

Abstract
Both environmental and genetic triggers factor into the etiology of autoimmune thyroid disease (AITD), including Graves' disease (GD) and Hashimoto's thyroiditis (HT). Although the exact pathogenesis and causative interaction between environment and genes are unknown, GD and HT share similar immune-mediated mechanisms of disease. They both are characterized by the production of thyroid autoantibodies and by thyroidal lymphocytic infiltration, despite being clinically distinct entities with thyrotoxicosis in GD and hypothyroidism in HT. Family and population studies confirm the strong genetic influence and inheritability in the development of AITD. AITD susceptibility genes can be categorized as either thyroid specific (Tg, TSHR) or immune-modulating (FOXP3, CD25, CD40, CTLA-4, HLA), with HLA-DR3 carrying the highest risk. Of the AITD susceptibility genes, FOXP3 and CD25 play critical roles in the establishment of peripheral tolerance while CD40, CTLA-4, and the HLA genes are pivotal for T lymphocyte activation and antigen presentation. Polymorphisms in these immune-modulating genes, in particular, significantly contribute to the predisposition for GD, HT and, unsurprisingly, other autoimmune diseases. Emerging evidence suggests that single nucleotide polymorphisms (SNPs) in the immunoregulatory genes may functionally hinder the proper development of central and peripheral tolerance and alter T cell interactions with antigen presenting cells (APCs) in the immunological synapse. Thus, susceptibility genes for AITD contribute directly to the key mechanism underlying the development of organ-specific autoimmunity, namely the breakdown in self-tolerance. Here we review the major immune-modulating genes that are associated with AITD and their potential functional effects on thyroidal immune dysregulation.
Keywords: Autoimmune thyroid disease, Graves' disease, Hashimoto's thyroiditis, autoimmunity, immunogenetics, polymorphism
1. Introduction
Autoimmune thyroid disease (AITD), which includes Graves' disease (GD) and Hashimoto's thyroiditis (HT), affects an estimated 5% of the general population, making it one of the most prevalent autoimmune diseases [1, 2]. Autoimmunity to the thyroid, defined by the presence of antibodies to thyroid antigens, is even more common and reported to be as high as 10-20% of all women. In addition to the generation of thyroid autoantibodies and abnormal thyroid hormone production, AITD histologically involves the infiltration of self-targeting T and B lymphocytes in the thyroid gland [3]. The etiology of AITD is currently understood to be multifactorial and is due to a complex interplay of specific susceptibility genes and environmental exposures. In fact, the influential role of susceptibility genes in the development of AITD is highlighted by epidemiological studies showing that approximately 50% of the siblings of GD patients test positive for thyroid antibodies and that up to 33% of those with AITD share the diagnosis with their siblings (resulting in a sibling risk ratio or λs as high as 16.9) [4-6]. Twin studies are even more convincing with higher concordance for AITD between monozygotic twins than dizygotic twins [7, 8] and have suggested that the overall heritable, genetic contribution to the development of GD is about 75% [7, 9]. Racial variations in AITD prevalence, as was demonstrated in a recent NHANES (the National Health and Nutritional Examination Surveys) study, further accentuate potential genetic differences and the role of genetic susceptibility in the etiology of GD and HT [10].
Of the susceptibility genes, the majority are general immune-regulatory genes involved in the complex process of ensuring robust immune responses against appropriate, foreign antigens while maintaining tolerance to self-antigens. These include genes involved in the proper progression from central tolerance, peripheral tolerance to antigen presentation and lymphocyte activation in the immunologic synapse. Aberrant activity of these immune-regulatory genes due to polymorphisms would logically and potentially lead to a breakdown in immune tolerance and ultimately autoimmunity. Through linkage and association analyses, genome screening, and genome wide association studies (GWAS), several single nucleotide polymorphisms (SNPs) in genes including FOXP3, CD25, CTLA-4, CD40, the HLA family, and others have been discovered to be associated with AITD. Interestingly, certain polymorphisms uniquely predispose to GD, HT or both while others are not thyroid-specific and increase the likelihood of autoimmunity in general (table 1).
Table 1.
Summary of major susceptibility genes for AITD and their point of action in the immunoregulatory pathway (central tolerance vs peripheral tolerance vs co-stimulatory signals for proper antigen presentation), chromosomal location, and associated autoimmune diseases (may vary by ethnicity; see references).
| Pathway/ Mechanism | GenGene | Chromosome | Associated Autoimmune Diseases | Ref. |
|---|---|---|---|---|
| Central Tolerance | TSHR | 14q31 | GD | [16, 17] |
| Peripheral Tolerance | FOXP3 | Xp11 | GD, HT IPEX T1D RA PBC Psoriasis, Vitiligo Autoimmune Hemolytic Anemia Autoimmune Cytopenia Autoimmune Hepatitis |
[33, 34, 36, 37, 40] |
| Peripheral Tolerance | IL-2Rα/ CD25 | 10p15 | GD T1D MS |
[41, 45] |
| Co-stimulation (APC and T cell activation) | CD40 | 20q11 | GD SLE RA MS Behcet's disease IBD |
[50, 55-58, 66-68, 113] |
| Co-stimulation (T cell activation) | CTLA-4 | 2q33 | GD, HT T1D Autoimmune Addison's Disease SLE RA MS PBC IBD Celiac disease Sjogren's disease Systemic sclerosis Myasthenia gravis |
[86, 114-120] |
| Co-stimulation (T cell activation) | PTPN22/LYP | 1p13 | GD, HT T1D SLE RA |
[121-124] |
| Co-stimulation (B Cell Activation) | FCRL3 | 1q21-22 | GD, HT SLE RA MS Behcet's disease NMO |
[125-128] |
| Antigen Presentation (T cell activation) | HLA | 6p21 | GD, HT T1D SLE RA MS Celiac disease Behcet's disease Myasthenia gravis |
[129, 130]* |
see references for complete list of HLA-associated autoimmune diseases
Abbreviations: TSHR (thyrotropin receptor), FOXP3 (forkhead box P3), CTLA-4 (cytotoxic T-lymphocyte-associated molecule-4), PTPN22 (protein tyrosine phosphatase nonreceptor type-22), LYP (lymphoid tyrosine phosphatase), FCRL3(Fc receptor-like 3), Graves' disease (GD), Hashimoto's thyroiditis (HT), immune dysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX), type I diabetes mellitus (T1D), rheumatoid arthritis (RA), primary biliary cirrhosis (PBC), systemic lupus erythematosus (SLE), multiple sclerosis (MS), inflammatory bowel disease (IBD), neuromyelitis optica (NMO)
2. Genetic Disruption of Central Tolerance – TSHR gene
An attractive hypothesis is that GD is triggered by a defect in negative selection of autoreactive T cells to the TSHR (thyrotropin receptor), either in the thymus or the peripheral immune system. Indeed, a number of genetic variants associated with GD were shown to impact central tolerance (TSHR) or peripheral tolerance (FOXP3 and CD25, see section 3.1). The hallmark of GD is the presence of the stimulating TSHR antibodies. Consequently, TSHR has been considered an important candidate gene predisposing to GD even before the era of GWAS studies. Subsequent GWAS and other association studies have confirmed TSHR as a disease specific locus [11-13]. Consecutive comprehensive sequence analyses of the TSHR gene/locus localized the causative variant(s) to a 40-kb region within intron 1 where at least five GD-associated SNPs were identified (rs179247; rs2284720; rs12101255; rs12101261; and rs2268458) [11, 14, 15]. Further functional analyses of TSHR intron 1 polymorphisms provided direct evidence of a link between central tolerance and TSHR intron 1 SNPs. Recently, our group showed that the disease-predisposing genotype (TT) of SNP rs12101261 was associated with decreased thymic expression levels of TSHR mRNA [16].
By mapping epigenetic modifications induced by interferon alpha (IFNα), a key cytokine secreted during viral infections that was previously shown to trigger autoimmunity, we showed that the disease-associated variant of rs12101261 (TT) interacts through chromatin remodeling with the transcriptional repressor, promyelocytic leukemia zinc finger protein (PLZF), to reduce TSHR gene expression [16]. We proposed that loss of adequate epigenetic interactions in the thymus due to micro-environmental influences (e.g., cytokines, viral infections) would affect TSHR gene expression through genetic variants. Decreased intra-thymic expression of TSHR would facilitate pathogenic T cell escape from central tolerance and increase the risk of autoimmunity to TSHR. In agreement with these findings, Colobran et al. showed a correlation between TSHR thymic mRNA levels and rs179247, a SNP in tight linkage disequilibium (LD) with rs12101261. They showed that individuals homozygous or heterozygous for the GD-associated allele at the rs179247 SNP (AA or AG) have significantly lower TSHR mRNA expression levels in the thymus than individuals homozygous for the protective allele (GG) [17]. This unbalanced allelic expression likely represents defective central tolerance, contributing to GD through the escape of T cell clones targeting the TSHR.
3. Genetic Disruption of Peripheral Tolerance – FOXP3 and CD25 genes
3.1. FOXP3
FOXP3 (forkhead box P3), an X-linked gene [18], belongs to the forkhead/winged-helix family of DNA-binding transcription factors [19, 20]. It can act as both a transcriptional repressor [21] and activator [22-24] for primarily immunological genes. For example, FOXP3 can bind to Runt-related transcription factors to inhibit IL-2 and IFNγ expression [25] or, by binding to other transcription factors, it can also activate CD25 [26]. To maintain immunological self-tolerance, regulatory T cells (Tregs) suppress peripheral self-reactive lymphocytes that have escaped central tolerance in the thymus [27, 28] and FOXP3 is a known crucial regulator of Treg differentiation and function. The role of FOXP3 in autoimmunity was first revealed by the mouse mutant Scurfy, a line defective in FOXP3. As expected, the scurfy mutant phenotype is characterized by massive hyperproliferation and multi-organ infiltration of CD4+ T cells and is lethal in hemizygous males [20]. In humans, mutations in FOXP3 lead to an X-linked syndrome characterized by immune dysregulation, polyendocrinopathy and enteropathy (IPEX) [29-33].
Various FOXP3 polymorphisms have been reported to be associated with autoimmune thyroiditis (AITD). For example, a DXS573 microsatellite that is in LD with FOXP3 was found to be associated with AITD in Caucasian female AITD patients [34]. An A/C polymorphism in position -3279 has been associated with the development of treatment-resistant GD [35] while the CC genotype at position -2383 has been associated with severe HT [35]. Our group found an association between the (TC)n microsatellite in intron 5 of the FOXP3 gene and AITD in Caucasian males (p-0.011) [24]. We also identified that this microsatellite is associated with a variant of autoimmune polyglandular syndrome type 3 (designated APS3v) [36], characterized by the co-occurrence of AITD and type 1 diabetes (T1D) [37].
Mechanistically, we hypothesized that the (TC)n microsatellite in intron 5 may affect splicing because of its location and size, as intronic microsatellites have been shown to be regulators of gene splicing [38, 39]. Although no significant difference in splicing efficiency was observed when human embryonic kidney cells (HEK 293) were transfected with the long or short repeats of the FOXP3 intron 5 (TC)n microsatellite, our study identified a new splice variant designated FOXP3Δ6 (Figure 1). FOXP3Δ6 was expressed in the thymus and lymph nodes, as well as in Tregs [40]. The role of this splice variant in thyroid autoimmunity warrants further investigation. Despite the fact that we did not find a difference in the splice variant levels associated with the long or short microsatellite repeats, epigenetic interactions and changes, which are known to regulate gene expression, can potentially influence splicing [16]. It is possible that different FOXP3 splice variants, including the novel splice variant FOXP3Δ6 that we identified to be expressed in Tregs, may modulate immune responses, although further evidence is needed.
Figure 1.
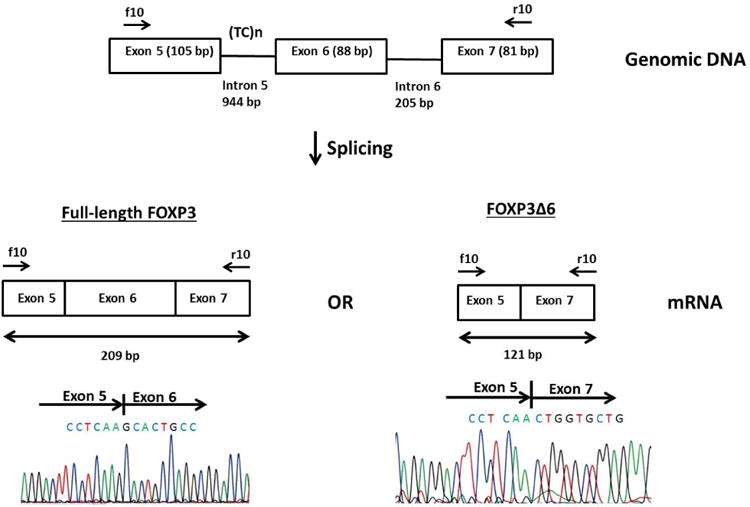
Schematic diagram of FOXP3 exon 5 through 7. The (TC)n microsatellite is located in intron 5. Primers FOXP3_f10 and FOXP3_r10 were used for amplification. The expected size of the PCR product with all 3 exons included is 209 bp. If exon 6 is skipped, the product size is 121 bp. The bottom part shows the sequencing results of the PCR products at the exon junctions (reproduced with permission from Gene 2015;556: 142-8).
3.2. CD25
CD25 (also known as IL-2Rα receptor or the α-subunit of the IL-2 receptor) is involved in the regulation of T cell function. More specifically, it is encoded by the CD25 region on chromosome 10p15.1, is highly expressed in Tregs, and mediates IL-2 signaling which is indispensable for CD25+CD4+ Treg survival and growth [41]. Similar to mice with impaired FOXP3, IL-2Rα deficient mice exhibit an analogous lethal lymphoproliferative disorder accompanied with severe autoimmunity [42]. Therefore, it is plausible that certain genetic variants in the CD25 gene predispose to autoimmunity by impairing Treg function and peripheral tolerance development. Indeed, a case-control study from the UK reported that CD25 was significantly associated with GD [43]. A GWAS study also from the UK reported similar results [44] and a study from Russia confirmed this association [45]. In the latter study, minor alleles of two SNPS in the IL-2Rα gene (rs41295061 and rs11594656) constituting the AA/AA haplotype were not only associated with increased risk of GD but also with elevated serum concentrations of sIL-2Rα in both GD patients and healthy controls compared to the protective GT/GT [45]. Our group further reported that CD25 was associated with only GD, but not with HT [15].
4. Genetic Disruption of Co-Stimulation – CD40 and CTLA-4 Genes
For proper T and B cell activation, antigen presenting cells (APCs) and T cells need appropriate positive and negative co-stimulatory signals, for which cytotoxic T lymphocyte-associated molecule-4 (CTLA-4) and CD40 are essential. [46]. As expected, they are both major susceptibility genes for AITD and autoimmunity in general [47, 48]. CTLA-4 carries an association with both GD and HT while CD40 is specific for GD.
4.1. CD40
CD40 is a tumor necrosis factor receptor (TNF-R) that plays a critical role in adaptive immunity and is predominantly present on APCs like B cells but also on thyroid epithelial cells [49]. Classically, the interaction between CD40 and its ligand CD154 on activated T lymphocytes provides co-stimulatory signals that are needed to activate APCs and T lymphocytes [50]. CD40 activation in B cells allows for immunoglobulin class switching, B cell proliferation, germinal center formation and generation of B cell memory [51]. CD40 also guides the development of immune-tolerant immature dendritic cells into potent immunostimulatory cells [48, 50]. And in T lymphocytes, CD40 enhances T cell priming and increases the expression of other co-stimulatory signals that ultimately result in potent cytokine production [52, 53]. Given CD40's pivotal role in coordinating antigen presentation and humoral immunity, it is not surprising that CD40 is associated with Graves' disease, a B cell mediated disease. In contrast, defective or absent CD40 activity results in a variety of profound immune system deficiencies including X-linked hyper-IgM syndrome (HIGM) in which the production of other immunoglobulin isotypes is greatly limited [54].
Through linkage and association studies and subsequent sequencing of the CD40 gene, several single nucleotide polymorphisms (SNPs) within the CD40 gene have emerged as causative genetic variants that predispose to GD. The most well-studied polymorphism, a -1C/T polymorphism (rs1883832) in the 5′ UTR Kozak sequence, was first identified by our group. It was found to have significant association with GD with a relative risk of 1.6 among individuals carrying the CC genotype [55]. These findings were replicated across various ethnicities including American and European Caucasians, Koreans, and Japanese with relative risks ranging from 1.22 to 1.93 [56-58]. Among treated GD patients, the C allele has been associated with higher persistent levels of thyroid peroxidase (TPO) and thyroglobulin (Tg) antibodies [59]. Indeed the T allele and TT genotype were found to carry a protective effect and even linked to later-age onset of GD. [60, 61]. In animal models of GD, blockade of CD40 stimulation has suppressed the progression to murine thyroiditis [53, 62, 63].
We have previously shown that the -1C/T CD40 SNP functionally affects the translation efficiency of CD40 and that the C allele correlated with 13-35% increased synthesis of translated CD40 protein [64]. This mechanistically could potentiate activation of APCs and B cells, thereby leading to a lower threshold for autoimmunity. Likewise upregulation of T cell co-stimulation would enhance an overall pro-inflammatory response. Increased CD40 expression on thyroid cells, in the setting of the aforementioned causative polymorphism, could additionally lead to autoimmunity by local, bystander mechanisms. In fact, overexpression of CD40 in the thyroids of transgenic mice heightened their susceptibility to experimental autoimmune Graves' disease (EAGD) [62]. Other less well-characterized CD40 SNPs (rs745307, rs11569309, and rs3765457) have been significantly associated with higher relapse of GD after withdrawal of antithyroid medications [65]. Various CD40 SNPs have been implicated in a wide range of autoimmune and immune-mediated diseases, including systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease, and IgE-mediated asthma [50, 66-69].
4.2. CTLA-4
CTLA-4 (CD152) is a transmembrane immunoregulatory protein classically present on activated T cells that functions as a competitive inhibitor to CD28 and is an important negative regulator of T cell activation. Upon T cell activation, CTLA-4 is rapidly expressed and binds to its ligands (B7-1 and B7-2), thereby blocking CD28 activation and leading to eventual T cell homeostasis [70]. CTLA-4 activation subsequently results in diminished T cell activation, reduced IL-2 production, and the arrest of T cell cycling and further activation [71, 72]. SNPs in CTLA-4 have been linked to both GD and HT, in addition to an array of other autoimmune diseases. Furthermore, in murine models, CTLA-4 activation suppressed experimental lupus, autoimmune nephritis, collagen-induced arthritis, and diabetes [73-76].
Several CTLA-4 SNPs have demonstrated risk for thyroid autoimmunity. An A/G SNP at position 49 in exon 1 [77-82], an (AT)n microsatellite polymorphism in the 3′ untranslated region of exon 4 [83, 84], and the CT60 polymorphism in the 3′UTR have consistently been associated with AITD across different ethnicities. Mechanistically, reduced CTLA-4 expression and/or activity would logically predispose to autoimmunity by way of decreased suppression of T cell activation and proliferation. Indeed, studies have shown that the A/G49 SNP leads to ineffective glycosylation and processing of CTLA-4 [85] while the longer (AT)n repeats lower the stability of the CTLA-4 mRNA transcript [83]. The GG genotype of the CT60 SNP has also been shown to correlate with decreased mRNA levels of the soluble form of CTLA-4 [86].
5. Genetic Disruption of Antigen Presentation – HLA-DR and Thyroglobulin Genes
The HLA-DR3 allele has a well established association with AITD (reviewed in [87]). HLA-DR is a class II HLA gene that plays a critical role in antigen presentation. We, therefore, hypothesized that specific sequence variants in HLA-DR3 predispose to AITD by creating unique pocket structures enabling the presentation of pathogenic thyroid-derived peptides to T cells. By sequencing the HLA-DR gene in a large cohort of AITD patients and controls, we discovered that the presence of arginine at position 74 of the HLA-DRβ chain (HLA-DRβ1-Arg74) is critical for the development of AITD while glutamine at this position (Gln-74) is protective [88, 89], a finding that was corroborated by others [90]. Further analysis, using molecular dynamic simulations, has shown that the HLA-DRβ1-Arg74 allele creates a narrow positively charged pocket P4 when compared to the pocket containing the protective Gln-74 allele [89]. These findings raised the question of which pathogenic peptides are presented within the HLA-DRβ1-Arg74 pocket to trigger AITD?
The major thyroid antigens that are targeted by the autoimmune response in AITD are Tg, TPO, and TSHR. A study by Muixi et al. [91] tested peptides that were bound to HLA-DR within thyroid tissues removed from GD patients. Only Tg peptides were found to be bound to HLA-DR, suggesting that presentation of Tg peptides by HLA-DR to T cells may be the initial trigger of AITD. Indeed, whole genome screening studies have shown that Tg is a major AITD susceptibility gene [92, 93]. In order to identify the specific Tg variants that predispose to AITD, we sequenced the Tg gene and identified several amino acid variants caused by non-synonymous SNPs that were significantly associated with AITD [94]. One amino acid variant, W1999R, showed an interaction with HLA-DRβ1-Arg74, conferring together a very high odds ratio for AITD [95].
In view of the interaction between the Tg gene and the HLA-DRβ1-Arg74 allele, we hypothesized that the HLA-DRβ1-Arg74 pocket may present pathogenic Tg peptides that can trigger AITD. Using recombinant HLA-DRβ1-Arg74 protein, we identified four Tg peptides that bound specifically to the HLA-DRβ1-Arg74 pocket and not to the Gln-74 pocket [96]. However, a link between the Tg amino acid variants and the production of the pathogenic Tg peptides has not yet been established.
6. Susceptibility Genes in Specific Subsets
6.1. Susceptibility to AITD in different ethnic groups
An NHANES database study that analyzed nearly 18,000 U.S. adults identified a significant difference in the prevalence of AITD by ethnicity and race. Although the study was limited by how GD was diagnosed, non-Hispanic blacks were noted to be hyperthyroid almost three-fold more than the non-Hispanic whites (odds ratio of 2.9; C.I. 1.5-5.7) [10]. This underscores the variability in AITD genetic susceptibility among different ethnicities. For example, HLA-DR3 is associated with AITD only in Caucasian populations [88]. A three-stage GWAS study in the Chinese Han population showed an association of a SNP (rs2474619) in intron 2 of BACH2 (BTB And CNC Homology 2) with GD [97]. BACH2 is highly expressed on B cells, suppresses the production of immunoglobulins [98], and has a known immune function as a regulator of nucleic acid-triggered antiviral responses in human cells [99]. A study from the UK also showed that the BACH2 locus was associated with an increased risk of AITD [44] although the exact susceptibility loci for each disease (GD, HT) were incompletely identified in this study. Another GWAS study from China reported an association between both GPR174-ITM2A (encoding G protein-coupled receptor 174) at Xq21.1 and SLAMF6 (signaling lymphocytic activation molecule) at chromosome 1q23.2 and GD [100]. The protein encoded by SLAMF6 gene is expressed on natural killer cells, T lymphocytes, and B lymphocytes and may be involved in the regulation of B cell receptor-mediated central tolerance [101].
Other genes identified to be associated with AITD in the aforementioned UK study were several non-HLA genes: MMEL1 (membrane metallo-endopeptidase-like 1) on chromosome 1p36, 16 kb upstream of TRIB2 (tribbles homolog 2) on chromosome 2p25, LPP (LIM domain containing preferred translocation partner in lipoma) on chromosome 3q27, PRICKLE1 (prickle homolog 1) on chromosome 12p12, 83 kb upstream of ITGAM (integrin, alpha M) on chromosome 16p11, and FGFR1OP (fibroblast growth factor receptor 1 oncogene partner, 118 kb upstream of CCR6) on chromosome 6 [44]. Most of these genes have been linked to other autoimmune diseases even though their precise immune-mediated functions are not yet known. The RNASET2-FGFR1OP-CCR6 region at chromosome 6q27 showed a significant association with GD in a GWAS study among the Chinese Han population [13]. In contrast, three SNPs in the CCR6 (CC chemokine receptor 6) gene were not associated with AITD in this same group despite CCR6's general role in the recruitment of dendritic cells and T helper 17 cells [102]. The association of two RNASET2 (ribonuclease T2) gene polymorphisms (SNPs rs3777722 and rs9355610) with GD but not with HT was further confirmed in other studies from China. And a trend for association between the rs9355610 SNP in the RNASET2 gene and GD was established in a different study among Polish Caucasians [103].
6.2. Susceptibility to AITD in different age groups
We studied a subset of Caucasian GD patients with young age of onset ([AO] < 30 years) and reported an association of BTNL2 (butyrophilin-like 2) with young AO GD [104]. BTNL2 modulates T cell activity by suppressing T cell proliferation and cytokine production and has been associated with other autoimmune diseases including rheumatoid arthritis and vitiligo. It is uncertain, however, if this association is strictly due to LD with the HLA genes due its location within the MHC class II region, as has also been reported by other groups [105]. Other genes found to be associated with young AO GD in our study were NOTCH4 (a member of the Type 1 transmembrane protein family), CXCR4 (a chemokine receptor), and TNFAIP3 (tumor necrosis factor α-induced protein 3) [104]. TNFAIP3, a negative regulator of inflammation that inhibits NF-kB (nuclear factor kappa-B) and TNF (tumor necrosis factor)-mediated pathways, has been linked to several immune-mediated diseases like T1D, systemic lupus erythematosus, and rheumatoid arthritis [106, 107]. Supporting these findings are results from a Chinese study that showed an association between TNFAIP3 polymorphisms (rs598493; rs610604; and rs661561) and GD, but not HT [107].
6.3. Subsetting by AITD phenotype
The development of thyroid antibodies (TAb) without clinical disease is a unique AITD phenotype. We performed a whole genome linkage study in multigenerational families in which the phenotype of TAb only clustered. Our study showed that three loci previously reported to be linked with clinical AITD also showed unique linkage with the TAb-only phenotype. These included the CTLA-4 locus on chromosome 2q, a locus on chromosome 6p close to the HLA region, and the Tg locus on chromosome 8q [108]. A meta-analysis of GWAS studies for TPO antibodies reported that MAGI3 (membrane associated guanylate kinase WW and PDZ domain containing 3) was associated with increased risk for both hypothyroidism and hyperthyroidism [109].
Another AITD phenotype we have investigated is the co-occurrence of AITD and T1D in the same individual, which is considered a variant of the autoimmune polyglandular syndrome type 3 (APS3v). In a genome wide study of North American Caucasian APS3v patients, our group recently reported associations between APS3v and PTPN22 (protein tyrosine phosphatase nonreceptor type-22), MAGI3, PHTF1 (putative homeodomain transcription factor 1), and GRP103 (G protein-coupled receptor 103) [110]. The GRP103 gene (also called QRFPR) is located on chromosome 4 and its ligand is expressed in the thyroid [111].
7. Discussion
The AITDs result from the complex interactions between susceptibility alleles and the environment. Although the exact, definitive underlying cause is unknown, evidence indicates that environmental factors (infections, medications, smoking, iodine, etc [82]) in a background of genetic predisposition trigger AITD. The immune-regulatory genes that predispose to AITD (FOXP3, CD25, CD40, CTLA-4, the HLA genes, PTPN22, among other emerging immunoregulatory genes) play critical parts in the development of an effective immune response including self-tolerance, cell-mediated immunity and humoral immunity. The various polymorphisms demonstrate that genetic triggers for autoimmunity can result from inherited abnormalities in the proper progression from central tolerance in the thymus and peripheral tolerance by Tregs to proper co-stimulation of T cells and APCs in the immunological synapse. Indeed many of these polymorphisms lead not only to higher risk of AITD but also to other systemic, autoimmune manifestations. With further advances in the techniques of genomic analysis, more genetic variants (both immune-modulating and thyroid-specific), will continue to be discovered and allow for the identification of high-risk individuals.
The exact functional effects of these polymorphisms, however, have not completely been elucidated. Whether the SNPs negatively affect sufficient thymic expression of self-antigens (TSHR), alter the function of Tregs (FOXP3, CD25), or lower the threshold for T cell activation (CD40, CTLA4, HLA), it is clear that there is ultimately a breakdown in tolerance that tips the balance towards autoimmunity. And evidence suggests that SNPs in more than one susceptibility gene may synergistically shift the balance even further [112]. Understanding the functional and mechanistic consequences of these susceptibility gene variants is essential for the development of new, targeted and preventative therapies for AITD.
Highlights.
Polymorphisms in immunoregulatory genes predispose to thyroid autoimmunity.
GD and HT can result from genetic disruptions in central tolerance, peripheral tolerance,co-stimulation, and antigen presentation.
Review of the potential mechanisms by which TSHR, FOXP3, CD25, CD40, CTLA-4,HLA, and other genes lead to a breakdown of self-tolerance.
Acknowledgments
This work was supported in part by National Institutes of Health Grants DK61659 and DK073681 and by the Department of Veterans Affairs, Veterans Health Administration.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clinical immunology and immunopathology. 1997;84:223–43. doi: 10.1006/clin.1997.4412. [DOI] [PubMed] [Google Scholar]
- 2.Antonelli A, Ferrari SM, Corrado A, Di Domenicantonio A, Fallahi P. Autoimmune thyroid disorders. Autoimmunity reviews. 2015;14:174–80. doi: 10.1016/j.autrev.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 3.Tomer Y. Mechanisms of autoimmune thyroid diseases: from genetics to epigenetics. Annual review of pathology. 2014;9:147–56. doi: 10.1146/annurev-pathol-012513-104713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villanueva R, Greenberg DA, Davies TF, Tomer Y. Sibling recurrence risk in autoimmune thyroid disease. Thyroid: official journal of the American Thyroid Association. 2003;13:761–4. doi: 10.1089/105072503768499653. [DOI] [PubMed] [Google Scholar]
- 5.Tomer Y, Davies TF. Searching for the autoimmune thyroid disease susceptibility genes: from gene mapping to gene function. Endocrine reviews. 2003;24:694–717. doi: 10.1210/er.2002-0030. [DOI] [PubMed] [Google Scholar]
- 6.Hall R, Stanbury JB. Familial studies of autoimmune thyroiditis. Clinical and experimental immunology. 1967;2(Suppl):719–25. [PMC free article] [PubMed] [Google Scholar]
- 7.Brix TH, Christensen K, Holm NV, Harvald B, Hegedus L. A population-based study of Graves' disease in Danish twins. Clinical endocrinology. 1998;48:397–400. doi: 10.1046/j.1365-2265.1998.00450.x. [DOI] [PubMed] [Google Scholar]
- 8.Brix TH, Hegedus L. Twin studies as a model for exploring the aetiology of autoimmune thyroid disease. Clinical endocrinology. 2012;76:457–64. doi: 10.1111/j.1365-2265.2011.04318.x. [DOI] [PubMed] [Google Scholar]
- 9.Brix TH, Kyvik KO, Christensen K, Hegedus L. Evidence for a major role of heredity in Graves' disease: a population-based study of two Danish twin cohorts. The Journal of clinical endocrinology and metabolism. 2001;86:930–4. doi: 10.1210/jcem.86.2.7242. [DOI] [PubMed] [Google Scholar]
- 10.McLeod DS, Cooper DS, Ladenson PW, Whiteman DC, Jordan SJ. Race/Ethnicity and the prevalence of thyrotoxicosis in young americans. Thyroid: official journal of the American Thyroid Association. 2015;25:621–8. doi: 10.1089/thy.2014.0504. [DOI] [PubMed] [Google Scholar]
- 11.Dechairo BM, Zabaneh D, Collins J, Brand O, Dawson GJ, Green AP, et al. Association of the TSHR gene with Graves' disease: the first disease specific locus. European journal of human genetics: EJHG. 2005;13:1223–30. doi: 10.1038/sj.ejhg.5201485. [DOI] [PubMed] [Google Scholar]
- 12.Brand OJ, Barrett JC, Simmonds MJ, Newby PR, McCabe CJ, Bruce CK, et al. Association of the thyroid stimulating hormone receptor gene (TSHR) with Graves' disease. Human molecular genetics. 2009;18:1704–13. doi: 10.1093/hmg/ddp087. [DOI] [PubMed] [Google Scholar]
- 13.Chu X, Pan CM, Zhao SX, Liang J, Gao GQ, Zhang XM, et al. A genome-wide association study identifies two new risk loci for Graves' disease. Nature genetics. 2011;43:897–901. doi: 10.1038/ng.898. [DOI] [PubMed] [Google Scholar]
- 14.Ploski R, Brand OJ, Jurecka-Lubieniecka B, Franaszczyk M, Kula D, Krajewski P, et al. Thyroid stimulating hormone receptor (TSHR) intron 1 variants are major risk factors for Graves' disease in three European Caucasian cohorts. PloS one. 2010;5:e15512. doi: 10.1371/journal.pone.0015512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomer Y, Hasham A, Davies TF, Stefan M, Concepcion E, Keddache M, et al. Fine mapping of loci linked to autoimmune thyroid disease identifies novel susceptibility genes. The Journal of clinical endocrinology and metabolism. 2013;98:E144–52. doi: 10.1210/jc.2012-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stefan M, Wei C, Lombardi A, Li CW, Concepcion ES, Inabnet WB, 3rd, et al. Genetic-epigenetic dysregulation of thymic TSH receptor gene expression triggers thyroid autoimmunity. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:12562–7. doi: 10.1073/pnas.1408821111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colobran R, Armengol Mdel P, Faner R, Gartner M, Tykocinski LO, Lucas A, et al. Association of an SNP with intrathymic transcription of TSHR and Graves' disease: a role for defective thymic tolerance. Human molecular genetics. 2011;20:3415–23. doi: 10.1093/hmg/ddr247. [DOI] [PubMed] [Google Scholar]
- 18.Allan SE, Passerini L, Bacchetta R, Crellin N, Dai M, Orban PC, et al. The role of 2 FOXP3 isoforms in the generation of human CD4+ Tregs. The Journal of clinical investigation. 2005;115:3276–84. doi: 10.1172/JCI24685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaufmann E, Knochel W. Five years on the wings of fork head. Mechanisms of development. 1996;57:3–20. doi: 10.1016/0925-4773(96)00539-4. [DOI] [PubMed] [Google Scholar]
- 20.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nature genetics. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 21.Schubert LA, Jeffery E, Zhang Y, Ramsdell F, Ziegler SF. Scurfin (FOXP3) acts as a repressor of transcription and regulates T cell activation. The Journal of biological chemistry. 2001;276:37672–9. doi: 10.1074/jbc.M104521200. [DOI] [PubMed] [Google Scholar]
- 22.Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–87. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 23.Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445:931–5. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445:936–40. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- 25.Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, et al. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–9. doi: 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- 26.Camperio C, Caristi S, Fanelli G, Soligo M, Del Porto P, Piccolella E. Forkhead transcription factor FOXP3 upregulates CD25 expression through cooperation with RelA/NF-kappaB. PloS one. 2012;7:e48303. doi: 10.1371/journal.pone.0048303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maloy KJ, Powrie F. Regulatory T cells in the control of immune pathology. Nature immunology. 2001;2:816–22. doi: 10.1038/ni0901-816. [DOI] [PubMed] [Google Scholar]
- 28.Sakaguchi S, Sakaguchi N, Shimizu J, Yamazaki S, Sakihama T, Itoh M, et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunological reviews. 2001;182:18–32. doi: 10.1034/j.1600-065x.2001.1820102.x. [DOI] [PubMed] [Google Scholar]
- 29.Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. The Journal of clinical investigation. 2000;106:R75–81. doi: 10.1172/JCI11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nature genetics. 2001;27:20–1. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 31.Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nature genetics. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 32.Wildin RS, Freitas A. IPEX and FOXP3: clinical and research perspectives. Journal of autoimmunity. 2005;25(Suppl):56–62. doi: 10.1016/j.jaut.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Katoh H, Zheng P, Liu Y. FOXP3: genetic and epigenetic implications for autoimmunity. Journal of autoimmunity. 2013;41:72–8. doi: 10.1016/j.jaut.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ban Y, Tozaki T, Tobe T, Ban Y, Jacobson EM, Concepcion ES, et al. The regulatory T cell gene FOXP3 and genetic susceptibility to thyroid autoimmunity: an association analysis in Caucasian and Japanese cohorts. Journal of autoimmunity. 2007;28:201–7. doi: 10.1016/j.jaut.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 35.Inoue N, Watanabe M, Morita M, Tomizawa R, Akamizu T, Tatsumi K, et al. Association of functional polymorphisms related to the transcriptional level of FOXP3 with prognosis of autoimmune thyroid diseases. Clinical and experimental immunology. 2010;162:402–6. doi: 10.1111/j.1365-2249.2010.04229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villano MJ, Huber AK, Greenberg DA, Golden BK, Concepcion E, Tomer Y. Autoimmune thyroiditis and diabetes: dissecting the joint genetic susceptibility in a large cohort of multiplex families. The Journal of clinical endocrinology and metabolism. 2009;94:1458–66. doi: 10.1210/jc.2008-2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jenkins RC, Weetman AP. Disease associations with autoimmune thyroid disease. Thyroid: official journal of the American Thyroid Association. 2002;12:977–88. doi: 10.1089/105072502320908312. [DOI] [PubMed] [Google Scholar]
- 38.Pagani F, Buratti E, Stuani C, Romano M, Zuccato E, Niksic M, et al. Splicing factors induce cystic fibrosis transmembrane regulator exon 9 skipping through a nonevolutionary conserved intronic element. The Journal of biological chemistry. 2000;275:21041–7. doi: 10.1074/jbc.M910165199. [DOI] [PubMed] [Google Scholar]
- 39.Gabellini N. A polymorphic GT repeat from the human cardiac Na+Ca2+ exchanger intron 2 activates splicing. European journal of biochemistry / FEBS. 2001;268:1076–83. doi: 10.1046/j.1432-1327.2001.01974.x. [DOI] [PubMed] [Google Scholar]
- 40.Li CW, Concepcion E, Tomer Y. Dissecting the role of the FOXP3 gene in the joint genetic susceptibility to autoimmune thyroiditis and diabetes: a genetic and functional analysis. Gene. 2015;556:142–8. doi: 10.1016/j.gene.2014.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cerosaletti K, Schneider A, Schwedhelm K, Frank I, Tatum M, Wei S, et al. Multiple autoimmune-associated variants confer decreased IL-2R signaling in CD4+ CD25(hi) T cells of type 1 diabetic and multiple sclerosis patients. PloS one. 2013;8:e83811. doi: 10.1371/journal.pone.0083811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bayer AL, Yu A, Adeegbe D, Malek TR. Essential role for interleukin-2 for CD4(+)CD25(+) T regulatory cell development during the neonatal period. The Journal of experimental medicine. 2005;201:769–77. doi: 10.1084/jem.20041179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brand OJ, Lowe CE, Heward JM, Franklyn JA, Cooper JD, Todd JA, et al. Association of the interleukin-2 receptor alpha (IL-2Ralpha)/CD25 gene region with Graves' disease using a multilocus test and tag SNPs. Clinical endocrinology. 2007;66:508–12. doi: 10.1111/j.1365-2265.2007.02762.x. [DOI] [PubMed] [Google Scholar]
- 44.Cooper JD, Simmonds MJ, Walker NM, Burren O, Brand OJ, Guo H, et al. Seven newly identified loci for autoimmune thyroid disease. Human molecular genetics. 2012;21:5202–8. doi: 10.1093/hmg/dds357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chistiakov DA, Chistiakova EI, Voronova NV, Turakulov RI, Savost'anov KV. A variant of the Il2ra / Cd25 gene predisposing to graves' disease is associated with increased levels of soluble interleukin-2 receptor. Scandinavian journal of immunology. 2011;74:496–501. doi: 10.1111/j.1365-3083.2011.02608.x. [DOI] [PubMed] [Google Scholar]
- 46.Reiser H, Stadecker MJ. Costimulatory B7 molecules in the pathogenesis of infectious and autoimmune diseases. The New England journal of medicine. 1996;335:1369–77. doi: 10.1056/NEJM199610313351807. [DOI] [PubMed] [Google Scholar]
- 47.Teft WA, Kirchhof MG, Madrenas J. A molecular perspective of CTLA-4 function. Annual review of immunology. 2006;24:65–97. doi: 10.1146/annurev.immunol.24.021605.090535. [DOI] [PubMed] [Google Scholar]
- 48.Munroe ME. Functional roles for T cell CD40 in infection and autoimmune disease: the role of CD40 in lymphocyte homeostasis. Seminars in immunology. 2009;21:283–8. doi: 10.1016/j.smim.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 49.Faure GC, Bensoussan-Lejzerowicz D, Bene MC, Aubert V, Leclere J. Coexpression of CD40 and class II antigen HLA-DR in Graves' disease thyroid epithelial cells. Clinical immunology and immunopathology. 1997;84:212–5. doi: 10.1006/clin.1997.4391. [DOI] [PubMed] [Google Scholar]
- 50.Toubi E, Shoenfeld Y. The role of CD40-CD154 interactions in autoimmunity and the benefit of disrupting this pathway. Autoimmunity. 2004;37:457–64. doi: 10.1080/08916930400002386. [DOI] [PubMed] [Google Scholar]
- 51.Bishop GA, Hostager BS. The CD40-CD154 interaction in B cell-T cell liaisons. Cytokine & growth factor reviews. 2003;14:297–309. doi: 10.1016/s1359-6101(03)00024-8. [DOI] [PubMed] [Google Scholar]
- 52.Bishop GA. The many faces of CD40: multiple roles in normal immunity and disease. Seminars in immunology. 2009;21:255–6. doi: 10.1016/j.smim.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 53.Carayanniotis G, Masters SR, Noelle RJ. Suppression of murine thyroiditis via blockade of the CD40-CD40L interaction. Immunology. 1997;90:421–6. doi: 10.1111/j.1365-2567.1997.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kehry MR. CD40-mediated signaling in B cells. Balancing cell survival, growth, and death. Journal of immunology. 1996;156:2345–8. [PubMed] [Google Scholar]
- 55.Tomer Y, Concepcion E, Greenberg DA. A C/T single-nucleotide polymorphism in the region of the CD40 gene is associated with Graves' disease. Thyroid: official journal of the American Thyroid Association. 2002;12:1129–35. doi: 10.1089/105072502321085234. [DOI] [PubMed] [Google Scholar]
- 56.Kurylowicz A, Kula D, Ploski R, Skorka A, Jurecka-Lubieniecka B, Zebracka J, et al. Association of CD40 gene polymorphism (C-1T) with susceptibility and phenotype of Graves' disease. Thyroid: official journal of the American Thyroid Association. 2005;15:1119–24. doi: 10.1089/thy.2005.15.1119. [DOI] [PubMed] [Google Scholar]
- 57.Ban Y, Tozaki T, Taniyama M, Tomita M, Ban Y. Association of a C/T single-nucleotide polymorphism in the 5′ untranslated region of the CD40 gene with Graves' disease in Japanese. Thyroid: official journal of the American Thyroid Association. 2006;16:443–6. doi: 10.1089/thy.2006.16.443. [DOI] [PubMed] [Google Scholar]
- 58.Kim TY, Park YJ, Hwang JK, Song JY, Park KS, Cho BY, et al. A C/T polymorphism in the 5′-untranslated region of the CD40 gene is associated with Graves' disease in Koreans. Thyroid: official journal of the American Thyroid Association. 2003;13:919–25. doi: 10.1089/105072503322511319. [DOI] [PubMed] [Google Scholar]
- 59.Jacobson EM, Huber AK, Akeno N, Sivak M, Li CW, Concepcion E, et al. A CD40 Kozak sequence polymorphism and susceptibility to antibody-mediated autoimmune conditions: the role of CD40 tissue-specific expression. Genes and immunity. 2007;8:205–14. doi: 10.1038/sj.gene.6364375. [DOI] [PubMed] [Google Scholar]
- 60.Mukai T, Hiromatsu Y, Fukutani T, Ichimura M, Kaku H, Miyake I, et al. A C/T polymorphism in the 5′ untranslated region of the CD40 gene is associated with later onset of Graves' disease in Japanese. Endocrine journal. 2005;52:471–7. doi: 10.1507/endocrj.52.471. [DOI] [PubMed] [Google Scholar]
- 61.Hsiao JY, Hsieh MC, Hsiao CT, Weng HH, Ke DS. Association of CD40 and thyroglobulin genes with later-onset Graves' disease in Taiwanese patients. European journal of endocrinology / European Federation of Endocrine Societies. 2008;159:617–21. doi: 10.1530/EJE-08-0410. [DOI] [PubMed] [Google Scholar]
- 62.Huber AK, Finkelman FD, Li CW, Concepcion E, Smith E, Jacobson E, et al. Genetically driven target tissue overexpression of CD40: a novel mechanism in autoimmune disease. Journal of immunology. 2012;189:3043–53. doi: 10.4049/jimmunol.1200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Resetkova E, Kawai K, Enomoto T, Arreaza G, Togun R, Foy TM, et al. Antibody to gp39, the ligand for CD40 significantly inhibits the humoral response from Graves' thyroid tissues xenografted into severe combined immunodeficient (SCID) mice. Thyroid: official journal of the American Thyroid Association. 1996;6:267–73. doi: 10.1089/thy.1996.6.267. [DOI] [PubMed] [Google Scholar]
- 64.Jacobson EM, Concepcion E, Oashi T, Tomer Y. A Graves' disease-associated Kozak sequence single-nucleotide polymorphism enhances the efficiency of CD40 gene translation: a case for translational pathophysiology. Endocrinology. 2005;146:2684–91. doi: 10.1210/en.2004-1617. [DOI] [PubMed] [Google Scholar]
- 65.Wang PW, Chen IY, Juo SH, Hsi E, Liu RT, Hsieh CJ. Genotype and phenotype predictors of relapse of graves' disease after antithyroid drug withdrawal. European thyroid journal. 2013;1:251–8. doi: 10.1159/000342621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koshy M, Berger D, Crow MK. Increased expression of CD40 ligand on systemic lupus erythematosus lymphocytes. The Journal of clinical investigation. 1996;98:826–37. doi: 10.1172/JCI118855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Filion LG, Matusevicius D, Graziani-Bowering GM, Kumar A, Freedman MS. Monocyte-derived IL12, CD86 (B7-2) and CD40L expression in relapsing and progressive multiple sclerosis. Clinical immunology. 2003;106:127–38. doi: 10.1016/s1521-6616(02)00028-1. [DOI] [PubMed] [Google Scholar]
- 68.Danese S, Katz JA, Saibeni S, Papa A, Gasbarrini A, Vecchi M, et al. Activated platelets are the source of elevated levels of soluble CD40 ligand in the circulation of inflammatory bowel disease patients. Gut. 2003;52:1435–41. doi: 10.1136/gut.52.10.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park JH, Chang HS, Park CS, Jang AS, Park BL, Rhim TY, et al. Association analysis of CD40 polymorphisms with asthma and the level of serum total IgE. American journal of respiratory and critical care medicine. 2007;175:775–82. doi: 10.1164/rccm.200609-1286OC. [DOI] [PubMed] [Google Scholar]
- 70.Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunological reviews. 2008;224:166–82. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- 71.Brunner MC, Chambers CA, Chan FK, Hanke J, Winoto A, Allison JP. CTLA-4-Mediated inhibition of early events of T cell proliferation. Journal of immunology. 1999;162:5813–20. [PubMed] [Google Scholar]
- 72.Vaidya B, Kendall-Taylor P, Pearce SH. The genetics of autoimmune thyroid disease. The Journal of clinical endocrinology and metabolism. 2002;87:5385–97. doi: 10.1210/jc.2002-020492. [DOI] [PubMed] [Google Scholar]
- 73.Finck BK, Linsley PS, Wofsy D. Treatment of murine lupus with CTLA4Ig. Science. 1994;265:1225–7. doi: 10.1126/science.7520604. [DOI] [PubMed] [Google Scholar]
- 74.Nishikawa K, Linsley PS, Collins AB, Stamenkovic I, McCluskey RT, Andres G. Effect of CTLA-4 chimeric protein on rat autoimmune anti-glomerular basement membrane glomerulonephritis. European journal of immunology. 1994;24:1249–54. doi: 10.1002/eji.1830240602. [DOI] [PubMed] [Google Scholar]
- 75.Knoerzer DB, Karr RW, Schwartz BD, Mengle-Gaw LJ. Collagen-induced arthritis in the BB rat. Prevention of disease by treatment with CTLA-4-Ig. The Journal of clinical investigation. 1995;96:987–93. doi: 10.1172/JCI118146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lenschow DJ, Herold KC, Rhee L, Patel B, Koons A, Qin HY, et al. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity. 1996;5:285–93. doi: 10.1016/s1074-7613(00)80323-4. [DOI] [PubMed] [Google Scholar]
- 77.Donner H, Rau H, Walfish PG, Braun J, Siegmund T, Finke R, et al. CTLA4 alanine-17 confers genetic susceptibility to Graves' disease and to type 1 diabetes mellitus. The Journal of clinical endocrinology and metabolism. 1997;82:143–6. doi: 10.1210/jcem.82.1.3699. [DOI] [PubMed] [Google Scholar]
- 78.Nithiyananthan R, Heward JM, Allahabadia A, Franklyn JA, Gough SC. Polymorphism of the CTLA-4 gene is associated with autoimmune hypothyroidism in the United Kingdom. Thyroid: official journal of the American Thyroid Association. 2002;12:3–6. doi: 10.1089/105072502753451896. [DOI] [PubMed] [Google Scholar]
- 79.Villanueva R, Inzerillo AM, Tomer Y, Barbesino G, Meltzer M, Concepcion ES, et al. Limited genetic susceptibility to severe Graves' ophthalmopathy: no role for CTLA-4 but evidence for an environmental etiology. Thyroid: official journal of the American Thyroid Association. 2000;10:791–8. doi: 10.1089/thy.2000.10.791. [DOI] [PubMed] [Google Scholar]
- 80.Braun J, Donner H, Siegmund T, Walfish PG, Usadel KH, Badenhoop K. CTLA-4 promoter variants in patients with Graves' disease and Hashimoto's thyroiditis. Tissue antigens. 1998;51:563–6. doi: 10.1111/j.1399-0039.1998.tb02993.x. [DOI] [PubMed] [Google Scholar]
- 81.Yanagawa T, Taniyama M, Enomoto S, Gomi K, Maruyama H, Ban Y, et al. CTLA4 gene polymorphism confers susceptibility to Graves' disease in Japanese. Thyroid: official journal of the American Thyroid Association. 1997;7:843–6. doi: 10.1089/thy.1997.7.843. [DOI] [PubMed] [Google Scholar]
- 82.Tomer Y, Huber A. The etiology of autoimmune thyroid disease: a story of genes and environment. Journal of autoimmunity. 2009;32:231–9. doi: 10.1016/j.jaut.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yanagawa T, Hidaka Y, Guimaraes V, Soliman M, DeGroot LJ. CTLA-4 gene polymorphism associated with Graves' disease in a Caucasian population. The Journal of clinical endocrinology and metabolism. 1995;80:41–5. doi: 10.1210/jcem.80.1.7829637. [DOI] [PubMed] [Google Scholar]
- 84.Kotsa K, Watson PF, Weetman AP. A CTLA-4 gene polymorphism is associated with both Graves disease and autoimmune hypothyroidism. Clinical endocrinology. 1997;46:551–4. doi: 10.1046/j.1365-2265.1997.1710996.x. [DOI] [PubMed] [Google Scholar]
- 85.Anjos S, Nguyen A, Ounissi-Benkalha H, Tessier MC, Polychronakos C. A common autoimmunity predisposing signal peptide variant of the cytotoxic T-lymphocyte antigen 4 results in inefficient glycosylation of the susceptibility allele. The Journal of biological chemistry. 2002;277:46478–86. doi: 10.1074/jbc.M206894200. [DOI] [PubMed] [Google Scholar]
- 86.Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–11. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 87.Jacobson EM, Huber A, Tomer Y. The HLA gene complex in thyroid autoimmunity: from epidemiology to etiology. Journal of autoimmunity. 2008;30:58–62. doi: 10.1016/j.jaut.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ban Y, Davies TF, Greenberg DA, Concepcion ES, Osman R, Oashi T, et al. Arginine at position 74 of the HLA-DR beta1 chain is associated with Graves' disease. Genes and immunity. 2004;5:203–8. doi: 10.1038/sj.gene.6364059. [DOI] [PubMed] [Google Scholar]
- 89.Menconi F, Monti MC, Greenberg DA, Oashi T, Osman R, Davies TF, et al. Molecular amino acid signatures in the MHC class II peptide-binding pocket predispose to autoimmune thyroiditis in humans and in mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14034–9. doi: 10.1073/pnas.0806584105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Simmonds MJ, Howson JM, Heward JM, Cordell HJ, Foxall H, Carr-Smith J, et al. Regression mapping of association between the human leukocyte antigen region and Graves disease. American journal of human genetics. 2005;76:157–63. doi: 10.1086/426947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Muixi L, Carrascal M, Alvarez I, Daura X, Marti M, Armengol MP, et al. Thyroglobulin peptides associate in vivo to HLA-DR in autoimmune thyroid glands. Journal of immunology. 2008;181:795–807. doi: 10.4049/jimmunol.181.1.795. [DOI] [PubMed] [Google Scholar]
- 92.Sakai K, Shirasawa S, Ishikawa N, Ito K, Tamai H, Kuma K, et al. Identification of susceptibility loci for autoimmune thyroid disease to 5q31-q33 and Hashimoto's thyroiditis to 8q23-q24 by multipoint affected sib-pair linkage analysis in Japanese. Human molecular genetics. 2001;10:1379–86. doi: 10.1093/hmg/10.13.1379. [DOI] [PubMed] [Google Scholar]
- 93.Tomer Y, Ban Y, Concepcion E, Barbesino G, Villanueva R, Greenberg DA, et al. Common and unique susceptibility loci in Graves and Hashimoto diseases: results of whole-genome screening in a data set of 102 multiplex families. American journal of human genetics. 2003;73:736–47. doi: 10.1086/378588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ban Y, Greenberg DA, Concepcion E, Skrabanek L, Villanueva R, Tomer Y. Amino acid substitutions in the thyroglobulin gene are associated with susceptibility to human and murine autoimmune thyroid disease. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15119–24. doi: 10.1073/pnas.2434175100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hodge SE, Ban Y, Strug LJ, Greenberg DA, Davies TF, Concepcion ES, et al. Possible interaction between HLA-DRbeta1 and thyroglobulin variants in Graves' disease. Thyroid: official journal of the American Thyroid Association. 2006;16:351–5. doi: 10.1089/thy.2006.16.351. [DOI] [PubMed] [Google Scholar]
- 96.Jacobson EM, Yang H, Menconi F, Wang R, Osman R, Skrabanek L, et al. Employing a recombinant HLA-DR3 expression system to dissect major histocompatibility complex II-thyroglobulin peptide dynamism: a genetic, biochemical, and reverse immunological perspective. The Journal of biological chemistry. 2009;284:34231–43. doi: 10.1074/jbc.M109.041574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu W, Wang HN, Gu ZH, Yang SY, Ye XP, Pan CM, et al. Identification of BACH2 as a susceptibility gene for Graves' disease in the Chinese Han population based on a three-stage genome-wide association study. Human genetics. 2014;133:661–71. doi: 10.1007/s00439-013-1404-2. [DOI] [PubMed] [Google Scholar]
- 98.Muto A, Hoshino H, Madisen L, Yanai N, Obinata M, Karasuyama H, et al. Identification of Bach2 as a B-cell-specific partner for small maf proteins that negatively regulate the immunoglobulin heavy chain gene 3′ enhancer. The EMBO journal. 1998;17:5734–43. doi: 10.1093/emboj/17.19.5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hong SW, Kim S, Lee DK. The role of Bach2 in nucleic acid-triggered antiviral innate immune responses. Biochemical and biophysical research communications. 2008;365:426–32. doi: 10.1016/j.bbrc.2007.10.183. [DOI] [PubMed] [Google Scholar]
- 100.Zhao SX, Xue LQ, Liu W, Gu ZH, Pan CM, Yang SY, et al. Robust evidence for five new Graves' disease risk loci from a staged genome-wide association analysis. Human molecular genetics. 2013;22:3347–62. doi: 10.1093/hmg/ddt183. [DOI] [PubMed] [Google Scholar]
- 101.Menard L, Cantaert T, Chamberlain N, Tangye SG, Riminton S, Church JA, et al. Signaling lymphocytic activation molecule (SLAM)/SLAM-associated protein pathway regulates human B-cell tolerance. The Journal of allergy and clinical immunology. 2014;133:1149–61. doi: 10.1016/j.jaci.2013.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen XJ, Gong XH, Yan N, Meng S, Qin Q, Jiang YF, et al. RNASET2 tag SNP but not CCR6 polymorphisms is associated with autoimmune thyroid diseases in the Chinese Han population. BMC medical genetics. 2015;16:11. doi: 10.1186/s12881-015-0150-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Szymanski K, Bednarczuk T, Krajewski P, Ploski R. The replication of the association of the rs6832151 within chromosomal band 4p14 with Graves' disease in a Polish Caucasian population. Tissue antigens. 2012;79:380–3. doi: 10.1111/j.1399-0039.2012.01854.x. [DOI] [PubMed] [Google Scholar]
- 104.Brown RS, Lombardi A, Hasham A, Greenberg DA, Gordon J, Concepcion E, et al. Genetic analysis in young-age-of-onset Graves' disease reveals new susceptibility loci. The Journal of clinical endocrinology and metabolism. 2014;99:E 1387–91. doi: 10.1210/jc.2013-4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Simmonds MJ, Heward JM, Barrett JC, Franklyn JA, Gough SC. Association of the BTNL2 rs2076530 single nucleotide polymorphism with Graves' disease appears to be secondary to DRB1 exon 2 position beta74. Clinical endocrinology. 2006;65:429–32. doi: 10.1111/j.1365-2265.2006.02586.x. [DOI] [PubMed] [Google Scholar]
- 106.Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. The Journal of biological chemistry. 1992;267:17971–6. [PubMed] [Google Scholar]
- 107.Song RH, Yu ZY, Wang Q, Muhali FS, Jiang WJ, Xiao L, et al. Polymorphisms of the TNFAIP3 region and Graves' disease. Autoimmunity. 2014;47:459–65. doi: 10.3109/08916934.2014.914504. [DOI] [PubMed] [Google Scholar]
- 108.Ban Y, Greenberg DA, Davies TF, Jacobson E, Concepcion E, Tomer Y. Linkage analysis of thyroid antibody production: evidence for shared susceptibility to clinical autoimmune thyroid disease. The Journal of clinical endocrinology and metabolism. 2008;93:3589–96. doi: 10.1210/jc.2008-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Medici M, Porcu E, Pistis G, Teumer A, Brown SJ, Jensen RA, et al. Identification of novel genetic Loci associated with thyroid peroxidase antibodies and clinical thyroid disease. PLoS genetics. 2014;10:e1004123. doi: 10.1371/journal.pgen.1004123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tomer Y, Dolan LM, Kahaly G, Divers J, D'Agostino RB, Jr, Imperatore G, et al. Genome wide identification of new genes and pathways in patients with both autoimmune thyroiditis and type 1 diabetes. Journal of autoimmunity. 2015;60:32–9. doi: 10.1016/j.jaut.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jiang Y, Luo L, Gustafson EL, Yadav D, Laverty M, Murgolo N, et al. Identification and characterization of a novel RF-amide peptide ligand for orphan G-protein-coupled receptor SP9155. The Journal of biological chemistry. 2003;278:27652–7. doi: 10.1074/jbc.M302945200. [DOI] [PubMed] [Google Scholar]
- 112.Chen X, Hu Z, Li W, Wu P, Liu M, Bao L, et al. Synergistic combined effect between CD40-1C>T and CTLA-4+6230G>A polymorphisms in Graves' disease. Gene. 2015;567:154–8. doi: 10.1016/j.gene.2015.04.074. [DOI] [PubMed] [Google Scholar]
- 113.Inal EE, Rustemoglu A, Inanir A, Ekinci D, Gul U, Yigit S, et al. Associations of rs4810485 and rs1883832 polymorphisms of CD40 gene with susceptibility and clinical findings of Behcet's disease. Rheumatology international. 2015;35:837–43. doi: 10.1007/s00296-014-3171-3. [DOI] [PubMed] [Google Scholar]
- 114.Vaidya B, Imrie H, Geatch DR, Perros P, Ball SG, Baylis PH, et al. Association analysis of the cytotoxic T lymphocyte antigen-4 (CTLA-4) and autoimmune regulator-1 (AIRE-1) genes in sporadic autoimmune Addison's disease. The Journal of clinical endocrinology and metabolism. 2000;85:688–91. doi: 10.1210/jcem.85.2.6369. [DOI] [PubMed] [Google Scholar]
- 115.Downie-Doyle S, Bayat N, Rischmueller M, Lester S. Influence of CTLA4 haplotypes on susceptibility and some extraglandular manifestations in primary Sjogren's syndrome. Arthritis and rheumatism. 2006;54:2434–40. doi: 10.1002/art.22004. [DOI] [PubMed] [Google Scholar]
- 116.Lee YH, Harley JB, Nath SK. CTLA-4 polymorphisms and systemic lupus erythematosus (SLE): a meta-analysis. Human genetics. 2005;116:361–7. doi: 10.1007/s00439-004-1244-1. [DOI] [PubMed] [Google Scholar]
- 117.Huang D, Liu L, Noren K, Xia SQ, Trifunovic J, Pirskanen R, et al. Genetic association of Ctla-4 to myasthenia gravis with thymoma. Journal of neuroimmunology. 1998;88:192–8. doi: 10.1016/s0165-5728(98)00119-2. [DOI] [PubMed] [Google Scholar]
- 118.Djilali-Saiah I, Schmitz J, Harfouch-Hammoud E, Mougenot JF, Bach JF, Caillat-Zucman S. CTLA-4 gene polymorphism is associated with predisposition to coeliac disease. Gut. 1998;43:187–9. doi: 10.1136/gut.43.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Agarwal K, Jones DE, Daly AK, James OF, Vaidya B, Pearce S, et al. CTLA-4 gene polymorphism confers susceptibility to primary biliary cirrhosis. Journal of hepatology. 2000;32:538–41. doi: 10.1016/s0168-8278(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 120.Song GG, Lee YH. The CTLA-4 and MCP-1 polymorphisms and susceptibility to systemic sclerosis: a meta-analysis. Immunological investigations. 2013;42:481–92. doi: 10.3109/08820139.2013.789910. [DOI] [PubMed] [Google Scholar]
- 121.Velaga MR, Wilson V, Jennings CE, Owen CJ, Herington S, Donaldson PT, et al. The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves' disease. The Journal of clinical endocrinology and metabolism. 2004;89:5862–5. doi: 10.1210/jc.2004-1108. [DOI] [PubMed] [Google Scholar]
- 122.Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nature genetics. 2004;36:337–8. doi: 10.1038/ng1323. [DOI] [PubMed] [Google Scholar]
- 123.Carlton VE, Hu X, Chokkalingam AP, Schrodi SJ, Brandon R, Alexander HC, et al. PTPN22 genetic variation: evidence for multiple variants associated with rheumatoid arthritis. American journal of human genetics. 2005;77:567–81. doi: 10.1086/468189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VE, et al. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. American journal of human genetics. 2004;75:504–7. doi: 10.1086/423790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chistiakov DA, Chistiakov AP. Is FCRL3 a new general autoimmunity gene? Human immunology. 2007;68:375–83. doi: 10.1016/j.humimm.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 126.Wang X, Yu T, Yan Q, Wang W, Meng N, Li X, et al. Significant Association Between Fc ReceptorLike 3 Polymorphisms (-1901A>G and -658C>T) and Neuromyelitis Optica (NMO) Susceptibility in the Chinese Population. Molecular neurobiology. 2015 doi: 10.1007/s12035-014-9036-7. [DOI] [PubMed] [Google Scholar]
- 127.Li K, Zhao M, Hou S, Du L, Kijlstra A, Yang P. Association between polymorphisms of FCRL3, a non-HLA gene, and Behcet's disease in a Chinese population with ophthalmic manifestations. Molecular vision. 2008;14:2136–42. [PMC free article] [PubMed] [Google Scholar]
- 128.Yuan M, Wei L, Zhou R, Bai Q, Wei Y, Zhang W, et al. Four FCRL3 Gene Polymorphisms (FCRL3_3, _5, _6, _8) Confer Susceptibility to Multiple Sclerosis: Results from a Case-Control Study. Molecular neurobiology. 2015 doi: 10.1007/s12035-015-9149-7. [DOI] [PubMed] [Google Scholar]
- 129.Svejgaard A, Platz P, Ryder LP. HLA and disease 1982--a survey. Immunological reviews. 1983;70:193–218. doi: 10.1111/j.1600-065x.1983.tb00715.x. [DOI] [PubMed] [Google Scholar]
- 130.Todd JA, Acha-Orbea H, Bell JI, Chao N, Fronek Z, Jacob CO, et al. A molecular basis for MHC class II--associated autoimmunity. Science. 1988;240:1003–9. doi: 10.1126/science.3368786. [DOI] [PubMed] [Google Scholar]