the BC1 iPSC line (Supplemental Fig. S1), with an enucleation rate of 4.7% ± 2.8% (n=21) and producing erythrocytes that express HBB as well as HBG proteins. Thus, we demonstrated that we can generate a population of human erythrocytes from blood cell-derived iPSCs using an improved (free-free and xeno-free) culture condition.
Efficient and specific targeting of the SCD HBB locus in human iPSCs derived from blood cells
Previously, we and others have used a pair of ZFNs or TALENs to target the SCD point mutation in human iPSCs and cancer lines [8–13]. Several groups have achieved HR-mediated gene correction of the SCD HBB allele in human iPSC lines that were derived from fibroblasts using genome-inserting vectors, although the HR efficiencies were typically low [8, 9, 13]. To improve the HBB gene targeting in blood-derived SCD iPSCs that were generated by transient plasmid expression and showed better erythroid differentiation (Fig. 1), we used the CRISPR/Cas9 endonuclease system [25]. Three different guide RNAs (gRNAs), targeting the HBB DNA sequence between the SCD mutation and an intended targeting insertion in intron 1, were made (Supplemental Fig. S2). In the preliminary studies using the 293T cell lines, guide gRNA-a and gRNA-c showed a high level of targeting at the HBB sequence (data not shown), and therefore were used for subsequent analyses. Because the gene targeting efficiency in human iPSCs is >10-fold lower than in 293T cells by either HR or NHEJ mechanisms, we employed a Next-Gen sequencing (MiSeq)-based method (Fig. S2C) as previously described [18, 25]. The level of CRISPR/Cas9 stimulated NHEJ events that often result in mis-repairs and indels can be measured as mutation (indel) rates (Fig. S2C). We also evaluated efficiency and specificity of gRNA in conjunction of Cas9 at targeting the endogenous HBB locus, in comparison with the ZFNs and TALENs published previously [8–10]. Although the ZFNs and TALENs targeted efficiently at the HBB reporter transgene and in 293T cells at the endogenous HBB locus, they were significantly less efficient in human iPSCs than gRNA-a/Cas9 in making a DSB that triggers NHEJ-mediated indel formation (Fig. S2D). Using the same assay, we also examined levels of off-targeting mediated by either gRNA-a or gRNA-c at the nearby HBD locus (Fig. S2E), which is highly homologous to the HBB sequence (Fig. S2B). Additionally we examined 14 bioinformatically predicted off-targets of gRNA-a (Fig. S2F–G). We found that gRNA-a is highly specific in both human iPSCs (Fig. S2F) and to a lesser extent in 293T cells (Fig. S2G).
Next, we targeted the endogenous HBB locus in SCD iPSCs, by using the Cas9 and the gRNA-a that targets HBB 20-bp downstream to the βs mutation (Fig. S2B and Fig. 2A). The TNC1 iPSCs containing the homozygous βs mutation were transfected with the donor plasmid (Fig. 2B), gRNA-a, and Cas9 vector. The donor vector provides the wild-type HBB DNA to correct the βs point mutation and a puromycin-resistance selection cassette that is inserted into the HBB intron 1 and useful in selecting a HR product (Fig. 2A). The drug-resistant iPSC colonies were picked and screened for a HR product by PCR amplification of genomic DNA using two pairs of primers (Fig. 2C). Each pair has one primer sequence inside the puromycin selection cassette (TP1R and TP2F) and the other at the genomic sequence outside the homologous arms (TP1F and TP2R) of the donor plasmid (Fig. 2F; Fig S2D–E). Of the ten clones first screened, four (40%) showed HR detected by both pairs of primers (Fig. 2F). More are shown in Fig. S3D–E. The targeted clones that were positive for both primer pairs were selected for further analysis (Table 1).
Figure 2. A strategy for site-specific correction of the sickle cell disease mutation in the HBB locus using homologous recombination stimulated by Cas9 and a guide RNA.
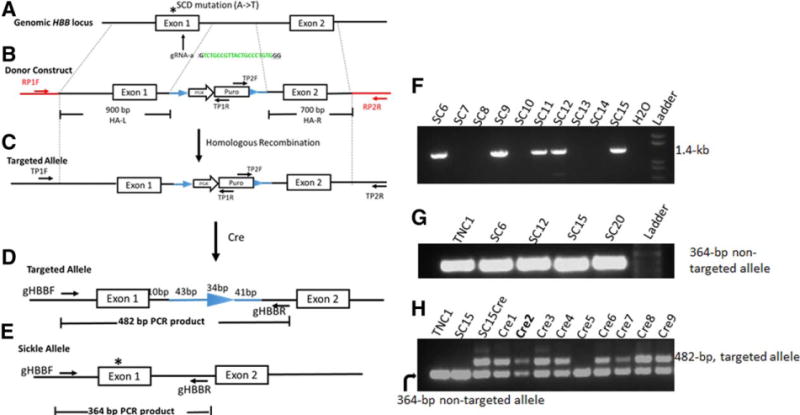
(A) The HBB genomic structure (first two exons), and locations of SCD mutation and gRNA-a targeting HBB exon 1. (B) The gene-targeting donor MGH vector with 2 homology arms (900-bp left arm and 700-bp right arm) introduces a homologous recombination (HR) template for T-to-A replacement in the sickle allele and a loxP-flanked drug-selection cassette PGK-puromycin (puro) to be inserted into the HBB intron 1. (C) Targeted allele following HR with corrected SCD mutation and inserted PGK-puro cassette in intron 1. We used 2 PCR primers (black arrows: TP1F/TP1R and TP2F/TP2R) for initial screening of targeted insertion (TI) indicative of correct HR. (D) Cre-mediated excision removes the PGK-puro cassette and leaves one copy of the loxP DNA (34-bp) and 84-bp linker DNA in HBB intron 1 of the corrected allele. Genomic DNA PCR with primers gHBB-F/gHBB-R produces a 482-bp product from the targeted allele of “Cre” clones. (E) Genomic DNA PCR with primers gHBB-F/gHBB-R produces a 364-bp product from the unintended sickle allele of original and “Cre” clones. (F) PCR screening for targeted integration (TI) by the primer set TP2F/TP2R that detect 3′ TI (1.4-kb). (G) Genomic PCR using the primer set gHBB-F/gHBB-R that amplify the unintended (sickle) allele but not the targeted allele (it is too long to be amplified by PCR with a short extension time) before Cre-mediated excision. (H) PCR screening of Cre-mediated excision of SC15 clones using primer set gHBB-F/gHBB-R. The 482-bp PCR product emerged after Cre-excision as shown in (D).
Table 1.
12 Clones (out of 24) with a targeted insertion (TI) as detected by primers for the 3′ junction
| Puromycin Resistant Clones | TI detected by 3′ PCR primers | TI detected by 5′ PCR primers | SCD mutation correction | Donor vector random insertion | Intact or disruption of the untargeted SCD allele by NHEJ |
|---|---|---|---|---|---|
| SC2 | + | + | + | + | / |
| SC3 | + | + | + | + | / |
| SC5 | + | + | + | + | / |
| SC6 | + | + | + | + | / |
| SC9 | + | + | + | + | / |
| SC11 | + | − | / | / | / |
| SC12 | + | + | + | − | − 5 bp deletion |
| SC15 | + | + | + | − | + 1 bp insertion |
| SC16 | + | + | Mixture of 2 clones? | / | / |
| SC17 | + | − | / | / | / |
| SC18 | + | − | / | / | / |
| SC20 | + | + | + | − | Intact (SCD mutation) |
“/”: discontinued in analysis or effort;
Abbreviations: SCD, sickle cell disease; NHEJ, non-homologous end joining
We further confirmed the correction of the SCD mutation in targeted and integrated allele by various methods, in 12 out of the first 24 clones that scored positive for at least one targeted insertion (Table 1). We also eliminated clones (such as SC2) that have a copy of undesired insertion of the donor vector. Among the four clones that showed at least one allele of targeted insertion and no sign of random vector insertion in the genome, we determine the presence and nature of the other allele, e.g., the un-intended or non-integrated allele in targeted TNC1 iPSCs (Fig. 2G). These 4 clones (SC6, SC12, SC15 and SC20) were heterozygous: each has one copy of the targeted insertion and one copy of the HBB locus similar to the wild-type or βs HBB allele. We sequenced the 364-bp PCR product of the non-integrated allele from the 4 clones to examine if there was any genetic modification. We found that SC15 has a single nucleotide insertion in the HBB exon 1 (Fig. S4A) in the gRNA-a recognition site. This insertion introduced a pre-mature stop codon in exon 1, so the predicted protein product is ~2.3-kD if it is ever produced. The clone SC20 has no genetic modification in the non-integrated HBB allele in this genomic region (Supplemental Fig. S4B). In our case, the disruption of the second βs HBB allele by the same Cas9/gRNA-a that also achieved a desired HR-mediated targeting is actually advantageous, because only the corrected allele that converts to the wild-type HBB gene can make a full-length HBB protein from iPSC clones such as SC15. We focused on the SC15 iPSC clone for subsequent analyses.
Because the PGK-puromycin gene selection cassette may interfere with expression of the corrected HBB allele [8], we excised the loxP-flanked PGK-puromycin cassette using transient transfection of a Cre-expressing plasmid [19]. We selected SC15 for Cre excision, and picked 9 individual clones to screen for excision of the LoxP DNA segment (Fig. 2H). Because the Cre excision will leave a 118-bp loxP-containing “footprint” sequence in the intron 1 of HBB locus, genomic PCR using the same primers that amply the 364-bp endogenous HBB allele (Fig. 2G) produced an additional and larger 482-bp product after Cre-mediated excision (Fig. 2H). We sequenced the 482-bp PCR product of several excised clones and confirmed the correction of SCD mutation and insertion of 118-bp “footprint” sequence in the first intron, but no other extra modifications (Supplemental Fig. 4C).
Production of human erythrocytes from corrected iPSC clones and parental TNC1 iPSCs
Using the three-step feeder-free and xeno-free procedure for differentiating human iPSCs to erythrocytes that we recently developed (Fig. 1A), we evaluated erythrocytes production of gene corrected iPSC clones such as SC15 and SC15-Cre2, in comparison with the parental TNC1 iPSCs containing the homozygous βs mutation. The SC15-Cre2 clone behaved similarly to the TNC1 iPSCs at various stages of differentiation. After the terminal differentiation (Fig. 3A), the cells gradually lost the markers of CD45 and CD49d, maintained CD235a expression, and gained the expression of Band3 as previously described for erythrocytes from primary CB CB34+ cells [22]. Among the mature erythrocytes that are Band3+ but CD45- and CD49d-, ~5–8% of cells lacked DNA staining by DRAQ5 (Fig. 3A and 3B). Many of the enucleated cells remained metabolic active with an intact cell membrane, because they were positive when probed by Calcein AM (a fluorogenic substrate of cellular esterases) (Fig. 3C). The enucleated cells (more abundant at day 8) also expressed hemoglobin as stained positive for benzidine (Fig. 3D).
Figure 3. Erythroid terminal maturation of erythroblasts that generated from SC15-Cre2 iPSC clone.
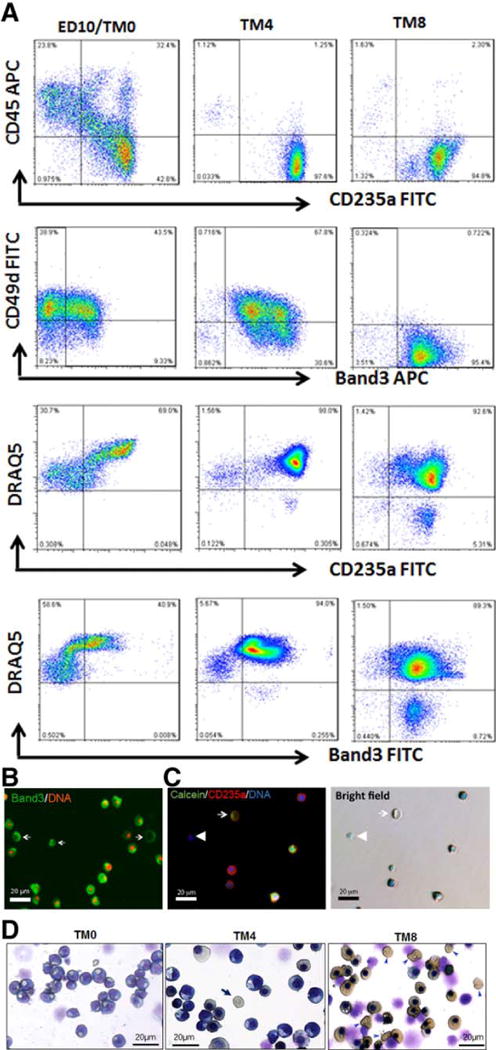
The phenotypes of erythroblasts (ED10) from SC15-Cre2 are similar those from TNC1 and BC1 iPSC lines. (A) Cell surface marker expression dynamics during 8 days of terminal maturation (TM). By day 8 of TM (TM8), nearly all the cells expressed CD235a and Band3, and lost the expression of CD45 and CD49d. About 9% of CD235a+ and Band3+ cells were stained negative for DNA by DRAQ5. (B) Fluorescent image of TM8 cells showing Band3 cell surface expression and ~10% lacking DNA staining (red). (C) Fluorescent images of TM8 cells showing cell surface expression CD235a in enucleated cells (lacking blue DAN staining) that are metabolically active based on Calcein AM staining (green, by arrows). (D) Histology staining and morphology of cells after terminal maturation at day 0 (ED10), day 4 (TM4) and day 8 (TM8) after Wright-Giemsa (blue) and benzidine (brown) staining. Enucleated and hemoglobinized cells are easily identified (arrow). Scale bar: 20μm.
Production of corrected HBB proteins in erythrocytes derived from genome-edited SCD iPSCs
We specifically measured the level of corrected HBB gene expression and protein in ex vivo generated erythrocytes (TM8) from the gene-edited SC15-Cre2 clones, in comparison to the parental TNC1 that expressed the βs protein (Fig 4). The RT-PCR primers and the HBB antibody would not distinguish the wild-type and βs forms, allowing us to quantitatively compare the level of HBB mRNAs or proteins in erythrocytes from the corrected SC15-Cre2 or parental TNC1 iPSCs. By RT-PCR, the level of HBB mRNA in SC15-Cre2 after terminal differentiation (TM8) increased further over those at earlier stages, but appeared >10-fold lower than those in erythrocytes from CB or TNC1 iPSCs (Fig. 4A). By Western blot, the HBB protein level in the erythrocyte culture from the SC15-Cre2 iPSCs is comparable to that from the parental TNC1 iPSCs (Fig. 4B). The HBB protein level in the erythrocytes derived from the corrected and excised SC15-Cre2 iPSCs appeared to be higher than that from the SC15 iPSCs, the genome-edited clones without the removal of the PGK-puromycin selection cassette located in the HBB intron 1. The data corroborated with the early observations that it is important to remove the expression cassette for selection even if it is located in an intron [26].
Figure 4. Analysis of globin gene and protein expressions the corrected erythrocytes, and their function.
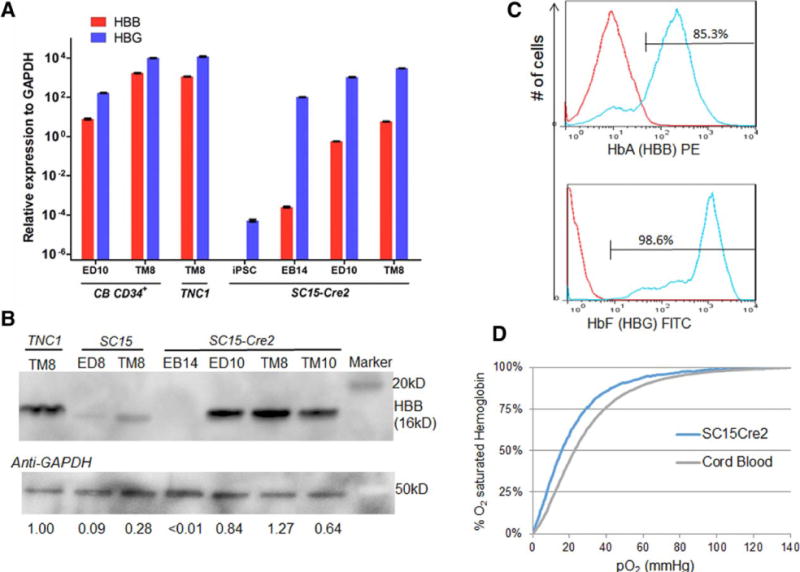
(A) Quantitative RT-PCR analysis of HBB and HBG gene expression in iPSCs, after hematopoietic differentiation (EB14), erythroid differentiation (ED10) and terminal maturation (TM8) from SC15-Cre2 and parental TNC1 iPSCs and a control of cord blood (CB) CD34+ cells. (B) Western blot analysis of adult hemoglobin beta (HBB) protein level during the differentiation of iPSC clones before and after correction and excision. The relative levels of full-length HBB proteins normalized to the GAPDH control are also indicated. (C) FACS analysis of intracellular adult and fetal hemoglobin protein expression of maturated erythrocytes generated from SC15-Cre2 iPSC clone. (D) Oxygen affinity curves of erythrocytes from SC15-Cre2 after terminal maturation and a cord blood control as measured by a Hemox Analyzer.
We also measured the HBB and HBG proteins in the native form of hemoglobin tetramers in individual cells by flow cytometric analysis (Fig. 4C). We detected >85% and >98% of cells expressed HBB and HBG proteins, respectively. However, the level of HBB proteins appears ~10-fold lower that that of HBG (Fig. 4C): nearly all the HBB-expressing cells also express HBG (Supplemental Fig. 1I).
We also measured the functionality of O2 binding by hemoglobin produced in the erythrocyte generated from iPSCs, in comparison with primary CB erythrocytes (Fig. 4D). The iPSC-generated cells have an O2-binding curve similar to that of cord blood, except that the affinity is slightly higher (p50 is 16.6 in SC15-Cre2 vs. 22.9 for CB). This may reflect the fact that HbF (containing HBG), which has a higher affinity for oxygen, are more abundant in the erythrocytes derived the iPSCs.
Discussion
In this study, we demonstrate that the sickle point mutation in the HBB allele in patient-specific iPSCs can be corrected efficiently by genome editing, which allows the expression of normal HBB proteins in erythrocytes after hematopoietic differentiation of gene-corrected iPSCs. This study has broad implications for studies using human iPSCs for disease modeling, and for developing combined gene and (stem) cell therapies. First, this study corroborates with other recent studies showing that CRISPR/Cas9 can be both efficient and specific in human iPSCs for gene targeting, even for correcting a point mutation [14, 27–29]. Second, the CRISPR/Cas9-mediated genome editing has little adverse effect on the integrity of targeted iPSCs, since they can differentiate into mature progeny such as enucleated erythrocytes that express the corrected HBB gene in SCD iPSCs. The level of HBB protein expression is similar to that of erythrocytes differentiated from untargeted iPSCs (Fig. 4).
Our study showing HBB protein expression in human erythrocytes differentiated from corrected iPSCs would not have been possible, if the HBB mRNA expression and protein synthesis had remained at low levels as previously observed [8, 9, 30–35]. Previously, HBB protein expression was not detected in erythrocytes generated from human iPSCs or ESCs ex vivo, unless transgene expression promoting erythroid formation [36–38] or co-culture with mouse stromal cell lines such as OP9 or MS5 were used [24, 33, 39]. Compared to these pioneering studies, we achieved much higher levels of HBB mRNA expression under the culture condition for producing erythrocytes generated from iPSCs such as TNC1 and their corrected progeny (Figure 4). In addition, we also observed the presence of enucleated erythrocytes under the feeder-free and xeno-free culture condition, although the percentage was not high (typically 5–10%) compared to that derived from CB CD34+ cell cultures (30% or higher as reported).
The much improved erythrocyte maturation and HBB protein expression from human iPSCs that we reported here could be due to at least the following two possibilities, which are not mutually exclusive. The first one is that we improved culture condition for terminal differentiation: we added human plasma that is known to promote enucleation, and omitted dexamethasone that promotes stressed erythropoiesis and fetal globin expression. The second one is that we used blood-derived iPSCs that have some residual epigenetic memory inherited from parental cells (TNC1 was derived from PB erythroblasts and BC1 was from adult BM hematopoietic cells). This notion is also supported by a recent study showing that blood-derived iPSCs tend to have better erythroid differentiation potential than fibroblast-derived iPSCs [37]. Together, these studies provide additional strong evidence that residual epigenetic memory found in these blood-derived iPSCs [40–42] favors differentiation back to erythrocytes, especially when the differentiation condition is still sub-optimal. According to this notion, blood MNC-derived iPSCs are a better choice for efficient erythroid differentiation than fibroblast-derived iPSCs and human ESC lines (especially those in the NIH registry that are derived some years ago and in high passage numbers). It is of note, however, that the fibroblast-derived iPSCs could generate enucleated cells and express the HBB proteins under enhanced conditions [23, 24], even though at an efficiency lower than that from blood-derived human iPSCs as we report here. We predict that with improved differentiation conditions, the origin of human iPS cells will be even less critical to the efficient formation of erythrocytes.
Under the current culture conditions, however, the level of HBB expression (at both mRNA and protein levels) is still >10-fold lower than that of the HBG. This may explain the fact that we and others did not observe sickling or defect in erythrocytes generated from iPSCs derived from SCD patients [23]. The high-level of HBG protein may prevent the sickling of α2βs2 complexes, especially under culture conditions with a normoxic level of O2. In the future, human differentiated erythrocytes generated ex vivo from human iPSCs (with or without genome editing) should also be tested using improved immune-deficient mice and conditioning, to examine in vivo properties of human erythrocyte in the circulation of mouse models.
For future therapeutic uses, it is desirable to correct at least one of the βs HBB allele in the iPSCs derived from SCD patients, before the iPSC-derived HPCs or erythrocyte can be used for autologous cell transplantation or transfusion. In addition to achieving the elusive objective to develop a robust and reproducible method for generating transplantable HPCs [43, 44], we could also use erythrocytes generated from patient-specific iPSCs, with or without genome-editing to correct the underlying genetic defect, for transfusion medicine. The enucleated erythrocytes generated from iPSC lines, which could be further irradiated to block DNA replication, may have less risk than other nucleated progeny in causing cancers [45–47]. We also envision that we could generate genetically modified erythrocytes from genome-engineered iPSCs, for providing a source of functionally enhanced erythrocytes for cell therapies, not just a source of replacement in transfusion medicine [47]. Future studies are warranted to test efficacy and safety of erythrocytes from genome-edited iPSCs in preclinical models and in clinical trials.
Summary
In this study, we used the human SCD model and demonstrated that a point mutation could be corrected in patient-specific iPSCs by genome editing, which allows the expression of the corrected gene after differentiation into biologically relevant cells. Our result is a significant step towards the goal of generating gene-edited and functionally repaired or enhanced cells after genome editing of patient-specific iPSCs and possible other stem cell types, for disease modeling and future gene therapies.
Supplementary Material
Supplemental Figure 1. Hematopoietic differentiation and erythroid differentiation and terminal maturation of BC1 iPSCs. The BC1 iPSCs, derived from a health adult bone marrow Cd34+ cells, were differentiated to erythrocytes as described in Figure 1 (for TNC1 iPSCs). (A) The yield of CD34+, CD45+, and CD34+CD45+ cells derived from one input iPSC, after EB formation and hematopoietic differentiation at day 14 (EB14). Means ± s.d., n = 4. (B, D, F) Representative flow cytometry analysis of BC1 cells on EB14, erythroid differentiation at day 10 (ED10), and terminal differentiation day 8 (TM8), respectively, as described in Figure 1. (C, E) Cell expansion during erythroid differentiation (ED) and terminal maturation (TM). Means ± s.d., n = 8. (G) Benzidine (gold brown color) and Wright-Giemsa staining of TM cells. (H) Wright-Giemsa staining of TM cells. 40× images. Scale bar: 50 mm. (I) Representative flow cytometry analysis of fetal (HbF) and adult (HbA) hemoglobin in TM8 BC1 cells. (J) Quantitative PCR analysis of gene expression of HBB (adult) and HBG (fetal) at different differentiation stages of BC1 cells, comparing to CB CD34+ cells. Means ± s.d., n = 3.
Supplemental Figure 2. Efficiency and specificity of the guide RNAs used in the CRIPSR/Cas9 system for targeting the endogenous HBB locus near the βspoint mutation. (A) A scheme of beta-globin cluster locus. The DNA sequence of the HBB and HBD sequence in the exon 1 near the βs point mutation (A>T). The recognition sequences of 3 tested guide RNA (gR), a, b and c for targeting the HBB sequence, were denoted from by arrow from 5′ to 3′, followed by the NGG PAM motif. The recognition sequences of the ZFNs [8, 9] and TALENs [10] that were used in previous studies to target the HBB sequences were also denoted and tested. (C) A strategy to measure the efficiency of each type designer nuclease to stimulate the targeting of the endogenous HBB locus is outlined. This strategy is based on the measurement of small insertion and deletion due to non-homologous-end-joining resulting from the double strand break induced by a designer nuclease. (D) The frequencies of each nuclease to induce indels in human iPSCs (TNC1) after transient transfection (nucleofection) of a designer nuclease. The indel calls are based on Miseq as compared to the DNA sequence of untransfected TNC1 iPSCs. (E) The indel frequencies induced by gRNA-a and gRNA-c in TNC1 iPSCs at both HBB and HBD loci were examined, after Miseq of different genomic PCR products specific for either HBB or HBD DNA sequences. (F, G) The indel frequencies at the additional 14 likely (off-) targeting sequences were also examined, in comparison with that of the HBB targeting in both human iPSCs (TNC1) and 293T cells (blue bars). As controls, the indel frequencies or basal DNA sequencing errors in each locus was also measured using the untransfected cells as controls (in red bars, but negligible). A low level of cut was observed in 203T cells at 6 sites in addition to the HBB sequence (G), which is significantly higher than the background (*). However, we did not observe this low-level background in these and other 8 sites in human iPSCs, which have an overall >10-fold lower efficiency by this assay (F).
Supplemental Figure 3. Strategies to screen and select clones with intended homologous recombination that result in a targeted insertion and correction of the sickle mutation. (A) Genomic HBB locus and locations of SCD mutation and gRNA-a targeting HBB exon 1. (B) The gene-targeting donor MGH vector with 2 homology arms (900-bp left arm and 700-bp right arm) introduces an HR template for T-to-A replacement in the sickle allele and a loxP-flanked drug-selection cassette PGK-puromycin to be inserted into the HBB intron 1. (C) Targeted allele following homologous recombination with the corrected SCD mutation and inserted PGK-puromycin cassette in intron 1. (D, E) We used 2 PCR primers (black arrows: TP1F/TP1R and TP2F/TP2R) for initial screening of targeted insertion (TI) indicative of correct HR. (F) The clones with both 5′ and 3′ targeted insertions were screened to against those that an undesired insertion of the vector sequences (such as RP1F and RP2R), likely due to random insertion of the whole vector DNA. The results of 24 screened iPSC clones we screened that have 3′ targeted insertion (TI) are summarized in Table 1.
Supplemental Figure 4. Analysis of the second (intended SCD) HBB allele in iPSC clones with intended gene correction and insertion. Genomic DNA PCR with primers gHBB-F/gHBB-R produces a 364-bp product from the untargeted (or unintended) sickle allele (with or without excision mediated by Cre). (A) The sequence of the clone SC15 in the unintended allele. One basepair deletion was detected within the recognition sequence of the guide RNA-a, indicating that the guide RNA induced a NHEJ resulting in the deletion. As a result, the unintended (SCD) HBB allele was disrupted, due to the acquisition of a stop codon (TGA) soon after a reading frame-shift. This result was further confirmed by sequencing the sequence of the untargeted allele in SC15 after Cre excision, in clones such as SC15-Cre2. (B) The DNA sequence from the clone SC20 perfectly matched to that in parental TNC1 cells, indicating that the unintended SCD allele is indeed intact and untargeted. (C) The sequence of the targeted and excised allele in the SC15-Cre2 was also sequenced (amplified by the same gHBB-F/gHBB-R primers, resulting in a 482-bp DNA fragment). The corrected allele indeed have a corrected A, to in replacing the βs point mutation (T). All the DNA sequences are normal as found in the wild-type HBB allele, except that a loxP-containing (in purple) in the intron 1.
Acknowledgments
We thank Dr. Xiuli An at NYBC for providing Band 3 antibodies, Dr. J. Keith Joung in MGH for providing the MGH donor vector, Drs. Evan Braunstein and Robert Brodsky for discussions and critical reading. This work was supported in part by grants from Maryland State Stem Cell Research Fund (2011-MSCRFII-0088 and 2011-MSCRFE-0087) and from NIH (2R01 HL-073781, U01 HL107446 and T32HL007525-31). L. C. is also supported by Edythe Harris Lucas and Clara Lucas Lynn Chair in Hematology of Johns Hopkins University.
Footnotes
Author contributions:
Xiaosong Huang: Conception and design, Collection and/or assembly of data, Data analysis and interpretation, Manuscript writing
Ying Wang: Collection and/or assembly of data, Data analysis and interpretation, Manuscript writing
Wei Yan, Cory Smith and Zhaohui Ye: Collection and/or assembly of data, Data analysis and interpretation
Jing Wang and Laurel Mendelsohn: Collection and/or assembly of data
Yongxing Gao: Collection and/or assembly of data, Administrative support
Linzhao Cheng: Conception and design, Data analysis and interpretation, Manuscript writing, Financial support, Final approval of manuscript
The authors indicate no potential conflicts of interest.
References
- 1.Porteus MH, Carroll D. Gene targeting using zinc finger nucleases. Nature biotechnology. 2005;23(8):967–973. doi: 10.1038/nbt1125. [DOI] [PubMed] [Google Scholar]
- 2.Porteus MH. Mammalian gene targeting with designed zinc finger nucleases. Molecular therapy : the journal of the American Society of Gene Therapy. 2006;13(2):438–446. doi: 10.1016/j.ymthe.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Hockemeyer D, Jaenisch R. Gene targeting in human pluripotent cells. Cold Spring Harbor symposia on quantitative biology. 2010;75:201–209. doi: 10.1101/sqb.2010.75.021. [DOI] [PubMed] [Google Scholar]
- 4.Collin J, Lako M. Concise review: putting a finger on stem cell biology: zinc finger nuclease-driven targeted genetic editing in human pluripotent stem cells. Stem cells. 2011;29(7):1021–1033. doi: 10.1002/stem.658. [DOI] [PubMed] [Google Scholar]
- 5.Mali P, Cheng L. Concise review: Human cell engineering: cellular reprogramming and genome editing. Stem cells. 2012;30(1):75–81. doi: 10.1002/stem.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zou J, Maeder ML, Mali P, Pruett-Miller SM, Thibodeau-Beganny S, Chou BK, Chen G, Ye Z, Park IH, Daley GQ, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell stem cell. 2009;5(1):97–110. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nature biotechnology. 2009;27(9):851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zou J, Mali P, Huang X, Dowey SN, Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood. 2011;118(17):4599–4608. doi: 10.1182/blood-2011-02-335554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sebastiano V, Maeder ML, Angstman JF, Haddad B, Khayter C, Yeo DT, Goodwin MJ, Hawkins JS, Ramirez CL, Batista LF, et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem cells. 2011;29(11):1717–1726. doi: 10.1002/stem.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan W, Smith C, Cheng L. Expanded activity of dimer nucleases by combining ZFN and TALEN for genome editing. Scientific reports. 2013;3:2376. doi: 10.1038/srep02376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voit RA, Hendel A, Pruett-Miller SM, Porteus MH. Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucleic acids research. 2014;42(2):1365–1378. doi: 10.1093/nar/gkt947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic acids research. 2013;41(20):9584–9592. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun N, Zhao H. Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnology and bioengineering. 2014;111(5):1048–1053. doi: 10.1002/bit.25018. [DOI] [PubMed] [Google Scholar]
- 14.Chou BK, Mali P, Huang X, Ye Z, Dowey SN, Resar LM, Zou C, Zhang YA, Tong J, Cheng L. Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell research. 2011;21(3):518–529. doi: 10.1038/cr.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dowey SN, Huang X, Chou BK, Ye Z, Cheng L. Generation of integration-free human induced pluripotent stem cells from postnatal blood mononuclear cells by plasmid vector expression. Nature protocols. 2012;7(11):2013–2021. doi: 10.1038/nprot.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen G, Gulbranson DR, Hou Z, Bolin JM, Ruotti V, Probasco MD, Smuga-Otto K, Howden SE, Diol NR, Propson NE, et al. Chemically defined conditions for human iPSC derivation and culture. Nature methods. 2011;8(5):424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Chou BK, Dowey S, He C, Gerecht S, Cheng L. Scalable expansion of human induced pluripotent stem cells in the defined xeno-free E8 medium under adherent and suspension culture conditions. Stem cell research. 2013;11(3):1103–1116. doi: 10.1016/j.scr.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith C, Abalde-Atristain L, He C, Brodsky B, Braunstein E, Chaudhari P, Jang Y, Cheng L, Ye Z. Efficient and allele-specific genome editing of disease loci in human iPSCs. Molecular therapy : the journal of the American Society of Gene Therapy. 2014 doi: 10.1038/mt.2014.226. online Nov 24, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woodhead GJ, Mutch CA, Olson EC, Chenn A. Cell-autonomous beta-catenin signaling regulates cortical precursor proliferation. The Journal of Neuroscience. 2006;26(48):12620–12630. doi: 10.1523/JNEUROSCI.3180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye Z, Liu CF, Lanikova L, Dowey SN, He C, Huang X, Brodsky RA, Spivak JL, Prchal JT, Cheng L. Differential sensitivity to JAK inhibitory drugs by isogenic human erythroblasts and hematopoietic progenitors generated from patient-specific induced pluripotent stem cells. Stem Cells. 2014;32(1):269–278. doi: 10.1002/stem.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang X, Shah S, Wang J, Ye Z, Dowey SN, Tsang KM, Mendelsohn LG, Kato GJ, Kickler TS, Cheng L. Extensive ex vivo expansion of functional human erythroid precursors established from umbilical cord blood cells by defined factors. Molecular therapy : the journal of the American Society of Gene Therapy. 2014;22(2):451–463. doi: 10.1038/mt.2013.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu J, Liu J, Xue F, Halverson G, Reid M, Guo A, Chen L, Raza A, Galili N, Jaffray J, et al. Isolation and functional characterization of human erythroblasts at distinct stages: implications for understanding of normal and disordered erythropoiesis in vivo. Blood. 2013;121(16):3246–3253. doi: 10.1182/blood-2013-01-476390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobari L, Yates F, Oudrhiri N, Francina A, Kiger L, Mazurier C, Rouzbeh S, El-Nemer W, Hebert N, Giarratana MC, et al. Human induced pluripotent stem cells can reach complete terminal maturation: in vivo and in vitro evidence in the erythropoietic differentiation model. Haematologica. 2012;97(12):1795–1803. doi: 10.3324/haematol.2011.055566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang CT, French A, Goh PA, Pagnamenta A, Mettananda S, Taylor J, Knight S, Nathwani A, Roberts DJ, Watt SM, et al. Human induced pluripotent stem cell derived erythroblasts can undergo definitive erythropoiesis and co-express gamma and beta globins. British journal of haematology. 2014;166(3):435–448. doi: 10.1111/bjh.12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis RP, Costa M, Grandela C, Holland AM, Hatzistavrou T, Micallef SJ, Li X, Goulburn AL, Azzola L, Elefanty AG, et al. A protocol for removal of antibiotic resistance cassettes from human embryonic stem cells genetically modified by homologous recombination or transgenesis. Nature protocols. 2008;3(10):1550–1558. doi: 10.1038/nprot.2008.146. [DOI] [PubMed] [Google Scholar]
- 27.Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, Musunuru K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell stem cell. 2013;12(4):393–394. doi: 10.1016/j.stem.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C, Wang Y, Brodsky RA, Zhang K, Cheng L, et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell stem cell. 2014;15(1):12–13. doi: 10.1016/j.stem.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H, Erdin S, Talkowski ME, Musunuru K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 2014;15(1):27–30. doi: 10.1016/j.stem.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiu C, Hanson E, Olivier E, Inada M, Kaufman DS, Gupta S, Bouhassira EE. Differentiation of human embryonic stem cells into hematopoietic cells by coculture with human fetal liver cells recapitulates the globin switch that occurs early in development. Experimental hematology. 2005;33(12):1450–1458. doi: 10.1016/j.exphem.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Lu SJ, Feng Q, Park JS, Vida L, Lee BS, Strausbauch M, Wettstein PJ, Honig GR, Lanza R. Biologic properties and enucleation of red blood cells from human embryonic stem cells. Blood. 2008;112(12):4475–4484. doi: 10.1182/blood-2008-05-157198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma F, Ebihara Y, Umeda K, Sakai H, Hanada S, Zhang H, Zaike Y, Tsuchida E, Nakahata T, Nakauchi H, et al. Generation of functional erythrocytes from human embryonic stem cell-derived definitive hematopoiesis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(35):13087–13092. doi: 10.1073/pnas.0802220105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dias J, Gumenyuk M, Kang H, Vodyanik M, Yu J, Thomson JA, Slukvin II. Generation of red blood cells from human induced pluripotent stem cells. Stem cells and development. 2011;20(9):1639–1647. doi: 10.1089/scd.2011.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang KH, Huang A, Hirata RK, Wang PR, Russell DW, Papayannopoulou T. Globin phenotype of erythroid cells derived from human induced pluripotent stem cells. Blood. 2010;115(12):2553–2554. doi: 10.1182/blood-2009-11-252650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie F, Ye L, Chang JC, Beyer AI, Wang J, Muench MO, Kan YW. Seamless gene correction of beta-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome research. 2014;24(9):1526–1533. doi: 10.1101/gr.173427.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ran D, Shia WJ, Lo MC, Fan JB, Knorr DA, Ferrell PI, Ye Z, Yan M, Cheng L, Kaufman DS, et al. RUNX1a enhances hematopoietic lineage commitment from human embryonic stem cells and inducible pluripotent stem cells. Blood. 2013;121(15):2882–2890. doi: 10.1182/blood-2012-08-451641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ochi K, Takayama N, Hirose S, Nakahata T, Nakauchi H, Eto K. Multicolor staining of globin subtypes reveals impaired globin switching during erythropoiesis in human pluripotent stem cells. Stem cells translational medicine. 2014;3(7):792–800. doi: 10.5966/sctm.2013-0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trakarnsanga K, Wilson MC, Lau W, Singleton BK, Parsons SF, Sakuntanaga P, Kurita R, Nakamura Y, Anstee DJ, Frayne J. Induction of adult levels of beta-globin in human erythroid cells that intrinsically express embryonic or fetal globin by transduction with KLF1 and BCL11A-XL. Haematologica. 2014;99(11):1677–1685. doi: 10.3324/haematol.2014.110155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi KD, Vodyanik MA, Togarrati PP, Suknuntha K, Kumar A, Samarjeet F, Probasco MD, Tian S, Stewart R, Thomson JA, et al. Identification of the hemogenic endothelial progenitor and its direct precursor in human pluripotent stem cell differentiation cultures. Cell reports. 2012;2(3):553–567. doi: 10.1016/j.celrep.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LI, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467(7313):285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim K, Zhao R, Doi A, Ng K, Unternaehrer J, Cahan P, Huo H, Loh YH, Aryee MJ, Lensch MW, et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nature biotechnology. 2011;29(12):1117–1119. doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang G, Zhang Y. Genetic and epigenetic variations in iPSCs: potential causes and implications for application. Cell stem cell. 2013;13(2):149–159. doi: 10.1016/j.stem.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaufman DS. Toward clinical therapies using hematopoietic cells derived from human pluripotent stem cells. Blood. 2009;114(17):3513–3523. doi: 10.1182/blood-2009-03-191304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slukvin II. Hematopoietic specification from human pluripotent stem cells: current advances and challenges toward de novo generation of hematopoietic stem cells. Blood. 2013;122(25):4035–4046. doi: 10.1182/blood-2013-07-474825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bouhassira EE. Concise review: production of cultured red blood cells from stem cells. Stem cells translational medicine. 2012;1(12):927–933. doi: 10.5966/sctm.2012-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Migliaccio AR, Whitsett C, Papayannopoulou T, Sadelain M. The potential of stem cells as an in vitro source of red blood cells for transfusion. Cell stem cell. 2012;10(2):115–119. doi: 10.1016/j.stem.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shah S, Huang X, Cheng L. Concise review: stem cell-based approaches to red blood cell production for transfusion. Stem cells translational medicine. 2014;3(3):346–355. doi: 10.5966/sctm.2013-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Hematopoietic differentiation and erythroid differentiation and terminal maturation of BC1 iPSCs. The BC1 iPSCs, derived from a health adult bone marrow Cd34+ cells, were differentiated to erythrocytes as described in Figure 1 (for TNC1 iPSCs). (A) The yield of CD34+, CD45+, and CD34+CD45+ cells derived from one input iPSC, after EB formation and hematopoietic differentiation at day 14 (EB14). Means ± s.d., n = 4. (B, D, F) Representative flow cytometry analysis of BC1 cells on EB14, erythroid differentiation at day 10 (ED10), and terminal differentiation day 8 (TM8), respectively, as described in Figure 1. (C, E) Cell expansion during erythroid differentiation (ED) and terminal maturation (TM). Means ± s.d., n = 8. (G) Benzidine (gold brown color) and Wright-Giemsa staining of TM cells. (H) Wright-Giemsa staining of TM cells. 40× images. Scale bar: 50 mm. (I) Representative flow cytometry analysis of fetal (HbF) and adult (HbA) hemoglobin in TM8 BC1 cells. (J) Quantitative PCR analysis of gene expression of HBB (adult) and HBG (fetal) at different differentiation stages of BC1 cells, comparing to CB CD34+ cells. Means ± s.d., n = 3.
Supplemental Figure 2. Efficiency and specificity of the guide RNAs used in the CRIPSR/Cas9 system for targeting the endogenous HBB locus near the βspoint mutation. (A) A scheme of beta-globin cluster locus. The DNA sequence of the HBB and HBD sequence in the exon 1 near the βs point mutation (A>T). The recognition sequences of 3 tested guide RNA (gR), a, b and c for targeting the HBB sequence, were denoted from by arrow from 5′ to 3′, followed by the NGG PAM motif. The recognition sequences of the ZFNs [8, 9] and TALENs [10] that were used in previous studies to target the HBB sequences were also denoted and tested. (C) A strategy to measure the efficiency of each type designer nuclease to stimulate the targeting of the endogenous HBB locus is outlined. This strategy is based on the measurement of small insertion and deletion due to non-homologous-end-joining resulting from the double strand break induced by a designer nuclease. (D) The frequencies of each nuclease to induce indels in human iPSCs (TNC1) after transient transfection (nucleofection) of a designer nuclease. The indel calls are based on Miseq as compared to the DNA sequence of untransfected TNC1 iPSCs. (E) The indel frequencies induced by gRNA-a and gRNA-c in TNC1 iPSCs at both HBB and HBD loci were examined, after Miseq of different genomic PCR products specific for either HBB or HBD DNA sequences. (F, G) The indel frequencies at the additional 14 likely (off-) targeting sequences were also examined, in comparison with that of the HBB targeting in both human iPSCs (TNC1) and 293T cells (blue bars). As controls, the indel frequencies or basal DNA sequencing errors in each locus was also measured using the untransfected cells as controls (in red bars, but negligible). A low level of cut was observed in 203T cells at 6 sites in addition to the HBB sequence (G), which is significantly higher than the background (*). However, we did not observe this low-level background in these and other 8 sites in human iPSCs, which have an overall >10-fold lower efficiency by this assay (F).
Supplemental Figure 3. Strategies to screen and select clones with intended homologous recombination that result in a targeted insertion and correction of the sickle mutation. (A) Genomic HBB locus and locations of SCD mutation and gRNA-a targeting HBB exon 1. (B) The gene-targeting donor MGH vector with 2 homology arms (900-bp left arm and 700-bp right arm) introduces an HR template for T-to-A replacement in the sickle allele and a loxP-flanked drug-selection cassette PGK-puromycin to be inserted into the HBB intron 1. (C) Targeted allele following homologous recombination with the corrected SCD mutation and inserted PGK-puromycin cassette in intron 1. (D, E) We used 2 PCR primers (black arrows: TP1F/TP1R and TP2F/TP2R) for initial screening of targeted insertion (TI) indicative of correct HR. (F) The clones with both 5′ and 3′ targeted insertions were screened to against those that an undesired insertion of the vector sequences (such as RP1F and RP2R), likely due to random insertion of the whole vector DNA. The results of 24 screened iPSC clones we screened that have 3′ targeted insertion (TI) are summarized in Table 1.
Supplemental Figure 4. Analysis of the second (intended SCD) HBB allele in iPSC clones with intended gene correction and insertion. Genomic DNA PCR with primers gHBB-F/gHBB-R produces a 364-bp product from the untargeted (or unintended) sickle allele (with or without excision mediated by Cre). (A) The sequence of the clone SC15 in the unintended allele. One basepair deletion was detected within the recognition sequence of the guide RNA-a, indicating that the guide RNA induced a NHEJ resulting in the deletion. As a result, the unintended (SCD) HBB allele was disrupted, due to the acquisition of a stop codon (TGA) soon after a reading frame-shift. This result was further confirmed by sequencing the sequence of the untargeted allele in SC15 after Cre excision, in clones such as SC15-Cre2. (B) The DNA sequence from the clone SC20 perfectly matched to that in parental TNC1 cells, indicating that the unintended SCD allele is indeed intact and untargeted. (C) The sequence of the targeted and excised allele in the SC15-Cre2 was also sequenced (amplified by the same gHBB-F/gHBB-R primers, resulting in a 482-bp DNA fragment). The corrected allele indeed have a corrected A, to in replacing the βs point mutation (T). All the DNA sequences are normal as found in the wild-type HBB allele, except that a loxP-containing (in purple) in the intron 1.