

Abstract
Purpose
To evaluate functional immunogenicity of CHrPfs25. a malaria transmission blocking vaccine antigen, using nanoemulsion and porous polymeric PLGA nanoparticles.
Methods
CHrPfs25 was formulated with nanoemulsions (NE) and poly(D,L-lactide-co-glycolide) nanoparticles (PLGA-NP) and evaluated via IM route in mice. Transmission blocking efficacy of antibodies was evaluated by standard mosquito membrane feeding assay using purified IgG from immune sera. Physicochemical properties and stability of various formulations were evaluated by measuring poly-dispersity index, particle size and zeta potential.
Results
Mice immunized with CHrPfs25 using alum via IP and IM routes induced comparable immune responses. The highest antibody response was obtained with CHrPfs25 formulated in 4% NE as compared to 8% NE and PLGA-NP. No further increases were observed by combining NE with MPL-A and chitosan. 100% transmission blocking activity was demonstrated at 400 μg/ml of IgG for alum groups (both routes IP and IM), 4% NE and NE-MPL-A. Purified IgG from various adjuvant groups at lower doses (100 μg/mL) still exhibited >90% transmission blocking activity, while 52-81% blocking was seen at 50 μg/mL.
Conclusion
Results suggest that CHrPfs25 delivered in various adjuvants / nanoparticles elicited strong functional immunogenicity in pre-clinical studies in mice. We are now continuing these studies to develop effective vaccine formulations for further evaluation of immune correlates of relative immunogenicity of CHrPfs25 in various adjuvants and clinical trials.
Keywords: Malaria, Vaccine, Nanoemulsion, PLGA nanoparticles
Introduction
Malaria caused by Plasmodium spp. is a mosquito-borne, life-threatening public health problem worldwide with an estimated 198 million cases and approximately 584,000 deaths annually (WHO, 2014). The infection begins with the inoculation of sporozoites by an anopheline mosquito during blood feeding process. Sporozoites quickly invade hepatocytes and develop over the next 7-10 days into hepatic schizonts. Merozoites released from hepatic schizonts invade red blood cells and begin erythrocytic schizogony (asexual development). During blood stage parasite development, some differentiate into intraerythrocytic sexual forms known as male and female gametocytes. While erythrocytic asexual stages are responsible for all the clinical symptoms, including death, the sexual forms are absolutely critical for continued malaria transmission. Gametocytes ingested by the mosquito during blood feeding, begin sexual life cycle of the malaria parasite consisting of formation of extracellular male and female gametes, fertilization and development of fertilized zygotes into ookinetes. Motile ookinetes traverse the midgut wall and develop into oocysts. Sporozoites produced in the oocysts travel to the salivary gland and remain there to be inoculated into the host during blood feeding process. Widespread resistance of parasites to anti-malaria drugs and of mosquitoes to insecticides continue to hamper current malaria control efforts (1). Efforts are underway to develop malaria vaccines targeting various stages of the complex life cycle of malaria parasites; however, none has been shown to be completely effective. Currently, RTS,S/ASO1 has advanced to phase III clinical trial but has shown only partial protective efficacy (2).
Malaria transmission blocking vaccines (TBV) targeting sexual stages of the parasite have been identified as a crucial tool for eventual elimination and eradication of the disease at the population level in endemic areas. Antibodies induced by TBV antigens, when ingested at the time of the blood meal, prevent sexual development of the parasite inside the mosquito vector and effectively interfere with malaria transmission (3-7). Pfs25, expressed on the surface of P. falciparum gametes, zygotes and ookinetes (8), as well as Pfs230 (9, 10) and Pfs48/45 (11-13), expressed on the surface of gametocytes and gametes, have been identified as primary TBV target antigens.
Pfs25, a 25 KDa antigen, consists of four tandem epidermal growth factor (EGF)-like repeat motifs requiring correct conformational folding by pairing of 22 cysteine residues (8, 14, 15). The TBV potential of Pfs25 has been previously reported by several studies including recombinant Pfs25 expressed in yeast (16, 17), cell-free translation using wheat germ (18), plants (19) and algal system (20). A Phase I clinical trial with Pfs25 expressed in P. pastoris and Montanide ISA51 demonstrated moderate transmission blocking immunity (21). Since Pfs25 was shown to exhibit poor functional immunogenicity, further attempts have been made to enhance the immunogenicity by chemical conjugation to the outer membrane protein or exoprotein of Neisseria mengiditis or Pseudomonas aeruginosa or by developing vaccine-like particles (22-25). In our previous approach, we demonstrated highly potent malaria transmission blocking efficacy of codon-harmonized recombinant Pfs25 (CHrPfs25) expressed in E. coli after successful refolding in an appropriate monomeric conformation (26).
Success of an effective TBV will depend upon the availability of well-characterized recombinant antigen in stable functional conformation and adjuvants capable of eliciting a long-lasting antibody response. Although highly purified recombinant antigens have many desirable qualities and reduce the risk of toxicity associated with live or attenuated vaccines, their ability to induce potent immune responses is weaker; therefore, overcoming these hurdles requires formulation development with safe and effective adjuvants, optimization of delivery systems and fine-tuning of vaccine particulate size (27). Our previous studies (26) employed evaluation of immunogenicity of CHrPfs25 in three different adjuvants, including complete Freund's adjuvant, Alum and Montanide ISA-51. One of the goals of vaccine research is to identify and evaluate novel adjuvants and vaccine delivery systems that are safe and immunologically more potent. Although the preferred adjuvants for most adjuvanted vaccines are aluminum salts (alum), they are not appropriate for all antigens and do not always induce a strong cell-mediated immune response. Several nanoparticulate carriers have been investigated in the last two decades as vaccine adjuvants (28). Most promising of these potential adjuvants are nanoemulsions and biodegradable nanoparticles of poly(lactide-co-glycolide) (PLGA). Nanoemulsions have a proven history to be safe for human use both for cosmetic and medicinal applications (29). Besides safety, nanoemulsions can be stable for a long storage life, and their creation is scalable to large scale manufacturing (29). Similarly, biodegradable nanoparticles of PLGA also have a long history of investigation as vaccine adjuvants (30-32). These nanoparticles are effective adjuvants due to their ability to efficiently target antigen presenting cells, and PLGA has already been approved by the U.S. FDA for human use as controlled release matrix. In view of these facts, we sought to evaluate transmission blocking potency of CHrPfs25 antigen delivered using nanoemulsion (NE) and poly(D,L-lactide-co-glycolide) nanoparticles (PLGA-NP) via IM route of vaccination. The study revealed that CHrPfs25 emulsified with squalane-containing NE elicited strong transmission blocking antibodies and can be further developed as an alternative to alum as a Pfs25 vaccine adjuvant.
Materials and Methods
Materials
Squalane, Span-80, Tween-80, dichloromethane, chitosan and polyvinyl alcohol (30,000-70,000 MW) were obtained from Sigma Aldrich (St. Louis, MO). Poly(D,L-lactide-co-glycolide) (50:50, ester terminated, MW 7,000) was obtained from Boehringer Ingelheim (Ingelheim am Rhein, Germany). L-α-phosphatidyl choline was obtained from Avanti Polar Lipids, Inc. (Alabaster, AL). Monophosphoryl lipid-A from S. minnesota R595 was obtained from InvivoGen (San Diego, CA).
Purification and characterization of CHrPfs25
Purification of CHrPfs25 protein expressed in E. coli was as described (26). The qualitative analysis of the purified protein was performed by SDS-PAGE and characterized by western blotting using anti-(His)6 and Pfs25 specific monoclonal (ID3) antibodies (26). BCA protein assay kit was used for quantitative analysis of protein (Thermo Scientific, Rockford, IL). Endotoxin levels were determined by using LAL chromogenic endotoxin quantitation kit (Thermo Scientific, Rockford, IL) and were found to be <7.2 EU/ml.
Preparation of Nanoemulsion (NE) and PLGA nanoparticles (PLGA-NP) Formulations and Characterization
Two different formulations were prepared as vaccine adjuvants, a NE formulation and porous PLGA-NP formulation. The NE was composed of 30.6% (w/v) squalane, 10.3% (w/v) surfactant/co-surfactant (1:1 ratio of Span-80 and Tween-80) in phosphate buffered saline (PBS, pH 7.4). The NE was formed by high pressure homogenization at 20,000 psi for 5 passes on an EmulsiFlex-C3 high pressure homogenizer (Avestin Inc., Ottawa, Ontario, Canada). The NE was diluted with PBS to obtain a stock of 16% (w/v, combined concentration of squalane and surfactant/co-surfactant) and passed through a 0.2 μm syringe filter. Four different NE formulations were prepared for testing: 4% NE, 8% NE, 4% NE with monophosphoryl lipid-A (MPL-A) and 4% NE with chitosan. MPL-A was dissolved in DMSO and added to 16% NE at a concentration of 800 ng/mL. The chitosan stock solution was prepared in 0.1 N HCl and added to the 16% NE at a concentration of 100 μg/mL. The NE+MPL-A and NE+chitosan formulations were stored at 4 °C for 12 weeks to assess the effect of MPL-A and chitosan on the particle size and zeta potential of the NE.
PLGA-NP were prepared using a variation of the double emulsion solvent evaporation technique (33). Briefly, 40 mg sodium bicarbonate was dissolved in 400 μL of deionized water and emulsified into an organic solution containing 200 mg of PLGA dissolved in 3 mL of dichloromethane. 0.5 mg L-α-phosphatidyl choline was added using an ultrasonic probe for 30 seconds at 40 W forming a water-in-oil emulsion. 1% polyvinyl alcohol (PVA) in water (1 mL) was added to the emulsion, and the sample was sonicated for an additional 30 seconds. This inverted oil-in-water emulsion was poured into 100 mL of a 0.5% PVA solution containing 1% acetic acid. The solution was sonicated for 15 minutes and allowed to stir overnight to allow complete evaporation of dichloromethane and hardening of the particles. After the particles were formed, the sample was centrifuged at 35,000 rpm for 15 minutes. The supernatant was removed and discarded, and the precipitate was re-dispersed in deionized water. The centrifugation cycle was repeated four additional times to allow complete removal of the PVA and acetic acid. After the final rinse cycle, the sample was dispersed in a minimal amount of deionized water, frozen, and lyophilized at -25°C for 48 hours. Prior to use, the powder was resuspended in PBS, pH 7.4, at 80 mg/mL PLGA-NP concentration. The final nanoparticle porosity is achieved when the bicarbonate core, emulsified in an acidic evaporation solution, converts to carbon dioxide creating pores in the particles.
The particle size and zeta potential were measured in triplicate, before and after the addition of CHrPfs25, using a DelsaNano C particle analyzer (Beckman Coulter, Inc., Fullerton, CA). All samples were diluted with deionized water immediately prior to particle size measurements with the Size Cell for an optimal intensity of 10,000 cps. The zeta potential measurements were performed in 1 mM KCl solution. Scanning electron microscopy (SEM) (S-4800, Hitachi High Technologies America Inc., Gaithersburg, MD) was used to confirm the morphology and size distribution of the NE droplets and NP. The PLGA-NP were mounted on SEM stubs with carbon tape and sputter-coated with gold at 2 mA for one minute (K550X Sputter Coater, Quorum Technologies Ltd, West Sussex, UK). Micrographs were taken at an accelerating voltage of 5.0 kV and a working distance of approximately 6.5 mm. The NE droplets were negatively-stained with phosphotungstic acid for three minutes at room temperature, placed on a Formvar®-coated copper grid on a dark field stage and imaged in STEM mode at 30 kV. Micrographs were taken before and after the addition of the CHrPfs25 antigen.
Animal immunizations
Female BALB/c mice (4-5 weeks old, n=5 per group) were immunized with 10 μg of CHrPfs25 in a total volume of 100 μL. For immunization with alum, ChrPfs25 was adsorbed to Alhydrogel (Brenntag Biosector, DN) at a 1:3.2 ratio by rocking for 2-3 hours at room temperature and delivered via IP and IM routes (50 μL each quadricep). For formulation in NE, the protein was incubated with NE on a rocker at room temperature for 2 hours and delivered via IM route (50 μL per quadricep). For preparation of formulation with PLGA-NP, the protein was mixed with PLGA-NP (10 and 20 mg/mL) by gentle vortexing for 10 minutes and incubation for 3 hours with intermittent gentle vortexing, and the PLGA-NP with antigen was delivered via IM route (50 μL per quadricep). Mice were boosted with the same quantity of Pfs25 antigen in respective adjuvant and formulations at 3 week intervals and bled on days 21, 31 and 52 to collect sera for further analysis. The research on animals adhered to the ”Principles of Laboratory Animal Care” (NIH publication #85-23, revised in 1985) and approved by institutional IACUC.
Evaluation of antibody response in mice by ELISA
The antibody end-point titers were determined by ELISA as described previously (26). Sera from test animals were tested individually (at each time point) at 2-fold serial dilutions. Standard ELISA was used to determine the IgG isotypes. After incubation with purified IgG (100 ng/ml) or pooled mice sera, wells were incubated with HRP-conjugated anti-mouse IgG1 or IgG2a (Invitrogen, Carlsbad, CA) followed by color development and absorbance measurement.
Standard Membrane Feeding Assay (SMFA)
Total IgG was purified from pooled mice sera using Protein A-Sepharose beads as reported (26). Transmission blocking efficacy of purified IgG was determined by SMFA at four different concentrations (400, 200, 100 and 50 μg/mL). Briefly, female An. gambiae mosquitoes (4-6 days old adult), starved for four hours, were fed for 15 minutes on a mixture of cultured P. falciparum (NF54) gametocytes (0.3% final gametocytemia), normal human serum (NHS) and human erythrocytes (∼50% heamatocrit). NHS, control IgG (purified from mice immunized with PBS-alum adjuvant alone), or test IgG were mixed with the diluted parasites for feeding to mosquitoes through parafilm membrane wrapped around jacketed glass mini feeders, maintained at 37°C using a circulating water bath. Blood-fed mosquitoes were maintained for 8-9 days at 26°C and 60-80% relative humidity. Midguts of individual mosquitoes were dissected and stained with 0.1% mercurochrome to determine the number of oocysts in individual midguts by microscopy (10×). Typically 20-30 mosquitoes were analyzed for each test antibody or control samples. Malaria transmission blocking activity of immune sera or purified IgG was expressed either as infectivity prevalence (number of mosquitoes with any oocysts in the test group versus number of mosquitoes with any oocyst in the control groups) or percent reduction in the median number of oocysts in the test group as compared to controls. The controls mosquitoes were fed in parallel with parasites and purified IgG from sera of mice immunized with adjuvant alone (without ChrPfs25 antigen).
Statistical analysis
GraphPad Instat3 software package (GraphPab Software, Inc., La Jolla, CA) was used for statistical analysis. Antibody endpoint titers were defined as serum dilutions giving an absorbance higher than the mean absorbance of pre-immune serum plus three standard deviations. Percent reduction in oocyst development was determined by the formula: 100 × (mean oocyst number in control IgG - mean oocyst number in test IgG) / mean oocyst number in control IgG. One way analysis of variance (ANOVA) Kruskal-Wallis test was applied to determine the statistical significance of differences between experimental groups. P values of <0.05 were considered significant.
Results
Physicochemical properties and stability of NE and PLGA-NP formulations
Since the conformational integrity of the CHrPfs25 antigen and the physicochemical properties of the formulations are critical for stability of the protein and for functional immunogenicity, particle size distributions of NE and PLGA-NP formulations were analyzed before and after addition of the protein antigen. The nano-sized emulsion droplets of NE are clearly visible before and after the addition of antigen (Fig 1 A, B). The particle size of the NE was centered around 80-90 nm; while after the addition of the vaccine antigen, the polydispersity index and zeta potential of the NE formulation increased without significant change in the median particle size of the formulation (Table I, Fig. 1 E,F,H). The larger particles in the NE micrographs are probably due to limited coalescence of NE droplets during the drying process before imaging. These larger droplets, if present in the liquid state, would be well within the measurement range of the particle analyzer, but no larger particles were detected. The coalescence of the nanoemulsion droplets during imaging may be reduced by freeze drying the sample on the grid prior to imaging, by additional dilution of the nanoemulsion prior to staining and evaporation, and/or by utilizing alternative imaging techniques such as transmission electron microscopy. Similar observations were obtained on the particle size distributions of NE-MPL-A and NE-chitosan formulations (Table I, Fig. 1 F, H). Furthermore, the stability of NE was assessed over 12 weeks to determine the effect of MPL-A or chitosan on the particle size over time at 4°C. No changes in polydispersity index, particle size or zeta potential were observed during three months storage at 4°C (Fig. 2).
Fig. 1.
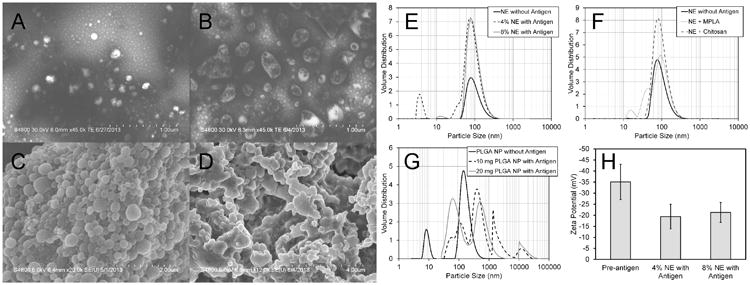
Physicochemical characterization of NE (A, B) and PLGA-NP (C, D) by scanning electron microscopy (SEM). (A) and (C) are representative images before the addition of the antigen, and (B) and (D) are after addition of the antigen. (E, F) show the particle size distributions of NE formulations, (G) shows the particle size distribution of PLGA-NP formulation and (H) shows the zeta potential measurements of NE formulations.
Table. I. Characterization of NE and PLGA-NP formulations.
| Formulation | Polydis-persity Index (mean ± SD) | D 10% (nm) (mean ± SD) | D 50% (nm) (mean ± SD) | D 90% (nm) (mean ± SD) | Zeta Potential (mV) (mean ± SD) |
|---|---|---|---|---|---|
| NE | 0.176 ± 0.017 | 62.8 ± 5.6 | 92.0 ± 6.7 | 157.9 ± 8.1 | -35.07 ± 7.99 |
| 4% NE + CHrPfs25 | 0.221 ± 0.011 | 52.1 ± 10.7 | 77.4 ± 13.7 | 138.5 ± 18.3 | -19.41 ± 5.58 |
| 8% NE + CHrPfs25 | 0.220 ± 0.022 | 57.3 ± 4.5 | 86.0 ± 4.9 | 150.8 ± 6.3 | -21.30 ± 4.50 |
| PLGA | 0.235 ± 0.048 | 101.4 ± 7.3 | 147.8 ± 8.8 | 252.9 ± 9.6 | |
| PLGA (10 mg) +CHrPfs25 | 0.433 ± 0.252 | 531.1 ± 1003.2 | 748.1 ± 1379.0 | 2124.3 ± 2980.5 | |
| NE+MPL-A +CHrPfs25 | 0.241 ± 0.021 | 39.0 ± 25.9 | 82.1 ± 27.3 | 144.8 ± 37.1 | -32.71 ± 4.73 |
| NE+Chitos an+ CHrPfs25 | 0.193 ± 0.018 | 55.4 ± 7.2 | 83.2 ± 9.0 | 147.2 ± 11.1 | -30.84 ± 2.64 |
Fig. 2.
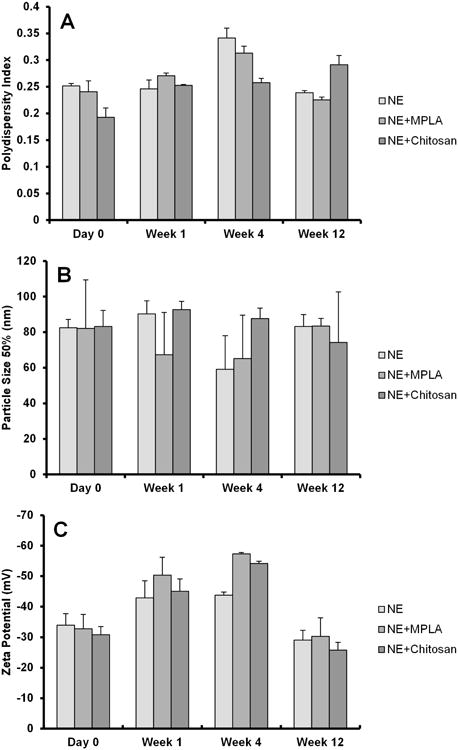
Stability of the NE formulations containing MPL-A and chitosan over 12 weeks. The addition of neither MPL-A nor chitosan had substantive effects on the particle size, size distribution, and zeta potential of the NE formulation over time.
The PLGA-NPs were smooth and spherical before the addition of the antigen. However, after addition of the antigen, the particles increased in size and revealed irregularly-shaped agglomerations (Fig. 1 C, D). The median particle size of the PLGA-NP prior to the addition of antigen was around 150 nm and had a monodispersed distribution (Fig.1 G). With the incorporation of the antigen, the particle size increased significantly – changing from a monodispersed, nanosized distribution to a multimodal distribution of particles as large as 3 μm (Fig. 1 G).
Evaluation of antibody response in mice sera after intramuscular immunization
In previous study, we had shown that CHrPfs25 adsorbed to alum was strongly immunogenic via IP route of vaccination(26). We next compared immunogenicity outcome for CHrPfs25 adsorbed to alum adjuvant administered via IP and IM routes. Immunization by both routes revealed comparable antibody titers (1:320,000 after the second vaccination) and titers increasing further (1:640,000) after the final vaccination (Table IIa).
Table II. Evaluation of anti-Pfs25 antibody titers in mice immunized with CHrPfs25.
| (a) CHrPfs25 adsorbed to alum and vaccinated via IP and IM routes | |||
|---|---|---|---|
| Antibody titers | |||
| Groups | Prime (Day 21) | 1st Boost (Day 31) | 2nd Boost (Day 52) |
| IP | 0 | 320,000 | 640,000 |
| IM | 800 | 320,000 | 640,000 |
| (b) CHrPfs25 admixed with NE and PGA-NPs administered via IM route | |||
| Antibody titers | |||
| Groups | Prime (Day 21) | 1st Boost (Day 31) | 2nd Boost (Day 52) |
| 8% NE | 400 | 160,000 | 320,000 |
| 4% NE | 800 | 320,000 | 640,000 |
| 4% NE+MPL-A | 400 | 80,000 | 640,000 |
| 4% NE+Chitosan | 0 | 20,000 | 640,000 |
| PLGA-NP (20 mg/mL) | 800 | 80,000 | 320,000 |
| PLGA-NP (10 mg/mL) | 800 | 20,000 | 160,000 |
Since the protein adsorbed to alum was equally immunogenic via IP and IM routes, we evaluated various NE and PLGA-NP formulations by IM route. Mice were immunized with CHrPfs25 formulated with NE (4% and 8% NE) and PLGA-NP (10 and 20 mg/mL). After final vaccination, CHrPfs25 formulated in 4% NE elicited the highest antibody response (antibody titer 1:1,280,000) as compared to 1:320,000 in 8% NE. We also evaluated CHrPfs25 with 4% NE formulations combined with MPL-A and chitosan, and the antibody titers with both (1:640,000) were not different from NE alone (Table II b). In contrast, the antibody titers with CHrPfs25 formulated with PLGA-NP (10 and 20 mg/mL) were only 1:160,000 and 320,000, respectively. These results established that the CHrPfs25 is strongly immunogenic by both the routes of administration and alum as well as 4% NE can be developed as effective ways to formulate and administer Pfs25 antigen.
Isotypes of antibodies in the immune sera or in the total IgG preparations after protein-A Sepharose purification were characterized to seek a correlation with differences in immunogenicity and transmission-blocking activity with various formulations. The ratio of IgG1/IgG2a in the purified total IgG preparation was 11.9, 16, 2.3, 7.5, 3.1 and 3 for 4% NE, 8%NE, NEM, NEC, 10mg/mL PLGA-NP and 20 mg/mL PLGA-NP, respectively. In order to rule out any purification bias, we also analyzed isotypes in the pooled immune sera and the ratios were 13.6, 57.5, 2.4, 9.2, 9.4 and 2.6, respectively.
Transmission blocking activity (TBA) of anti-Pfs25 antibodies in mice sera
TBA of induced antibodies in immunized mice was assessed by standard membrane feeding assay (MFA). Total IgG was evaluated at four concentrations (400, 200, 100 and 50 μg/mL) using female An. gambiae mosquitoes (Fig. 3-5). Normal human serum (NHS) and purified IgG from adjuvant alone immunized mice were used as controls. Total IgG from mice immunized with CHrPfs25 in alum adjuvant via IP and IM routes exhibited potent TBA with 100% blocking at a concentration of 400 μg/mL of IgG and 96-98% blocking at 200 μg/mL via both IP and IM routes (Fig. 3A). Dose dependent inhibition is also evident from data in Fig. 3, and even a lower concentration of IgG (50 μg/mL) continued to block parasite development in MFA by 74 to 82% (Fig. 3B).
Fig. 3.
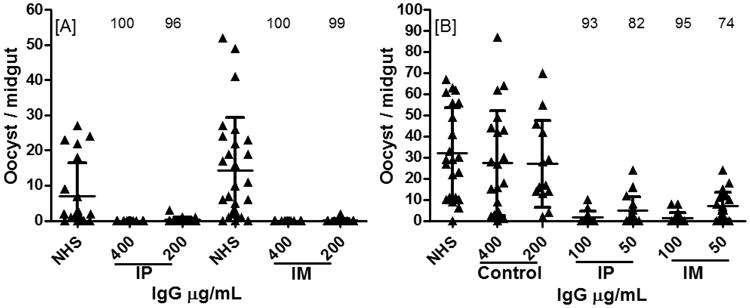
Transmission blocking activity (TBA) with total IgG in An. gambiae mosquitoes by standard membrane feeding assay (MFA). Mature gametocytes of P. falciparum (NF54) were mixed with control or purified total IgG from adjuvant alone or anti-Pfs25 serum from CHrPfs25 in alum immunized sera via IP and IM routes. TBA with total IgG concentration of (A) 400 and 200 μg/mL and (B) 100 and 50 μg/mL are presented. All data points represent the number of oocyst per individual mosquito and the horizontal line shows the median number of oocysts. Statistically significant differences in transmission blocking activity (TBA) based on mean values between groups were analyzed with one way analysis of variance (ANOVA) Kruskal-Wallis test. Statistical significance was P<0.0001.
Fig. 5.
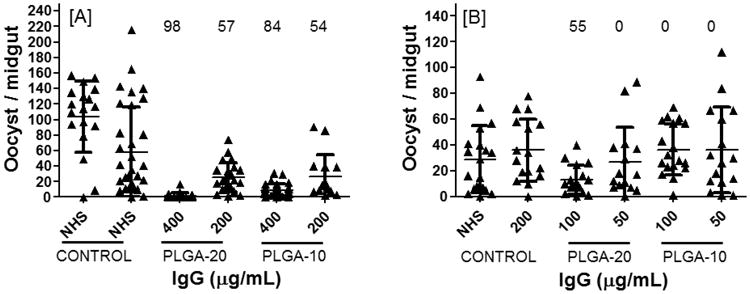
Evaluation of TBA at four concentrations (400 to 50 μg/mL) of total IgG from mice sera immunized with CHrPfs25 in PLGA-NP. Mature gametocytes of P. falciparum (NF54) were mixed with control or purified total IgG from adjuvant alone or anti-Pfs25 serum (A) PLGA (20 mg/mL) and (B) PLGA (10 mg/mL). Statistical analysis as described in Fig. 3.
Purified IgG from mice immunized with NE formulations were likewise tested in MFA. IgG from mice immunized with CHrPfs25 in 4% and 8% NE (Fig. 4 A, B) revealed potent (100%) blocking at 400 μg/mL. IgG from the 4% NE group at 200 μg/mL continued to exhibit strong blocking compared to slightly less potent blocking by the 8% NE group. Evaluation of lower IgG concentrations suggested relatively stronger blocking activity of IgG from the 4% NE group as compared to the 8% NE group. Addition of MPL-A (NEM groups in Fig. 4 A, B) or chitosan (NEC group in Fig. 4 C, D) to 4% NE did not reveal any significant benefit as the transmission blocking was generally comparable among NE, NEM and NEC at all IgG concentrations; the only exception was NEM which revealed higher percent blocking at 50 μg/mL IgG concentrations (Fig. 4 D).
Fig. 4.
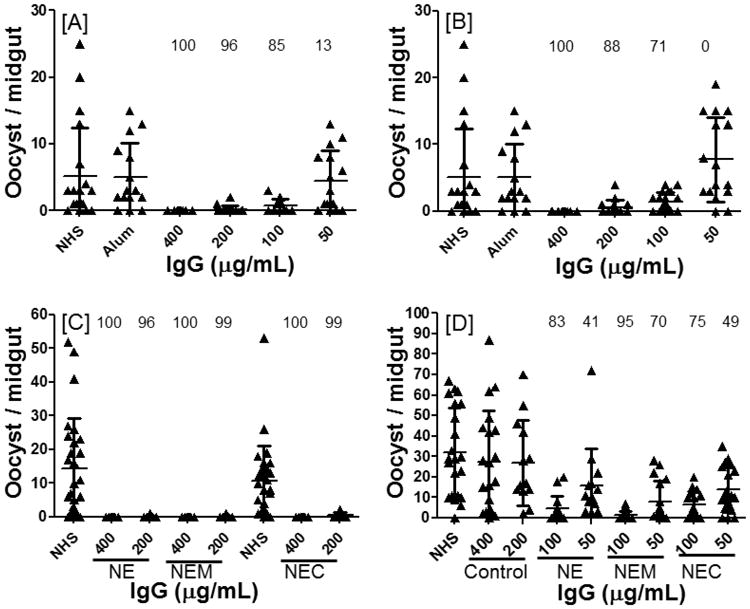
Comparative analysis of TBA at four concentrations (400 to 50 μg/mL) of total IgG from mice sera immunized with CHrPfs25 in [A] 8% NE, [B] 4% NE, [C] NE-MPL-A or [D] NE-Chitosan. Mature gametocytes of P. falciparum (NF54) were mixed with control or purified total IgG from adjuvant alone or anti-Pfs25 serum. Statistical analysis as described in Fig. 3. Please note that both 8% NE and 4% NE SMFAs [A and B] were done at the same time and the controls (NHS or Alum adjuvant group) are common to data shown in {a] and [B]. Similarly, SMFAs with NE and NEM panel [C] were done along with the SMFAs of alum (IM) group shown in Fig. 3 [A], and SMFAs in [D] were done along with SMFAs shown in Fig. 3 [B], resulting in common control groups for these respective SMFAs.
The TBA of CHrPfs25 in PLGA-NP (20 and 10 mg/mL) particles was 98 and 85%, respectively, at the higher concentration of 400 μg/mL total IgG (Fig. 5). The TBA was reduced when lower concentrations of IgG were tested with no blocking at 50 μg/mL. Comparing TBA between 10 and 20 mg/mL PLGA-NP formulations, the 20 mg/mL group revealed higher blocking activity as compared to the 10 mg/mL concentration of PLGA-NP. In general, TBA with PLGA were found to be less effective than that with alum alone or the 4% NE groups.
Discussion
Current global malaria burden and recent emergence of resistance to the anti-malarial drug used as first line treatment underscore the urgent need for the development of an effective malaria vaccine. The complex life cycle of malaria in the vertebrate host and mosquito vector presents several stages of the parasite for vaccine development. Vaccines targeting sexual stages of the parasite in the mosquito vector, in particular, are considered as critical tools for reducing transmission and eventual elimination of malaria. Pfs25 protein expressed in the mosquito midgut stages of the parasite is one of the most promising and a leading candidate for the development of a malaria TBV. Reproducible expression of recombinant Pfs25, present in an appropriate conformation with correct pairing of 22 cysteine residues forming eleven disulfide bonds, and safer adjuvants, suitable for clinical trials, are needed to develop an effective TBV.
Our recent success in expressing recombinant Pfs25 in E. coli, as a monomeric purified product after simple glutathione-mediated refolding steps retaining functional conformational epitopes and effective immunogenicity using CFA, Montanide ISA51 or alum as adjuvants, has provided a promising way to move forward with further vaccine development efforts (26). Acknowledging that CFA is unsuitable for use in humans, and that Montanide ISA51 has revealed strong reactogenicity in previous vaccine trials, we wished to evaluate additional ways to formulate and deliver Pfs25 for effective immunogenicity outcomes. One of the challenges in the development of vaccines is the selection of an adjuvant formulation that can induce robust functional immunogenicity. Currently, aluminum salts (alum), a relatively weak adjuvant for recombinant vaccine antigens, and oil-in-water emulsion MF59 are licensed for use in humans as vaccine adjuvants (34, 35). In the current study, NE (a squalane-based formulation) and PLGA-NP were evaluated as adjuvant formulations via IM route of immunization. The IM route was evaluated because it is the route commonly used for administering numerous vaccines in humans. To compare IP versus IM routes, Pfs25 in alum was first evaluated and showed highly comparable functional immunogenicity irrespective of the routes of immunization used.
In order to optimize formulation of CHrPfs25 with NE and PLGA-NP, two different concentrations of adjuvants were employed for evaluation in mice. CHrPfs25 formulated with 4% NE elicited higher antibody titers in comparison with 8% NE. Superiority of 4% NE alone was further evident from the fact that no significant improvement was seen after combining 4% NE with MPL-A or chitosan. Similar immunogenicity studies with PLGA-NP also revealed that PLGA-NP at 20 mg/mL were more effective than 10 mg/mL; however, the maximum antibody titers obtained were lower than that elicited by 4% NE. The reduced immunogenicity of the PLGA-NP could be due to inefficient uptake and presentation of antigen resulting in part from observed agglomerations of PLGA-NP after addition of antigen. It is well established that the optimum size of particles for antigen presentation is <100 nm (36, 37), while the size of PLGA-NP after formulation with antigen was as high as 3 μm.
Physicochemical properties of vaccine formulations are known to play an important role in the stability of immunogens and induction of a high quality immune response (27, 36). In addition to evaluation of immunogenicity parameters, we also conducted detailed characterization of the physicochemical parameters of the NE and PLGA-NP formulations before and after addition of the antigen. The particle size and distribution can have profound effects on vaccine efficacy because changes in particle size or distribution are indicative of instability of the formulation as well as greatly influence the uptake of vaccine antigens by antigen presenting cells (36). In our study, the particle size of NE was found to be uniform, and no significant changes were noticed before and after addition of the antigen. The polydispersity index of NE was found to increase slightly after addition of the antigen as evidenced by an additional peak (<20 nm) and a broader distribution. While this may be a sign of instability of the formulation, no large particles were detected, and the 90th percentile values were still around 150 nm. The small particles detected in the NE formulations with antigen may be attributed to aggregates of free antigen and/or aggregation of surfactant micelles. The polydispersity index, a measure of the size distribution, is typically defined as the standard deviation divided by the mean and is utilized as a measure of homogeneity. A polydispersity index of ≤ 0.2 is generally regarded as a monodispersed distribution [24]. In the present study while the polydispersity index was slightly higher, further optimization of the homogenization process during preparation may further improve the polydispersity of the NE formulation.
Another important characteristic of NE is the zeta potential, a measure of the electrical potential between the internal phase and the interfacial layer of external phase associated around the droplet. Zeta potential is a measure of stability because a high repulsive force among emulsion droplets reduces the occurrence of Ostwald ripening and coalescence, and a reduction in these forces can contribute to instability and phase separation of droplets in the emulsion. The zeta potential of the NE alone (∼ -35 mV) without the antigen indicates that the NE droplets are stable in dispersion; however, the addition of the antigen caused a reduction of the zeta potential (∼ -20 mV) probably due to the positive surface charge of the antigen. A reduction in charge, while expected due to the positive charge of the antigen, can also destabilize the NE. Even with the change in zeta potential, no evidence of phase separation or coalescence was observed over the time course of the study, even in the presence of MLP-A or chitosan, suggesting that the stability of NE formulations was not affected significantly. The zeta potential measurements did fluctuate over time, specifically becoming more negative at week 4, but remained more negative than -29 mV at all time points, except for NE+chitosan at week 12. Slight variations in pH and/or concentration can cause variability in zeta potential measurements. Further investigation is required to ensure that the zeta potential variability is not due to inhomogeneity within the formulations. However, the stability of the particle size and polydispersity index measurements indicates that the variation in the zeta potential measurement is more likely attributable to procedural variability than formulation inhomogeneity or instability.
PLGA, a biodegradable polymer, is approved for human use by the FDA and European Medicine Agency in various drug delivery systems (38-41). The particle size and surface charges of PLGA nanoparticles can have important implications in their interaction with cells and on their uptake (37, 41). The particle size of the PLGA-NP employed in this study was homogenous and monodispersed before addition of the antigen. However, after adding the antigen, the particles size increased substantially and revealed a polydispersed distribution of particles. These changes in the particle properties could be attributed to interaction between the nanoparticles and the antigen or an artifact of the time delay between mixing the antigen and particle size measurements. Further investigation must be done to determine if and how the increased polydispersity of the particles affects antigen delivery and/or effectiveness. If the polydispersity is due to aggregation of the nanoparticles, inclusion of a dispersant could improve the formulation.
CHrPfs25 produced in E. coli has been previously shown to induce transmission blocking antibodies when formulated with CFA, Montanide ISA-51 or alum (26). Current studies were conducted to evaluate additional adjuvant formulations to develop an effective CHrPfs25-based vaccine. CHrPfs25 formulations prepared with squalane-based NE and PLGA-NP delivered via IM route clearly demonstrated functional activity of antibodies elicited. Initially MFAs using 400 μg/mL IgG demonstrated 100% transmission blocking efficacy of NE (4% and 8%) formulated vaccines administered by IM route, results similar to those obtained with alum (used as a control for comparison). PLGA-NP at 10 mg/mL, which had revealed lower antibody titers, showed reduced TBA (only 84%) as expected. In order to compare superiority of functional potency of different NE and NP formulations, TBA was evaluated at varying concentrations (400, 200, 100 and 50 μg/mL) of purified IgG. Even though antibody titers among 4% NE alone, NE-MPL-A and NE-chitosan groups were comparable, higher blocking by the MPL-A group, especially comparing results at 50 μg/mL, seemed to suggest a slight advantage of including MPL-A to an already effective NE formulation. Additional studies are needed to identify if higher blocking activity of NE-MPL-A is due to differences in the epitope specificity and/or in the binding affinity/avidity of antibodies. We tried to seek a correlation between IgG isotypes and the blocking activity (SMFA) of IgG from immunized mice. In mice IgG1 and IgG2 isotypes reprersent either Th2 or Th1 biased IgG isotypes, respectively and a ratio of IgG1/IgG2 is often used to evaluate the relative immune bias. Although our analysis revealed differences in the relative IgG bias among various immunization groups, this immune bias alone is not sufficient to explain the differences in functional blocking activity of IgG among the various groups.
A malaria TBV targeting sexual development in the mosquito can provide an effective tool to eliminate malaria transmission in various endemic areas. Moreover, such vaccines targeting not only P. falciparum but also P. vivax can lead to gradual elimination of low intensity malaria transmission at the population level from regions where the two species are co-endemic. In this study, we evaluated vaccine adjuvant potential of squalane-based NE and PLGA-NP via IM route of administration. Antibodies elicited by 4% NE, in particular, were strongly immunogenic, and purified IgG from immune sera exhibited extremely potent TBA. The comparable potency of TBA of antibodies elicited by CHrPfs25 formulated in alum and NE provide a compelling rationale for developing these formulations as effective vaccine adjuvants for further preclinical evaluation in non-human primates leading to future human clinical trials.
Acknowledgments
Studies were supported by NIH R21 AI101427 and RO1-AI47089 (NK lab) and NIH 2G12MD007595-06 (TM lab).
Abbreviations
- NE
nano-emulsion
- PLGA-NP
poly (D,L-lactide-co-glycolide) nanoparticles
- ELISA
enzyme-linked immunosorbent assay
- MFA
standard membrane feeding assay
- TBA
transmission-blocking activity
- TBV
Transmission-blocking vaccine
References
- 1.Greenwood B. Control to elimination: Implication for malaria research. Trends Parasitol. 2008;24:449–54. doi: 10.1016/j.pt.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. The Lancet. doi: 10.1016/S0140-6736(15)60721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carter R, Chen D. Malaria transmission blocked by immunisation with gametes of the malaria parasite. Nature. 1976;263:57–60. doi: 10.1038/263057a0. [DOI] [PubMed] [Google Scholar]
- 4.Kaslow DC, Bathurst IC, Barr PJ. Malaria transmission-blocking vaccines. Trends in Biotechnology. 1992;10:388–91. doi: 10.1016/0167-7799(92)90280-9. [DOI] [PubMed] [Google Scholar]
- 5.Nunes JK, Woods C, Carter T, Raphael T, Morin MJ, Diallo D, et al. Development of a transmission-blocking malaria vaccine: Progress, challenges, and the path forward. Vaccine. 2014;32(43):5531–9. doi: 10.1016/j.vaccine.2014.07.030. [DOI] [PubMed] [Google Scholar]
- 6.Gwadz R. Successful immunization against the sexual stages of Plasmodium gallinaceum. Science. 1976;193:1150–1. doi: 10.1126/science.959832. [DOI] [PubMed] [Google Scholar]
- 7.Kumar N. A vaccine to prevent transmission of human malaria: a long way to travel on a dusty and often bumy road. Current science. 2007;92(11):1535–44. [Google Scholar]
- 8.Kaslow DC, Quakyi IA, Syin C, Raum MG, Keister DB, Coligan JE, et al. A vaccine candidate from the sexual stage of human malaria that contains EGF-like domains. Nature. 1988;333(6168):74–6. doi: 10.1038/333074a0. [DOI] [PubMed] [Google Scholar]
- 9.Quakyi IA, Carter R, Rener J, Kumar N, Good MF, Miller LH. The 230-kDa gamete surface protein of Plasmodium falciparum is also a target for transmission-blocking antibodies. J Immunol. 1987;139(12):4213–7. [PubMed] [Google Scholar]
- 10.Williamson KC. Pfs230: from malaria transmission-blocking vaccine candidate toward function. Parasite Immunology. 2003;25(7):351. doi: 10.1046/j.1365-3024.2003.00643.x. [DOI] [PubMed] [Google Scholar]
- 11.Rener J, Graves P, Carter R, Williams J, Burkot T. Target antigens of transmission-blocking immunity on gametes of Plasmodium falciparum. J Exp Med. 1983;158:976–81. doi: 10.1084/jem.158.3.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Outchkourov NS, Roeffen W, Kaan A, Jansen J, Luty A, Schuiffel D, et al. Correctly folded Pfs48/45 protein of Plasmodium falciparum elicits malaria transmission-blocking immunity in mice. Proceedings of the National Academy of Sciences. 2008;105(11):4301–5. doi: 10.1073/pnas.0800459105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chowdhury DR, Angov E, Kariuki T, Kumar N. A Potent Malaria Transmission Blocking Vaccine Based on Codon Harmonized Full Length Pfs48/45 Expressed in Escherichia coli. PloS one. 2009;4(7):e6352. doi: 10.1371/journal.pone.0006352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saxena AK, Saul A, Garboczi DN. Crystallization and preliminary X-ray analysis of the Plasmodium vivax sexual stage 25 kDa protein Pvs25, a transmission-blocking vaccine candidate for malaria. Acta Crystallographica: Section D. 2004;60(4):706–8. doi: 10.1107/S0907444904001398. [DOI] [PubMed] [Google Scholar]
- 15.Saxena AK, Singh K, Hua-Poo S, Klein MM, Stowers AW, Saul AJ, et al. The essential mosquito-stage P25 and P28 proteins from Plasmodium form tile-like triangular prisms. Nature Structural & Molecular Biology. 2006;13(1):90–1. doi: 10.1038/nsmb1024. [DOI] [PubMed] [Google Scholar]
- 16.Kaslow DC, Bathurst IC, Lensen T, Ponnudurai T, Barr PJ, Keister DB. Saccharomyces cerevisiae recombinant Pfs25 adsorbed to alum elicits antibodies that block transmission of Plasmodium falciparum. Infection and immunity. 1994;62(12):5576–80. doi: 10.1128/iai.62.12.5576-5580.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barr PJ, Green KM, Gibson HL, Bathurst IC, Quakyi IA, Kaslow DC. Recombinant Pfs25 protein of Plasmodium falciparum elicits malaria transmission-blocking immunity in experimental animals. J Exp Med. 1991;174(5):1203–8. doi: 10.1084/jem.174.5.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsuboi T, Takeo S, Iriko H, Jin L, Tsuchimochi M, Matsuda S, et al. Wheat Germ Cell-Free System-Based Production of Malaria Proteins for Discovery of Novel Vaccine Candidates. Infection and immunity. 2008;76(4):1702–8. doi: 10.1128/IAI.01539-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farrance CE, Chichester JA, Musiychuk K, Shamloul M, Rhee A, Manceva SD, et al. Antibodies to plant-produced Plasmodium falciparum sexual stage protein Pfs25 exhibit transmission blocking activity. Human vaccines. 2011;7(sup1):191–8. doi: 10.4161/hv.7.0.14588. [DOI] [PubMed] [Google Scholar]
- 20.Gregory JA, Li F, Tomosada LM, Cox CJ, Topol AB, Vinetz JM, et al. Algae-Produced Pfs25 Elicits Antibodies That Inhibit Malaria Transmission. PloS one. 2012;7(5):e37179. doi: 10.1371/journal.pone.0037179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Y, Ellis RD, Shaffer D, Fontes E, Malkin EM, Mahanty S, et al. Phase 1 Trial of Malaria Transmission Blocking Vaccine Candidates Pfs25 and Pvs25 Formulated with Montanide ISA 51. PloS one. 2008;3(7):e2636. doi: 10.1371/journal.pone.0002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Y, Przysiecki C, Flanagan E, Bello-Irizarry SN, Ionescu R, Muratova O, et al. Sustained high-titer antibody responses induced by conjugating a malarial vaccine candidate to outer-membrane protein complex. Proceedings of the National Academy of Sciences. 2006;103(48):18243–8. doi: 10.1073/pnas.0608545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kubler-Kielb J, Majadly F, Wu Y, Narum DL, Guo C, Miller LH, et al. Long-lasting and transmission-blocking activity of antibodies to Plasmodium falciparum elicited in mice by protein conjugates of Pfs25. Proceedings of the National Academy of Sciences. 2007;104(1):293–8. doi: 10.1073/pnas.0609885104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones RM, Chichester JA, Mett V, Jaje J, Tottey S, Manceva S, et al. A Plant-Produced Pfs25 VLP Malaria Vaccine Candidate Induces Persistent Transmission Blocking Antibodies against Plasmodium falciparum in Immunized Mice. PloS one. 2013;8(11):e79538. doi: 10.1371/journal.pone.0079538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimp RL, Jr, Rowe C, Reiter K, Chen B, Nguyen V, Aebig J, et al. Development of a Pfs25-EPA malaria transmission blocking vaccine as a chemically conjugated nanoparticle. Vaccine. 2013;31(28):2954–62. doi: 10.1016/j.vaccine.2013.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar R, Angov E, Kumar N. Potent Malaria Transmission-Blocking Antibody Responses Elicited by Plasmodium falciparum Pfs25 Expressed in Escherichia coli after Successful Protein Refolding. Infection and immunity. 2014;82(4):1453–9. doi: 10.1128/IAI.01438-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat Med. 2013;19(12):1597–608. doi: 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- 28.Oyewumi MO, Kumar A, Cui Z. Nano-microparticles as immune adjuvants: correlating particle sizes and the resultant immune responses. Expert Review of Vaccines. 2010;9(9):1095–107. doi: 10.1586/erv.10.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ledet G, Bostanian LA, Mandel T. Nanoemulsions as a Vaccine Adjuvant. CRC Press; Boca Raton: 2013. [Google Scholar]
- 30.Igartua M, Hernández RM, Esquisabel A, Gascón AR, Calvo MB, Pedraz JL. Enhanced immune response after subcutaneous and oral immunization with biodegradable PLGA microspheres. Journal of Controlled Release. 1998;56(1–3):63–73. doi: 10.1016/s0168-3659(98)00077-7. [DOI] [PubMed] [Google Scholar]
- 31.Spiers ID, Eyles JE, Baillie LWJ, Williamson ED, Alpar HO. Biodegradable Microparticles with Different Release Profiles: Effect on the Immune Response After a Single Administration via Intranasal and Intramuscular Routes. Journal of Pharmacy and Pharmacology. 2000;52(10):1195–201. doi: 10.1211/0022357001777324. [DOI] [PubMed] [Google Scholar]
- 32.Akagi T, Baba M, Akashi M. Biodegradable Nanoparticles as Vaccine Adjuvants and Delivery Systems: Regulation of Immune Responses by Nanoparticle-Based Vaccine. In: Kunugi S, Yamaoka T, editors. Polymers in Nanomedicine Advances in Polymer Science. Vol. 247. Springer; Berlin Heidelberg: 2012. pp. 31–64. [Google Scholar]
- 33.Graves RA, Pamujula S, Moiseyev R, Freeman T, Bostanian LA, Mandal TK. Effect of different ratios of high and low molecular weight PLGA blend on the characteristics of pentamidine microcapsules. International Journal of Pharmaceutics. 2004;270(1–2):251–62. doi: 10.1016/j.ijpharm.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 34.Kane CM, Cervi L, Sun J, McKee AS, Masek KS, Shapira S, et al. Helminth Antigens Modulate TLR-Initiated Dendritic Cell Activation. J Immunol. 2004;173(12):7454–61. doi: 10.4049/jimmunol.173.12.7454. [DOI] [PubMed] [Google Scholar]
- 35.Kenney JS, Hughes BW, Masada MP, Allison AC. Influence of adjuvants on the quantity, affinity, isotype and epitope specificity of murine antibodies. Journal of Immunological Methods. 1989;121(2):157–66. doi: 10.1016/0022-1759(89)90156-7. [DOI] [PubMed] [Google Scholar]
- 36.Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10(11):787–96. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 37.Foged C, Brodin B, Frokjaer S, Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. International Journal of Pharmaceutics. 2005;298(2):315–22. doi: 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 38.Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids and Surfaces B: Biointerfaces. 2010;75(1):1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Prokop A, Davidson JM. Nanovehicular intracellular delivery systems. Journal of Pharmaceutical Sciences. 2008;97(9):3518–90. doi: 10.1002/jps.21270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vert M, Mauduit J, Li S. Biodegradation of PLA/GA polymers: increasing complexity. Biomaterials. 1994;15(15):1209–13. doi: 10.1016/0142-9612(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 41.Vasir JK, Labhasetwar V. Quantification of the force of nanoparticle-cell membrane interactions and its influence on intracellular trafficking of nanoparticles. Biomaterials. 2008;29(31):4244–52. doi: 10.1016/j.biomaterials.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]