

Abstract
Cell-encapsulating devices can play an important role in advancing the types of tissue available for transplantation and further improving transplant success rates. To have an effective device, encapsulated cells must remain viable, respond to external stimulus, and be protected from immune responses, and the device itself must elicit a minimal foreign body response. To address these challenges, we developed a micro- and a nanoporous thin-film cell encapsulation device from polycaprolactone (PCL), a material previously used in FDA-approved biomedical devices. The thin-film device construct allows long-term bioluminescent transfer imaging, which can be used for monitoring cell viability and device tracking. The ability to tune the microporous and nanoporous membrane allows selective protection from immune cell invasion and cytokine-mediated cell death in vitro, all while maintaining typical cell function, as demonstrated by encapsulated cells' insulin production in response to glucose stimulation. To demonstrate the ability to track, visualize, and monitor the viability of cells encapsulated in implanted thin-film devices, we encapsulated and implanted luciferase-positive MIN6 cells in allogeneic mouse models for up to 90 days. Lack of foreign body response in combination with rapid neovascularization around the device shows promise in using this technology for cell encapsulation. These devices can help elucidate the metrics required for cell encapsulation success and direct future immune-isolation therapies.
Keywords: cell-encapsulation, polycaprolactone, immunoisolation, nanoporous, microporous
Graphical abstract
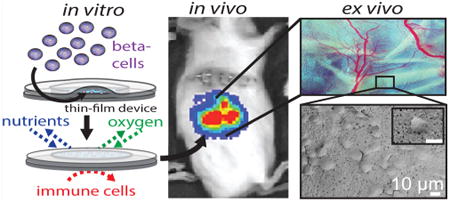
Cell replacement therapy has seen unprecedented progress in the past few years, including the ability to achieve insulin independence in humans through islet transplantation.1–3 Advancements in stem-cell technology holds potential to overcome donor shortages for many patients, who can benefit from islet replacement therapy. In particular, stem-cell-derived beta cells offer a promising new cell source for achieving insulin independence. Unfortunately, life-long systemic immunosuppression is required to protect transplanted cells from being rejected, putting patients at risk of organ damage, infection, and malignancies.4,5 Cell encapsulation provides an alternative approach to protect transplanted cells without the complications associated with immunosuppression. While a number of strategies are being investigated,6–11 there are several challenges associated with these approaches: retrievability, control over pore dimensions, biocompatibility, scalability, and reproducible fabrication methods.
The key function of an encapsulation device is to create an environment that allows for normal insulin secretion in response to fluctuating blood glucose while maintaining cell viability through effective nutrient exchange, effective waste exchange, and sequestration from the immune system. With the goal of creating immune-protected beta cells, a variety of micro- and macroencapsulation approaches have been developed over the past several decades.12–15 The fundamental distinction between micro- and macro-devices is a matter of scale: microencapsulation approaches encapsulate a single cell or islet, which maximizes surface area to volume ratios and promotes improved nutrient exchange;16,17 however, there is limited control of membrane thickness and pore size with microencapsulation. Additionally, because islets are individually encapsulated, thousands of microdevices are required for each transplant, and capsule size makes live imaging and tracking a significant challenge. Conversely, macroencapsulation devices house many cells or islets.18 These larger devices allow for greater control over membrane parameters, such as pore size and porosity, but are plagued by limited nutrient diffusion and cell response due to the device thickness and large device reservoir. In addition to these challenges, the sharp rigid structures typically associated with macro-encapsulation devices can lead to a foreign body response and subsequent device failure from fibrotic encapsulation.19,20
In this work, we fabricate and characterize poly-caprolactone (PCL) thin-film macroencapsulation devices as an innovative strategy to address the challenges of existing micro- and macroencapsulation approaches. A thin compliant design allows diffusion and flexibility similar to microencapsulation approaches, while the larger device surface area allows precise membrane control and retrievability, features associated with larger macroencapsulation technologies. Encapsulated cells demonstrated viability, function, protection from immune-cell intrusion, protection from cytokine-mediated cell death, and neovascularization. PCL has been used in FDA-approved medical devices and has demonstrated long-term biocompatibility in multiple animal models.21–26 Additionally, PCL degradation can be tuned to match the lifetime of the encapsulated cells, eliminating the need for device removal.27,28 The use of porous PCL thin films allows for a thin and flexible device to be designed with either micro- or nanoscaled features, leading to better nutrient exchange, precise membrane control, and device tracking. In this study, the MIN6 cell line, a well-established mouse insulinoma cell line known to respond to glucose with insulin secretion, was used as a model for islet beta cells. Using MIN6 cells provides a sustainable and consistent source of cells across experiments. Primary islets were also used to demonstrate long-term viability of encapsulated cells.
Results and Discussion
We describe the fabrication of microporous and nanoporous PCL thin-film cell-encapsulation devices, cell behavior in these devices, and in vivo integration of these devices in allogeneic mouse models. To design these encapsulation devices, we engineered the geometry to combine the advantages of the precise membrane control of macroencapsulation devices with improved nutrient exchange of microencapsulation devices. Furthermore, the choice of PCL was based on its range of molecular weights, tunable degradation profile, flexibility, and use as a nontoxic material in FDA-approved medical devices. Two different methods were used to create micro- and nanoporous membranes for thin-film devices. The microporous films utilize phase separation of PEG and PCL in solution. In this method, after films are cast, the pore forming agent (PEG) is dissolved, leaving a microporous film.27 By tuning the concentration ratio and composition of the two polymers, films can be tailored for a variety of porosities and architectures.22,27,29–34 Nanoporous films were created from a zinc oxide nanorod template and backed with a microporous support layer. Zinc oxide nanorod dimensions can be readily tuned, allowing a wide range of pores sizes and giving the ability to further refine these devices.35,36 Figure 1A schematically details the method for heat-sealing two thin films to generate a single device. Two-step sealing decouples device shape from cell encapsulation. A first heat-sealing step controls the device size. Once the device outline is sealed, cells are inserted into the lumen of the thin-film device, and a second heat-sealing step encapsulates the cells. Device geometry can be arbitrarily selected based on the shape of the nichrome wire that defines the device seal, typically from 1 to 5 cm in diameter, allowing devices to be scaled to contain more cells as necessary.
Figure 1.
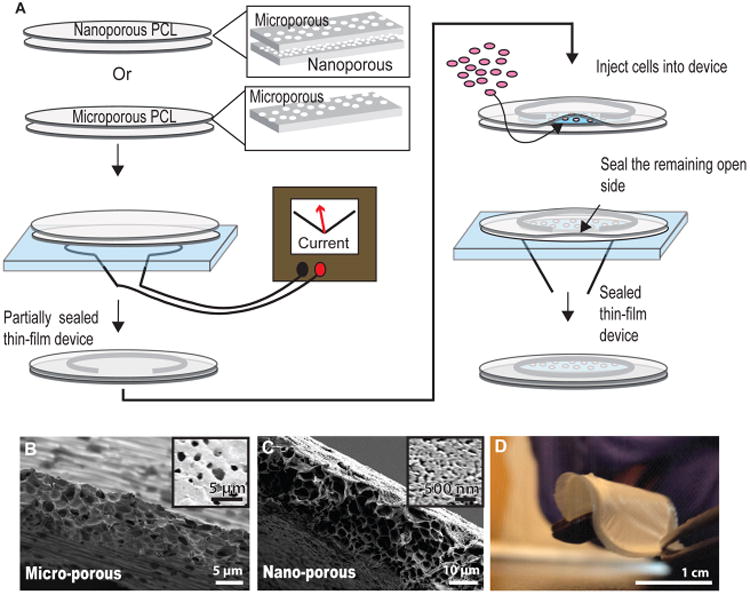
PCL micro- and nanoporous thin-film fabrication for cell encapsulating devices. (A) Schematicof the device two-step heat-sealing and cell encapsulation. (B) Cross-section SEM of the microporous thin-film and (inset) top down image of the film surface. (C) Cross-section SEM of the nanoporous thin-film and (inset) top-down image of the nanoporous film surface. (D) Image of an assembled device, demonstrating device flexibility.
Scanning electron microscopy (SEM) was used to visualize the microporous thin films, which had ∼2μm sized pores and a membrane thickness of ∼10 μm (Figure 1B). Similarly, an SEM image cross-section and top-down image of a nanoporous thin-film with a microporous backing showed a membrane thickness of 10μm and nanopores ranging from 30 to 100 nm (Figure 1C). The thin design, flexibility, compliance of the material, and structure of the device as a whole create a cell-encapsulating device that is easy to handle with precise membrane control (Figure 1D). Noting that oxygen diffusion in aqueous solutions is 100 to 200 μm, these thin-film devices with membrane thicknesses of 10 μm decrease the proximity to vasculature needed for adequate oxygen consumption.37,38 Given the thin-film nature of the devices, the total cell content scales with device area, while the average distance of cells from the nutrient source at the device exterior is maintained, bridging the advantages of both micro- and macroencapsulation technologies. We expect the thin-film design of the device, coupled to rapid device vascularization, to provide sufficient oxygen for encapsulated cells.
mCherry-expressing MIN6 cells encapsulated in either micro- or nanoporous devices maintain viability in vitro through 6 days, as defined by the persistence in mCherry signal, and are able to maintain glucose stimulated insulin secretion (Figure 2A). The glucose stimulation index is a metric to quantify beta cell function by comparing the ratio of insulin release in a high glucose state relative to a resting state. MIN6 cells encapsulated in either micro- or nanodevices demonstrate no statistically significant changes in their glucose stimulation index (Figure 2B). Furthermore, freshly isolated mouse islets encapsulated in these devices maintain their glucose stimulation index over a period of 20 days in vitro, which is significantly improved over free islets alone, which have a >25% decrease in the glucose stimulation index from day 1 (Figure 2C). This demonstrates that beta cell insulin response to glucose is maintained within both nano-and microporous thin-film devices. Additionally, glucose sensing and insulin secretion, a major function of beta cells, is unaffected by encapsulation in either micro- or nanodevices.
Figure 2. In vitro.
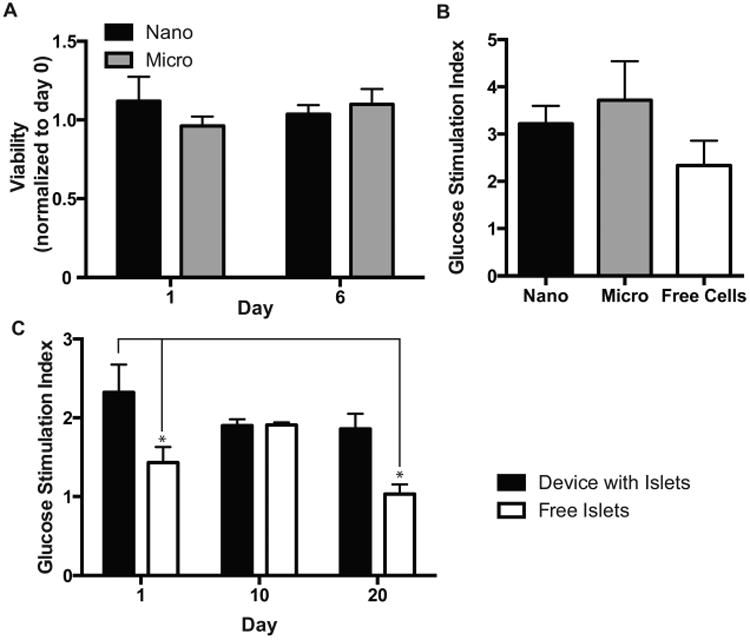
device function. (A) In vitro device viability of encapsulated MIN6 cells as measured with mCherry fluorescence. (B) Glucose stimulation index of MIN6 cells encapsulated in either micro- or nanoporous devices. (C) Glucose stimulation of primary islets encapsulated in microporous devices (p ≤ 0.5, n ≥ 3).
Viability and persistence of transplanted cells can be monitored in recipient mice in real time using bioluminescence imaging (Figure 3). This technique was used to monitor in vivo Luciferase-expressing MIN6. LUC encapsulated into thin-film devices implanted under the abdomen above the liver (Figure 3A) or over the muscle layer in the subcutaneous space of the mouse dorsal flank (Figure 3B) or unencapsulated cells implanted into the kidney capsule (Figure 3C) of syngeneic B6 mice. The bioluminescent signal decreases with device implant depth, and both implanted device locations were visually brighter than the no device kidney capsule control. The persistence of the bioluminescent signal demonstrates maintained viability though 90 days of implantation (Figure 3D–F). As the bioluminescent signal tracks with device location, it also provides a noninvasive method to track device movement. Because the encapsulated cells are not fixed within the device and the device itself is not sutured or tethered to any tissue, cellular reorganization of the encapsulated cells or daily movement of the mouse can result in the movement of the bioluminescent signal.
Figure 3. In vivo.
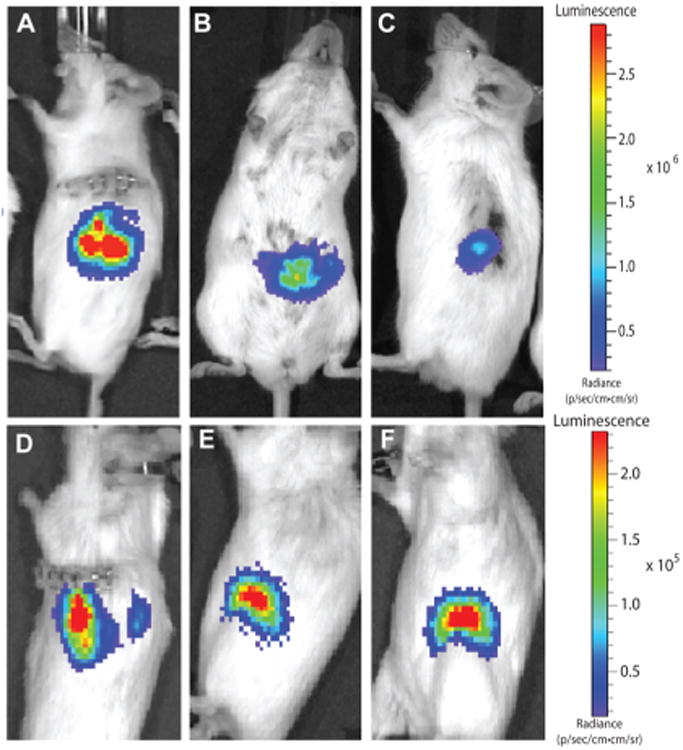
device image and tracking. (A) Device with encapsulated MIN6 cells implanted in the subcutaneous space of the mouse dorsal. (B) Device with encapsulated MIN6 cells implanted under the mouse skin and muscle over the liver. (C) No device control with cells implanted directly into the kidney capsule. Device with encapsulated MIN6 cells implanted after (D) 1, (E) 30, and (F) 90 days.
Ideal immune protection requires physically excluding immune cells as well as restricting diffusion of immune mediators such as cytokines that are toxic to beta cells. By encapsulating cells in microporous devices, cell-contact-mediated immune protection may be achieved, and additional cytokine-mediated immune protection may be accomplished with the nanoporous devices. Cells encapsulated in thin-film devices are physically compartmentalized from the in vivo environment, as clearly seen in Figure 4A, where cells are attached to the outer surface of the device but no infiltration into the device lumen was found. Despite cell adhesion on device surfaces, pores remain unclogged (Figure S1 in the Supporting Information) most likely due to the limited fibrotic response of the surrounding tissue. Figure 4B shows a SEM cross-section, with a cell attached to the external surface of a device. No cellular processes are seen extending into the device, further confirming the ability of the device to prevent cell-contact-mediated interaction by isolating the encapsulated cells from the surrounding in vivo tissue. By further controlling the porosity of the membrane, cytokine-mediated immune protection may additionally be achieved. Tumor necrosis factor α (TNFα), interleukin 1 β (IL1/β), and interferon γ (IFNγ) inflammatory cytokines are known to kill beta cells individually and act synergistically when present in combination. They were chosen to test the devices' ability to protect from cytotoxic cytokines39–42 (Figure S2 in the Supporting Information).43–45 Interestingly, whereas microporous thin-film devices failed to maintain cell viability (Figure 5A), the use of a nanoporous layer in these thin-film devices mitigated the cytokine-mediated decrease in viability (Figure 5B). It is unclear if cytokines are completely isolated from the lumen of devices; given the size of cytokines in relation to the nanopores, a portion of cytokines are expected to pass through the membrane. The protection by nanoporous devices would result from limited transport and diffusion of cytokines though the membrane, such that the cells are unresponsive to the reduced cytokine concentrations. Considering that the cytokine cocktail concentration used exceeds known cytotoxic concentrations by 10-fold, we expect the majority of the cytokines to be limited by the nanoporous barrier. This further highlights how microporous and nanoporous membranes can be used to control desired cell responses.
Figure 4.
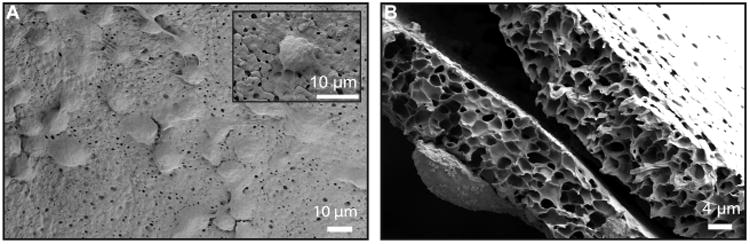
Microporous barrier inhibits cell invasion. (A) Top-down SEM image of cells attached to the exterior surface of the microporous thin-film device after 1 month in vivo. (B) Cross-section SEM image of the microporous thin-film device after 1 month in vivo, demonstrating membrane integrity and isolation of internal and external cells.
Figure 5.
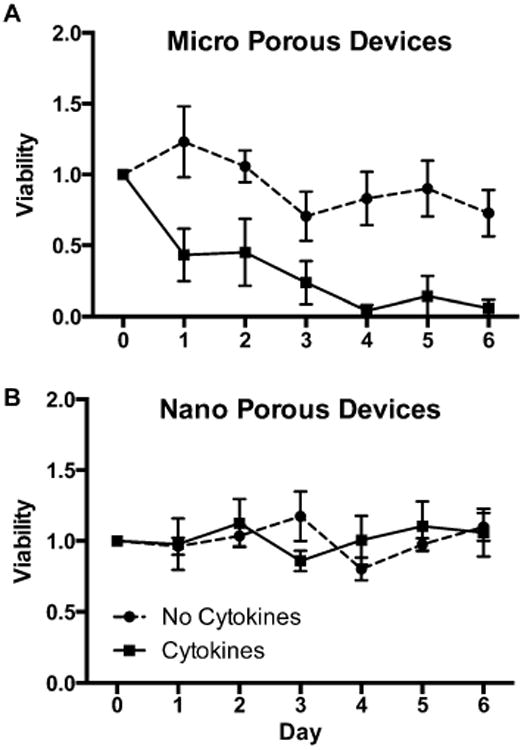
Cytokine protection.Viability of cells within (A) a microporous device and (B) a nanoporous device over 1 week, with (solid line) and without (dashed lines) cyto-kines. (n ≥ 4).
Device vascularization in vivo is imperative for long-term survival of encapsulated cells. Vascularization surrounding cells encapsulated in thin-film devices is important for function and survival of encapsulated cells. Devices were implanted, then removed and imaged at 7, 14, 30, and 90 days to monitor the state of device vascularization (Figure 6A–D). The first visible signs of vascularization of cell-encapsulated thin-film devices, were observed 14 days after implantation (Figure 6B). These devices demonstrate a steady increase in vivo vascularization of 1.5% daily over a 2 month period (Figure 6E). Vascularization of these PCL devices occurs without any supplementary additional proangiogenic factors, as shown with implanted cell-free devices with similar vascularization (Figure S3A, B in the Supporting Information). When compared with common polymeric implant materials PLGA (Figure S3C in the Supporting Information) and PVDF (Figure S3D in the Supporting Information), PCL cell-free devices exhibit noticeably more developed and branched vasculature. Furthermore, we believe the combination of thin size of the PCL devices, their flexibility, and the structure of the devices provides a relatively minimal foreign body response (Figure S4 in the Supporting Information).
Figure 6.
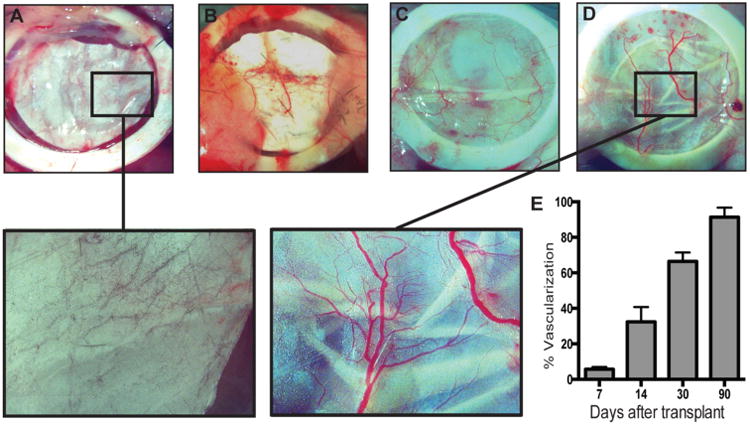
Device vascularization. Bright-field images of devices implanted after (A) 7, (B) 14, (C) 30, and D) 90 days, with magnified images at days 7 and 90. (E) Quantification of device vascularization from days 7 to 90 (n = 3).
Conclusions
Here we demonstrate the successful fabrication of an innovative cell-encapsulating device that combines some of the benefits of both micro- and macro-encapsulation strategies. A flexible thin-film geometry allows precise membrane porosity selection to direct desired cellular responses and interactions while maintaining a normal glucose response of encapsulated beta cells. A small reservoir volume allows a rapid response to external stimuli, limiting dilutional interference from the device reservoir. Similar to microencapsulation devices with large surface area to volume ratios, our thin-film device structure is uninhibited by device thickness. Moreover, cells encapsulated in either micro- or nanodevices demonstrate a glucose stimulation index consistent with unencapsulated cells, indicating glucose sensing and responsive insulin secretion is successfully preserved. These devices allow sufficient bioluminescence transmission through the device membrane tobe measured with in vivo imaging systems. As demonstrated in vivo, these device membranes create a physical barrier between encapsulated cells and the host environment, physically preventing cell contact initiated signaling. Furthermore, incorporation of a nanoporous membrane enables these devices to obstruct cytokine passage and protect encapsulated cells from cytokine-mediated cell death. Additionally, in vivo studies show vascularization around the devices with limited fibrosis, which displays great promise for this device as a long-term cell encapsulation device.
This technology platform can be used to directly investigate the cell contact-dependent or soluble factor-mediated signaling by controlling pore dimension-inhibiting specific interactions. These devices have the capacity to prevent immune cell contact with encapsulated cells, and the nanoporous device can protect encapsulated cells from cytokine-induced cell death. Future directions include using these devices in vivo to investigate modes of immune attack, whether contact-or soluble factor-mediated. Given the nature of these thin-film cell-encapsulation devices, future generations could be scaled for humans as alternative treatments for Type 1 Diabetes.
Materials and Methods
Chemicals were purchased from Sigma-Aldrich unless noted, and cell culture materials were purchased from the UCSF cell culture facility. All films were spuncast onto silicon wafers at 1000 rpm for 30 s, followed by 2000 rpm for 30 s. Devices were characterized with a Carl Zeiss Ultra 55 field-emission scanning electron microscope using an in-lens secondary electron detector.
Microporous Thin-Film Fabrication
Microporous PCL thin-films were spuncast from a solution of 150 mg/mL PCL (70–90 kDa Mn) and polyethylene glycol (PEG, 2 kDa Mn) in 2,2,2-trifluoroethanol, which was prepared by stirring at 65 °C until dissolved. Following spin-casting, the PEG was dissolved by soaking in water for 1 h, resulting in a microporous PCL films with pores ∼2 μm in diameter. Devices were 1 cm in diameter, resulting in a surface area of 1.57 cm2 per side, with 67.5 ± 1.3% porosity and 0.37 ± 0.02 density.
Nanoporous Thin-Film Fabrication
Nanoporous PCL films were formed using an established template-based approach reported elseware.24 In brief, a 0.5 M solution of zinc acetate dihydrate and ethanolamine in 2-methoxyethanol was spuncast onto silicon wafers and annealed at 300 °C on a hot plate to generate a zinc oxide (ZnO) seed layer. From this seed layer, ZnO nanorods were hydrothermally grown in a 5 mM zinc acetate solution at 85-90 °C for 2 h. A 150 mg/mL PCL solution was then spuncast onto the nanorods, followed by a 150 mg/mL PEG:PCL solution to provide a microporous support, creating a nanoporous film with a microporous backing support layer. The film was soaked in a dilute sulfuric acid solution to etch away the ZnO nanorods and also dissolve the PEG, resulting in a nanoporous membrane with pores ranging from 30 to 100 nm supported by a microporous backing. Membrane characterizations and ZnO nanorod morphology were previously measured.23,24,27
Nonporous Membrane Fabrication
Nonporous PCL films were spuncast from a solution of 150 mg/mL PCL (70-80 kDa Mn) in 2,2,2-trifluoroethanol, which was prepared by stirring at 65 °C until dissolved. Nonporous poly(lactic-co-glycolic acid) (PLGA) films were spun cast from a solution of 300 mg/mL PLGA (85:15 LA:GA 45 kDa Mn) in 2,2,2-trifluoroethanol. Polyvinylidene fluoride (PVDF) film were prefabricated from Sigma and cut to shape.
Assembly of Thin-Film Devices
Devices consisted of two PCL thin-films heat-sealed together using resistive heating of a nichrome wire. A two-step heat-sealing method was used where 1.2 Amp current ran through a nichrome wire outlining the regions to be sealed. For the first sealing step, two films were placed over a U-shaped nichrome wire embedded in PDMS (Sylgard 184), 1 cm in diameter. To secure the membranes a PDMS weight was placed over the films holding them flat. A 1.2 Amp current ran through the wire for 30 s and sealed the devices in the shape of a U, defining the device lumen shape and leaving an open side for cell injection. 1.5 Million MIN6 cells in high glucose Dulbecco's modified Eagle's (DME) were injected into the devices through the remaining open side. Second, the remaining side of the device was sealed by placing the open edge over a straight nichrome wire embedded in PDMS and heat-sealed with a 1.2 Amp current for 30 s.
Characterization Using Scanning Electron Microscopy of Films and Devices
Micro- and nanoporous thin PCL films were mounted on a flat SEM mount with colloidal graphite (Ted Pella). Cross sections were flash-dipped in isopropanol, followed by liquid nitrogen freeze fracture and then mounted. Devices from in vivo experiments were fixed with 3.7% formaldehyde for 30 min, washed in deionized water three times, then sequentially dehydrated in increasing ethanol concentrations and mounted.
Cell Culture
MIN6 cells were cultured using standard media conditions.46 Genes for mCherry and puromycin resistance were introduced using a lentivirus construct designed by the Lentiviral Core at UCSF. The cells were transduced using standard protocol with a multiplicity of infection of 2, and transduced cells were selected using puromycin. Genes encoding firefly luciferase and green fluorescence protein were similarly introduced into MIN6 cells. Primary islets were isolated by the Islet Core at UCSF using standard islet isolation protocols.47
Glucose-Stimulated Insulin Secretion
Insulin secretion was analyzed using a glucose-stimulated insulin secretion assay. Cells were rested for30 mininmedium-containing5mMglucose and then stimulated using medium-containing 15 mM glucose. Culture supernatant was collected at 30 and 60 min after the addition of high glucose. Insulin protein content in the culture supernatant was measured using an enzyme linked immunosorbant assay (Mercodia). The ratio of insulin secreted at high-to-low glucose conditions was used to calculate the glucose stimulation index.
Cytokine Assay
To determine the effect of cytokines on the viability of encapsulated beta cells, we cultured 250 000 cells in micro- or nanoporous devices in a cytokine cocktail consisting of TNFα (300 ng/mL; VWR), IL1β (110 ng/mL; VWR), and IFNγ (200 ng/mL; Fisher) in high glucose DME media, with 10% fetal bovine serum, 1% penicillin, and 1% streptomycin. The devices were imaged daily for the mCherry signal using a standard spectro-photometer. The signal intensity was measured for each respective device for 7 days and normalized against the initial signal.
Bioluminescent Imaging
Thin-film devices with luciferase-expressing MIN6 (MIN6.LUC) cells were implanted, in either the subcutaneous space on the dorsal aspect or the abdominal cavity between the muscle wall and the liver of MOD.Cg-Prkdcscid III2rgtm1Wjl/SxJ (NSG) or BALB/C mice. Persistence of the encapsulated cells in vivo was assessed by monitoring luciferase activity using a Xenogene IVIS 200 imaging system (PerkinElmer). The animals transplanted with MIN6.LUC cells were injected IP with D-luciferin solution (Goldbio, St. Louis, MO) at the dose of 150 mg/kg 8 min before imaging to capture the peak in bioluminescent intensity, as previously described.48 The mice were anesthetized with an isoflurane mixture (2% in 98% O2) and imaged by using a Xenogen IVIS 200 imaging system. Bioluminescence images were acquired for 1 min and then analyzed using the Living Image analysis software (Xenogen, Alameda, CA). Regions of interest (ROI) were centered over the bioluminescence regions. Photons were counted within the ROI over the acquisition time. Adherence to the same imaging protocol ensured consistent signal detection and allowed us to compare data acquired over a period of at least 3 months.
Histology
Mouse tissue samples were collected and fixed in 4% paraformaldehyde for 24 h and washed with phosphate buffered saline at 4 °C for 48 h, then 30% sucrose for 24 h. Samples were then taken to the Mouse Pathology Core at UCSF and Optimal Cutting Temperature (OCT) embedded, sliced, and hematoxylin- and eosin-stained or Masson's-trichrome-stained by either the Mouse Pathology Core or the Histology and Imaging Core at UCSF.
Vasculature
At 7, 14, 30, and 90 days after transplantation, PCL device-bearing mice were anesthetized with an intraperitoneal injection of Avertin solution 2.5% (Sigma) and subjected to optical imaging using a Leica MZ16F microscope (Leica Biosystems, Wetzlar, Germany). The animals were euthanized by cervical dislocation, and the encapsulated devices were collected for further analysis. The images of the encapsulated grafts were analyzed using ImageJ software (NIH; http://rsb.info.nih.gov/ij/). Vessel density was measured by automated counting of red pixels divided by the area of the ROI within the device; a threshold was previously set for the red channel to subtract background.
Supplementary Material
Acknowledgments
We thank Jessica L. Allen, Erica B. Schlesinger, Laura A. Walsh, and Miquella G. Chavez for critiquing the manuscript and members of the Desai laboratories for insightful discussion and conversations. We thank Erica B. Schleshinger additionally for her insight and expertise in material properties. We thank the Nikon Center at UCSF for Imaging and the Center for Advanced Technologies at UCSF. We acknowledge funding from the Juvenile Diabetes Research Foundation, NIH P30DK063720 for UCSF Diabetes and Endocrinology Research Center and Larry L Hillblom Foundation fellowship (G.F.).
Footnotes
Supporting Information Available: Device exterior SEMs, affect of cytokines on cell number quantified by a cyquant assay, cell-free device controls for device vascularization, and histology of devices. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/ acsnano.5b00679.
Conflict of Interest: The authors declare no competing financial interest.
References and Notes
- 1.Atkinson MA, Eisenbarth GS, Michels AW. Type 1 Diabetes. Lancet. 2014;383:69–82. doi: 10.1016/S0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orlando G, Gianello P, Salvatori M, Stratta RJ, Soker S, Ricordi C, Domínguez-Bendala J. Cell Replacement Strategies Aimed at Reconstitution of the B-Cell Compartment in Type 1 Diabetes. Diabetes. 2014;63:1433–1444. doi: 10.2337/db13-1742. [DOI] [PubMed] [Google Scholar]
- 3.Hatziavramidis DT, Karatzas TM, Chrousos GP. Pancreatic Islet Cell Transplantation: An Update. Ann Biomed Eng. 2013;41:469–476. doi: 10.1007/s10439-012-0676-3. [DOI] [PubMed] [Google Scholar]
- 4.Ludwig B, Reichel A, Steffen A, Zimerman B, Schally AV, Block NL, Colton CK, Ludwig S, Kersting S, Bonifacio E, et al. Transplantation of Human Islets without Immunosuppression. Proc Natl Acad Sci U S A. 2013;110:19054–19058. doi: 10.1073/pnas.1317561110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, Kneteman NM, Rajotte RV. Islet Transplantation in Seven Patients with Type 1 Diabetes Mellitus Using a Glucocorticoid-Free Immunosuppressive Regimen. N Engl J Med. 2000;343:230–238. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- 6.Huang H, Ding Y, Sun XS, Nguyen TA. Peptide Hydrogelation and Cell Encapsulation for 3D Culture of MCF-7 Breast Cancer Cells. PLoS One. 2013;8 doi: 10.1371/journal.pone.0059482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah K. Encapsulated Stem Cells for Cancer Therapy. Biomatter. 2013;3:1–7. doi: 10.4161/biom.24278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song K, Yang Y, Li S, Wu M, Wu Y, Lim M, Liu T. In Vitro Culture and Oxygen Consumption of NSCs in Size-Controlled Neurospheres of Ca-Alginate/gelatin Micro-bead. Mater Sci Eng, C. 2014;40:197–203. doi: 10.1016/j.msec.2014.03.028. [DOI] [PubMed] [Google Scholar]
- 9.De Faveri S, Maggiolini E, Miele E, De Angelis F, Cesca F, Benfenati F, Fadiga L. Bio-Inspired Hybrid Microelec-trodes: A Hybrid Solution to Improve Long-Term Performance of Chronic Intracortical Implants. Front Neuroeng. 2014;7:7. doi: 10.3389/fneng.2014.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robles L, Storrs R, Lamb M, Alexander M, Lakey JR. Current Status of Islet Encapsulation. Cell Transplant. 2013;23:1321–1348. doi: 10.3727/096368913X670949. [DOI] [PubMed] [Google Scholar]
- 11.Tomei Aa, Manzoli V, Fraker Ca, Giraldo J, Velluto D, Najjar M, Pileggi A, Molano RD, Ricordi C, Stabler CL, et al. Device Design and Materials Optimization of Conformal Coating for Islets of Langerhans. Proc Natl Acad Sci U S A. 2014;111:10514–10519. doi: 10.1073/pnas.1402216111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scharp DW, Marchetti P. Encapsulated Islets for Diabetes Therapy: History, Current Progress, and Critical Issues Requiring Solution. Adv Drug Delivery Rev. 2014;67-68:35–73. doi: 10.1016/j.addr.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 13.Weir GC. Islet Encapsulation: Advances and Obstacles. Diabetologia. 2013;56:1458–1461. doi: 10.1007/s00125-013-2921-1. [DOI] [PubMed] [Google Scholar]
- 14.Buder B, Alexander M, Krishnan R, Chapman DW, Lakey JR. Encapsulated Islet Transplantation: Strategies and Clinical Trials. Immune Network. 2013;13:235–239. doi: 10.4110/in.2013.13.6.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Julien S, Nyitray C, Desai TA. Membranes to Achieve Immunoprotection of Transplanted Islets. Front Biosci, Landmark Ed. 2014;19:49–76. doi: 10.2741/4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cala R, Basta G. Clinical Application of Microencapsulated Islets: Actual Prospectives on Progress and Challenges. Adv Drug Delivery Rev. 2014;68:84–92. doi: 10.1016/j.addr.2013.09.020. [DOI] [PubMed] [Google Scholar]
- 17.Cornolti R, Figliuzzi M, Remuzzi A. Effect of Micro-and Macroencapsulation on Oxygen Consumption by Pancreatic Islets. Cell. 2009;18:195–201. doi: 10.3727/096368909788341252. [DOI] [PubMed] [Google Scholar]
- 18.Lathuilière A, Cosson S, Lutolf MP, Schneider BL, Aebischer P. A High-Capacity Cell Macroencapsulation System Supporting the Long-Term Survival of Genetically Engineered Allogeneic Cells. Biomaterials. 2014;35:779–791. doi: 10.1016/j.biomaterials.2013.09.071. [DOI] [PubMed] [Google Scholar]
- 19.Ward WKA. Review of the Foreign-Body Response to Subcutaneously-Implanted Devices: The Role of Macrophages and Cytokines in Biofouling and Fibrosis. J Diabetes Sci Technol. 2008;2:768–777. doi: 10.1177/193229680800200504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ward WK, Slobodzian EP, Tiekotter KL, Wood MD. The Effect of Microgeometry, Implant Thickness and Poly-urethane Chemistry on the Foreign Body Response to Subcutaneous Implants. Biomaterials. 2002;23:4185–4192. doi: 10.1016/s0142-9612(02)00160-6. [DOI] [PubMed] [Google Scholar]
- 21.Rentsch B, Bernhardt R, Scharnweber D, Schneiders W, Rammelt S, Rentsch C. Embroidered and Surface Coated Polycaprolactone-Co-Lactide Scaffolds: A Potential Graft for Bone Tissue Engineering. Biomatter. 2012;2:158–165. doi: 10.4161/biom.21931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernards DA, Desai TA. Nanoscale Porosity in Polymer Films: Fabrication and Therapeutic Applications. Soft Mater. 2011;6:1621–1631. doi: 10.1039/B922303G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernards DA, Bhisitkul RB, Wynn P, Steedman MR, Lee OT, Wong F, Thoongsuwan S, Desai TA. Ocular Biocompatibility and Structural Integrity of Micro- and Nanostructured Poly(caprolactone) Films. J Ocul Pharmacol Ther. 2013;29:249–257. doi: 10.1089/jop.2012.0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernards DA, Desai TA. Nanotemplating of Biodegradable Polymer Membranes for Constant-Rate Drug Delivery. Adv Mater. 2010;22:2358–2362. doi: 10.1002/adma.200903439. [DOI] [PubMed] [Google Scholar]
- 25.Abedalwafa M, Wang F, Wang L, Li C. Biodegradable Poly-Epsilon-Caprolcaction (PCL) for Tissue Engineering Applications: A Review. Rev Adv Mater Sci. 2013;34:123–140. [Google Scholar]
- 26.Angius D, Wang H, Spinner RJ, Gutierrez-Cotto Y, Yaszemeski MJ, Windebank AJ. A Systematic Review of Animal Models Used to Study Nerve Regereration in Tissue-Engineered Scaffolds. Biomaterials. 2013;33:8034–8039. doi: 10.1016/j.biomaterials.2012.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernards DA, Lance KD, Ciaccio NA, Desai TA. Nanostructured Thin Film Polymer Devices for Constant-Rate Protein Delivery. Nano Lett. 2012;12:5355–5361. doi: 10.1021/nl302747y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mendelsohn AD, Bernards Da, Lowe RD, Desai Ta. Patterning of Mono- and Multilayered Pancreatic Beta-Cell Clusters. Langmuir. 2010;26:9943–9949. doi: 10.1021/la1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu F, Lei L, Shen YY, Hou JW, Chen WL, Li YG, Guo SR. Effects of Amphiphilic PCL-PEG-PCL Copolymer Addition on 5-Fluorouracil Release from Biodegradable PCL Films for Stent Application. Int J Pharm. 2011;419:77–84. doi: 10.1016/j.ijpharm.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 30.Lin WJ, Lee HG. Design of a Microporous Controlled Delivery System for Theophylline Tablets. J Controlled Release. 2003;89:179–187. doi: 10.1016/s0168-3659(03)00090-7. [DOI] [PubMed] [Google Scholar]
- 31.Rong HJ, Chen WL, Guo SR, Lei L, Shen YY. PCL Films Incorporated with paclitaxel/5-Fluorouracil: Effects of Formulation and Spacial Architecture on Drug Release. Int J Pharm. 2012;427:242–251. doi: 10.1016/j.ijpharm.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Anzai R, Murakami Y. Poly(ε-Caprolactone) (PCL)-polymeric Micelle Hybrid Sheets for the Incorporation and Release of Hydrophilic Proteins. Colloids Surf, B. 2015;127:292–299. doi: 10.1016/j.colsurfb.2015.01.028. [DOI] [PubMed] [Google Scholar]
- 33.Lei L, Liu X, Shen YY, Liu JY, Tang MF, Wang ZM, Guo SR, Cheng L. Zero-Order Release of 5-Fluorouracil from PCL-Based Films Featuring Trilayered Structures for Stent Application. Eur J Pharm Biopharm. 2011;78:49–57. doi: 10.1016/j.ejpb.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Ledeuil JB, Uhart A, Allouche J, Dupin JC, Martinez H. New Insights into Micro/Nanoscale Combined. Nanoscale. 2014;6:11130–11140. doi: 10.1039/c4nr03211j. [DOI] [PubMed] [Google Scholar]
- 35.Kim YJ, Kim SS, Park JB, Sohn BH, Yi GC. Controlled Growth of Inorganic Nanorod Arrays Using Graphene Nanodot Seed Layers. Nanotechnology. 2014;25:135609. doi: 10.1088/0957-4484/25/13/135609. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, Yu H, Shao X, Han M. Near-Room-Temperature Production of Diameter-Tunable ZnO Nanorod Arrays through Natural Oxidation of Zinc Metal. Chemistry. 2005;11:3149–3154. doi: 10.1002/chem.200401153. [DOI] [PubMed] [Google Scholar]
- 37.Wendt D, Riboldi Sa, Cioffi M, Martin I. Potential and Bottlenecks of Bioreactors in 3D Cell Culture and Tissue Manufacturing. Adv Mater. 2009;21:3352–3367. doi: 10.1002/adma.200802748. [DOI] [PubMed] [Google Scholar]
- 38.Martin Y, Vermette P. Bioreactors for Tissue Mass Culture: Design, Characterization, and Recent Advances. Biomaterials. 2005;26:7481–7503. doi: 10.1016/j.biomaterials.2005.05.057. [DOI] [PubMed] [Google Scholar]
- 39.Tracey KJ. The Inflammatory Reflex. Nature. 2002;420:853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 40.Bastiaens P. When It Is Time to Die. Nature. 2009;459:334–335. doi: 10.1038/459334a. [DOI] [PubMed] [Google Scholar]
- 41.Libert C. Inflammation: A Nervous Connection. Nature. 2003;421:328–329. doi: 10.1038/421328a. [DOI] [PubMed] [Google Scholar]
- 42.Wang L, Du F, Wang X. TNF-Alpha Induces Two Distinct Caspase-8 Activation Pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 43.Fonseca FN, Papanicolaou G, Lin H, Lau CBS, Kennelly EJ, Cassileth BR, Cunningham-Rundles S. Echinacea Purpurea (L.) Moench Modulates Human T-Cell Cytokine Response. Int Immunopharmacol. 2014;19:94–102. doi: 10.1016/j.intimp.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roff SR, Noon-Song EN, Yamamoto JK. The Significance of Interferon-Γ in HIV-1 Pathogenesis, Therapy, and Prophylaxis. Front Immunol. 2014;4:498. doi: 10.3389/fimmu.2013.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang YHC, Vilin YY, Roberge M, Kurata HT, Johnson JD. Multi-Parameter Screening Reveals a Role for Na(+) Channels in Cytokine-Induced Beta-Cell Death. Mol Endocrinol. 2014:me20131257. doi: 10.1210/me.2013-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyazaki J, Araki K, Yamato E, Ikegami H, Yamamura K. Establishment of a Pancreatic/Beta Cell Line That Retains Glucose-Inducible Insulin Secretion: Special Reference to Expression of Glucose Transporter Isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- 47.Szot GL, Koudria P, Bluestone Ja. Murine Pancreatic Islet Isolation. J Vis Exp. 2007;1640:255. doi: 10.3791/255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fowler M, Virostko J, Chen Z, Poffenberger G, Radhika A, Brissova M, Shiota M, Nicholson WE, Shi Y, Hirshberg B, et al. Assessment of PancreaticIslet Mass after Islet Transplantation Using in vivo Bioluminescence Imaging. Transplantation. 2005;79:768–776. doi: 10.1097/01.tp.0000152798.03204.5c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.