

Abstract
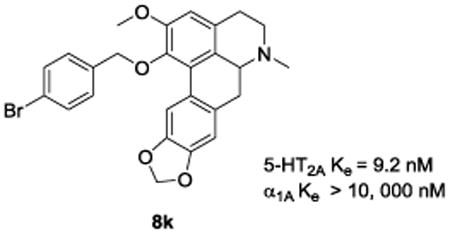
A series of C1 aporphine analogues related to compound 5 and that contain substituted allylic, alkynyl, nitrile, ester and benzyl groups was synthesized and evaluated for affinity at h5HT2A and α1A receptors in functional activity assays that measure calcium release. The presence of branched allylic substituent groups diminished affinity for the h5HT2A receptor. Likewise, the alkynyl, nitrile and ester derivatives evaluated displayed lower 5-HT2A receptor affinity as compared to 5. Hydrophobic, steric and electronic effects impact the affinity of p-substituted benzyl derivatives 8i – 8k but in different ways. High hydrophobicity and size favor 5-HT2A affinity whereas, high electronegativity disfavors 5-HT2A affinity. p-bromobenzyl analogue 8k was identified as a 5-HT2A receptor selective ligand, with the highest 5-HT2A receptor affinity of any aporphine known to date. Most of the other analogues were selective for the 5-HT2A versus the α1A receptor. ChemScore binding energies from docking studies correlated qualitatively with the observed trends in affinity for 8i - 8k, although the binding energies were not well differentiated quantitatively.
Keywords: aporphine, 5-HT2A, antagonist, adrenergic, nantenine
Graphical abstract
The neurotransmitter serotonin (5-HT) plays a significant role in a variety of central nervous system (CNS) processes such as mood, anxiety, cognition, feeding behavior, aggression, thermoregulation, sleep, pain and stimulant abuse.1 Peripherally, 5-HT is involved in the regulation of smooth muscle contraction, gastrointestinal function and cardiovascular function. These central and peripheral functions are mediated by a group of fourteen 5-HT receptors, comprising seven distinct families: 5-HT1 - 5-HT7. Some of these receptor families are divided further into subtypes; the 5-HT2 family has three subtypes, namely, 5-HT2A, 5-HT2B and 5-HT2C.
A handful of highly selective and potent 5-HT2A antagonists have been developed as potential sleep disorder medicaments and have shown promising results in clinical trials.2-5 Representatives from this group - eplivanserin (1), pruvanserin (2) and ritanserin (3) are shown in Figure 1. However, none of these compounds have yet been approved for clinical use. In light of the preceding, the identification of new, selective 5-HT2A antagonists would be a valuable addition to the pipeline of therapeutics germane to the treatment of sleep disorders. Furthermore, such compounds could serve as pharmacologically interesting research tools and potential therapeutics for other CNS-related disorders where 5-HT2A blockade is therapeutically beneficial.
Figure 1. Structures of eplivanserin (1), pruvanserin (2) and ritanserin (3).

Aporphine alkaloids have been extensively explored as ligands for dopamine D1 and D2 receptors.6 That being said, aporphines are a “privileged template” and representatives of this structural class have also shown good affinity for 5-HT receptors.7-9 This makes the scaffold an attractive one for the development of novel, selective 5-HT receptor ligands.
In contrast to structure-activity relationship (SAR) studies done on aporphines at dopamine D1 and D2 receptors, there are comparatively few such studies on aporphines with the 5-HT2A receptor as the focus. A major thrust of our research has been to understand the molecular features of aporphines that impact affinity, activity and selectivity to 5-HT2A receptors as a prerequisite for the design of novel, potent and selective 5-HT2A antagonist tools and therapeutics. In that context, our previous SAR studies based on the aporphine alkaloid nantenine (4, Figure 2) have resulted in the identification of new aporphinoid 5-HT2A receptor antagonists with significantly improved affinity and selectivity as compared to nantenine.10-13 In continuing efforts in that vein, we wish to report an SAR study that has resulted in the identification of the most potent aporphine-derived 5-HT2A receptor antagonist known to date.
Figure 2. Structures of nantenine (4) and compound 5.

Our prior SAR studies have been largely directed towards the C1 and C2 positions of the aporphine scaffold of nantenine.11,13,14 These studies have shown that substituents at the C1 position of nantenine may be manipulated to improve 5-HT2A affinity and selectivity. n-Alkyl substituents were particularly well tolerated. Among several derivatives synthesized and evaluated, the C1 allyl analogue 5 is one of the most potent that was identified (Ke = 70 nM). Compound 5 showed twelve-fold improvement in 5-HT2A receptor affinity as compared to nantenine.14 Moreover, unlike nantenine, compound 5 was devoid of affinity for the α1A adrenergic receptor. We considered whether the improvement in 5-HT2A receptor affinity and selectivity was due to the electron-rich nature of the allyl group or to other effects. In order to help to clarify this issue, we decided to synthesize and evaluate a series of C1 nantenine analogues related to 5, with diverse electronic, steric and hydrophobic characteristics.
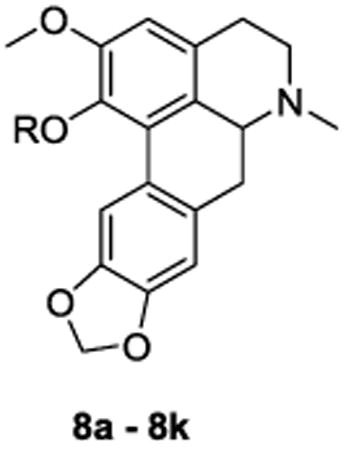
The analogues were synthesized from compound 6 as depicted in scheme 1 Compound 6 was obtained as we have detailed in previous publications.15,10 Reaction of 6 with various alkyl halides provided phenol ether derivatives 7a - 7k. Thereafter, removal of the Boc protecting group with zinc bromide ensued. The secondary amine thus produced, was methylated via reductive amination with formaldehyde/NaBH(OAc)3 to afford C1 analogues 8a - 8k.
Scheme 1. Synthesis of analogs 8a - 8k.

Analogues 8a - 8k were then evaluated for 5-HT2A receptor affinity in a functional assay. In order to gauge selectivity of the compounds, the analogues were also evaluated at the α1A adrenergic receptor (since nantenine itself is selective for this receptor subtype). Briefly, the analogs were screened at 10μM in multi-well format for intrinsic (agonist) and antagonist activity at the human 5-HT2A receptor using FLIPR-based (Molecular Devices, Sunnydale, CA) functional assays that detect receptor-mediated mobilization of internal calcium with a calcium sensitive fluorescent dye. Compounds that showed no intrinsic activity in the functional assay and inhibited the increase in basal fluorescence elicited by EC80 of 5-HT by at least 50%, had their Ke (apparent affinity in a functional assay) determined. Ke values were determined by running an 8-point half-log 5-HT concentration response curve in the presence and absence of a single concentration of antagonist. EC50 values were calculated for 5-HT (A) and 5-HT + test compound (A′), and these values used to calculate the test compound Ke using the formula: Ke = [L]/(DR-1), where [L] equals the concentration of test compound in the assay and DR equals the dose ratio or A′/A. A similar set of assays was performed for the α1A - adrenergic receptor. Data from these evaluations is presented in Table 1.
Table l. Apparent affinity (Ke) and selectivity of compounds 8a - 8k at 5-HT2A and α1A receptors.
| ||||
|---|---|---|---|---|
|
| ||||
| Compound | R | Affinity (Ke in nM)a | Selectivity | |
|
|
|
|||
| 5-HT2A | α1A | α1A/5-HT2A | ||
| 8a | -CH2CH=CHCH3 (E) | 723 ± 68 | 1,980 ± 455 | 2.7 |
| 8b | -CH2C(CH2)CH3 | 172 ± 71 | >10,000 | > 58 |
| 8c | -CH2CH=C(CH3)2 | 2,074 ± 230 | >10,000 | > 5 |
| 8d | -CH2C≡CCH3 | 2,690 ± 806 | 557 ± 111 | 0.2 |
| 8e | -CH2CH=CHPh (E) | >10,000 | >10,000 | - |
| 8f | -CH2CH2CH2CH=CH2 | >10,000 | >10,000 | - |
| 8g | -CH2CN | >10,000 | 711 ± 64 | < 0.07 |
| 8h | -CH2CH2OCOCH3 | 1,913 ± 288 | 591 ± 81 | 0.3 |
| 8i | -CH2C6H4-p-Cl | 6,046 ± 3076 | >10,000 | > 1.7 |
| 8j | -CH2C6H4-p-CF3 | 2,458 ± 184 | >10,000 | > 4.1 |
| 8k | -CH2C6H4-p-Br | 9.2 ± 2.6 | >10,000 | >1,086 |
| 5 | -CH2CH=CH2 | 70±15 | >10,000 | >143 |
| 4 | CH3 | 850 ± 5.8 | 36 ± 7 | 0.04 |
| ketanserin | 32b,c | ndd | ||
| prazosin | ndd | 1.1 ± 0.4 | ||
Ke (apparent affinity) values are means of at least two experiments carried out in triplicate;
Experiment run once;
IC50 determined in the presence of 5-HT EC80;
not determined – compounds used as positive controls.
Compounds 8a - 8c all possessed an allylic moiety directly attached to C1. In the case of 8a, which has an (E)-2-butenyl moiety, a reduction in affinity for the 5-HT2A receptor was observed. The methallyl analog 8b, had improved affinity as compared to 8a but was still 2-fold lower in affinity than 5. The 2,2-dimethyl allyl analog 8c had the lowest affinity of all three compounds, being 30-fold lower in 5-HT2A receptor affinity than 5. These results indicate that there is a preference for an unsubstituted allyl group at the C1 position. The steric bulk introduced by the additional methyl groups in 8a - 8c may be partially responsible for this trend. Electronic effects might not play a large role here, since if that was the case it would be expected that the electronically similar 8a and 8b would have similar affinities. However, it appears that the additional steric bulk is better accommodated at the internal alkene carbon rather than the terminal alkene carbon of the allyl group (i.e. compare 8b with 8a/8c). Analog 8d with a 2-butynyl substituent, showed diminished 5-HT2A receptor affinity when compared to 8a. This might be due to increased electron density in the alkyne moiety of 8d. It is also possible that the geometry of the substituent has some effect - the terminal methyl group being linear in 8d. No affinity was seen for the C1 (E)-cinnamyl analogue 8e. As was the case with 8c, this result with 8e is perhaps attributable to a steric intolerance for groups (methyl in 8c; phenyl in 8e) attached to the terminal alkene carbon of the allyl moiety. Compound 8f lacked affinity for the 5-HT2A receptor, suggesting that the double bond unit is better tolerated at an allylic position, as in 5. Replacing the alkene moiety of 5 with a nitrile group (8g), also decimated affinity at the 5-HT2A receptor. Given the similarity in size of the C1 substituent groups in 5 and 8g, and based on their different electronic character, we surmise that there is an electronic factor operating in the reduced affinity of 8g. It seems reasonable to speculate that the addition of a nitrogen atom in 8g, leads to a reduction in hydrophobic contacts of the C1 substituent and thus a reduced 5-HT2A receptor affinity. The ester analogue 8h had low affinity for the 5-HT2A receptor. When the data from 8g and 8h are considered together with data from compounds 8a-8c, it may be reasoned that polar groups are less tolerated at the C1 position than similar sized allyl groups.
In a previous SAR study, we found that a nantenine analogue with a C1 benzyl group had moderate 5-HT2A receptor affinity.11 Since benzyl groups contain an allyl substructure, we were interested in ascertaining to what extent substituted benzyl groups could serve as surrogates for an allyl group as it pertains to 5-HT2A receptor affinity. We reasoned that perhaps substituents on the phenyl ring of a benzyl moiety might be appropriately placed to allow for optimal electronic character of the phenyl ring for binding to the 5-HT2A receptor. It was with this motive in mind that compounds 8i - 8k were synthesized and evaluated. The p-chlorobenzyl analog 8i lacked any appreciable affinity for the 5-HT2A receptor. This situation was made slightly better when a CF3 group replaced the chloro group (i.e. compound 8j). Compound 8k with a p-bromo group however, showed a dramatic increase in 5-HT2A receptor affinity (9.2 nM, representing a seven-fold increase as compared to 5), superseding any known aporphine in this regard. The reason for the significant increase in 5-HT2A receptor affinity of 8k was not absolutely clear, especially when one compares the chasm in affinities between 8i and 8k (bearing electronically similar halogen groups). Thus, the 5-HT2A receptor affinity in the 8i - 8k subset of analogues, does not seem to be due to the electron-withdrawing power of the p-substituents alone. Bromine has a higher hydrophobicity and is larger than chlorine, and we presume that the combination of these effects is favorable for 5-HT2A affinity i.e. the larger and more hydrophobic the p-substituent, the higher is the 5-HT2A affinity. This would explain the higher affinity of compound 8k as compared to 8i. However, this logic disintegrates when one considers compound 8j, which has a more hydrophobic and larger CF3 group, and would thus be expected to have the highest 5-HT2A receptor affinity of the three. The powerful electron-withdrawing effects of the CF3 group may be the reason for the lower than “expected” affinity of 8j. That is the stronger the electron-withdrawing effect of the phenyl substituent, the lower is the affinity.
The analogues assayed were inactive or had only moderate affinity for the α1A receptor. In the 8a - 8c series, compound 8a displayed the highest (though moderate) α1A receptor affinity; this suggests that branching on the allyl system is not desirable for α1A affinity, probably due to steric intolerance. Analogues with allyl or benzyl substituents showed selectivity for the 5-HT2A receptor; however analogues with alkynyl, nitrile or ester groups (8d, 8g and 8h) showed selectivity for the α1A receptor. It seems that size, while important, is not the only important factor that controls selectivity of these analogues since the allyl containing 5 is 5-HT2A selective while the similar sized nitrile containing 8g is α1A selective. It may be that in the case of 8g and 8h, the presence of polar functionalities capable of functioning as hydrogen bond acceptors or which disfavor hydrophobic interactions with the receptor, favors this reversal in selectivity. It is less clear what factors favor the switch to α1A selectivity in 8d, although it seems plausible that differences in electronic density play a role (compare 8c - with an alkene functionality and 8d). Further SAR studies will be required to obtain a clearer picture. Compounds 8i - 8k all lacked affinity for the α1A receptor, probably a consequence of the size of the benzyl group in these analogues. When one considers the smaller allyl-containing (8a – 8c) and the benzyl group-containing (8i – 8k) analogues it appears that these functionalities are worse for α1A affinity than for 5-HT2A affinity. As a consequence smaller allyl moieties as well as benzyl moieties confer 5-HT2A selectivity.
To determine the broader selectivity of 8k at other CNS receptor sites, the compound was submitted for screening at the Psychoactive Drug Screening Program (PDSP). Details of the assay protocols may be found in the PDSP assay protocol book available online (https://pdspdb.unc.edu/html/tutorials/UNC-CH%20Protocol%20Book.pdf). This screening confirmed the selectivity of 8k for the 5-HT2A receptor. 8k retained affinity for the 5-HT2A receptor (185 nM) but had no affinity (less than 50% inhibition in a primary assay) at the following receptor sites: 5-HT1A, 5-HT5A, α1A α1B α1D, β1, β2, β3, BZP rat brain site, D2, D4, D5, DOR, GABAA, H1, H3, H4, KOR, M1, M2, M3, M4, MOR, NET, SERT. Poor affinity (Ki > 900 nM) was seen for the following sites: 5-HT1B, 5-HT1D, 5-HT1E, 5-HT2B, 5-HT3, 5-HT7, α2B, α2C, D1, D3, H2, M5, PBR. Moderate affinity (300 nM < Ki < 900 nM) was seen at 5-HT2C, 5-HT6, α2A, DAT, sigma-1 and sigma-2 receptor sites. The discrepancy between the Ki values and Ke values at the 5-HT2A receptor for 8k (9.2 vs 185 nM) is likely attributable to differences in the assay protocols. The Ke assay is a functional assay and gives an indirect measurement of affinity (i.e. calculated based on blockade of calcium release by the compound). Nevertheless, both assays are in agreement with regards to the 5-HT2A versus α1A selectivity of 8k.
Docking of compounds 8i, 8j and 8k at the 5-HT2A receptor was performed in order to unravel the binding interactions among this group of substituted benzyl analogues. Analysis of the docking outcomes and the nature of the key protein-ligand interactions provides insights into the diverse affinities of the three compounds that are essentially in agreement with the above deductions derived from the experimental data. In the current work, the relevant ligands were docked into a homology model of the human 5-HT2A receptor that we constructed previously. Full details of the model construction and evaluation can be found in a prior publication9. Briefly, the 5-HT2A receptor homology model was built using a human β2-adrenergic receptor structure (PDB code: 2RH1) as a template. The 2RH1 crystal structure was solved at high resolution (2.40Å) and the sequence identity of the 5-HT2A receptor and the β2-adrenergic template is close to 30%. Furthermore, the two receptors have functional similarities as they are both categorized as neurotransmitter GPCRs. The sequences were aligned using the ClustalX16 and ICM Pro sequence alignment tools17 and the homology model was constructed with the ICM Pro model building program17. As secondary structure elements are highly conserved in these sequences, the alignments were manually ameliorated to achieve complete alignment of the residues known to be well conserved in the GPCR superfamily18. The model was subsequently relaxed by 500 steepest descent energy minimization steps via the Charmm molecular modeling package.
The docking experiments were performed using our in-house developed drug discovery platform. The algorithm invokes a simulated annealing protocol to find the best energetic fit between the ligand and the protein binding site19, 20. 3D atomic coordinates of the homology model serve as input to the program as well as the definition of the rectangular binding site box, which encompasses residues assumed to be important for ligand binding based on our previous docking studies of a series of nantenine analogs to the same 5-HT2A receptor homology model9. The annealing simulation evolves the ligand through a series of random moves, including rigid body translations and bond rotations. A score is calculated at each step based on the empirically derived ChemScore estimate of the binding energy21, as well as receptor-ligand clashes. The Metropolis condition is applied to either accept or reject the modified structure. The length of the simulation is defined by the Markov chain length and the number of chains, and the temperature is slowly lowered after each chain. A single run yields a single docked structure and for each ligand, thirty docked structures were generated, clustered by ligand rmsd and ranked according to the ChemScore binding energy value. Receptor flexibility is incorporated into the docking protocol by allowing for conformational rearrangements of the amino acid side chain χ torsion angles. This represents an additional type of transition that is invoked during the simulated annealing process. Several levels of side-chain flexibility were exploited involving all residues within 4 or 5Å of the evolving ligand or by specifying a subset of side chains that typically clash with the evolving ligand structure in rigid receptor simulations. The docking outputs were found to be very similar in all these flexible side-chain scenarios.
A representation of the docked poses for the ligands 8i, 8j and 8k within the 5-HT2A receptor binding pocket is depicted in Figure 3. Overall, each ligand generates three different binding modes. They are not differentiated particularly well energetically, nevertheless, the structures depicted in the Figure correspond to the top ranked poses according to ChemScore. The estimated binding energies are -30.6, -31.6 and -34.4 kJ/mol for 8i, 8j and 8k, respectively. This qualitatively correlates well with the observed affinity measurements, although quantitatively the binding energy differences are relatively small compared to the wide variation in affinity, particularly between compounds 8i and 8k. This is not particularly unexpected as no single scoring function can correctly rank order and quantitatively differentiate all protein-ligand complexes. Hence, docking scores are typically exploited to simply categorize ligands as active or inactive, rather than rank ordering. Furthermore, in this work, the tolerance of the binding site for multiple ligand binding modes and the relatively small set of compounds assessed in biological assays are other potential reasons for the lack of clear correlation between the docking results and the measured affinities measurements are needed for a larger, more structurally. It is clear that further computational simulations and affinity diverse library of compounds to gain binding energy for the latter analog is primarily due to better hydrophobic interactions with the receptor pocket involving the bromine substituent (-30.2 kJ/mol for the lipophilic term for 8k) compared to chlorine (-27.2 kJ/mol for the lipophilic term for 8i) as postulated above based on hydrophobicity and atomic size. For compound 8j with the relatively large, hydrophobic CF3 substituent, the hydrophobic contribution to the estimated binding energy is very similar to that for ligand 8k with the bromine atom. This appears reasonable based on the close similarity of the binding poses and the distance from hydrophobic residues. The lipophilic term is however, slightly less favorable for 8j presumably due to the relatively close proximity of the electron withdrawing CF3 group and the protonated Lys350 side chain, making it difficult for the simulation to identify an orientation for H-bonding interactions while eliminating clashes. Overall, this translates to a binding energy for 8j that is intermediate to that of compounds 8k and 8i.
Figure 3.
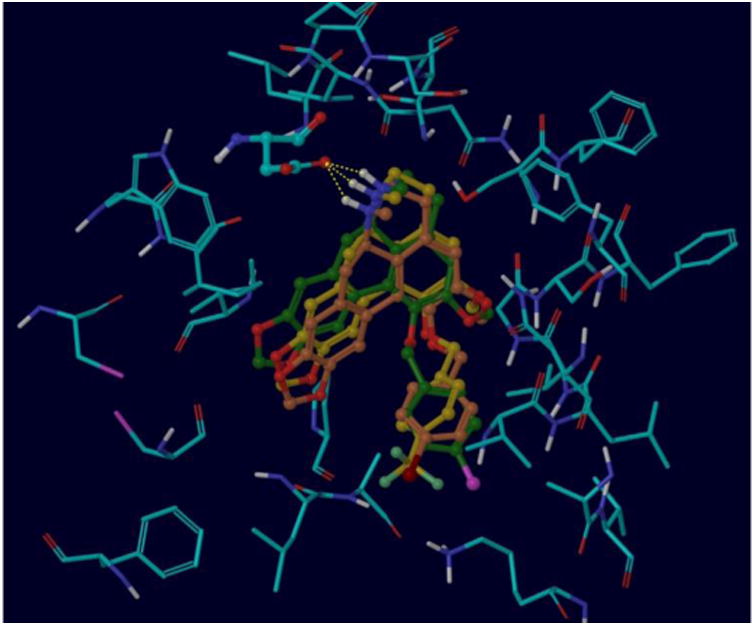
Superimposed binding poses for compounds 8i (C atoms in green), 8j (C atoms in yellow) and 8k (C atoms in brown). The key H-bonding interaction between the ligand quaternary N and Asp155 is depicted by the dashed line.
Finally, compounds 5 and 8g were docked into the 5-HT2A receptor binding site in order to explore the huge difference in affinity for these systems with similar sized C1 substituents in terms of their receptor-ligand contacts. The top-ranked docking outcomes for these ligands are illustrated in Figure 4. As above for the benzyl substituent groups, the estimated binding energies are in qualitative accordance with the affinity measurements (-32.4 and -30.1 kJ/mol for compounds 5 and 8g, respectively), however, the predicted energetic difference is too small compared to the diverse affinity data to provide definitive quantitative insight. The reasons postulated for this are similar to those explicated above for compounds 8i, 8j and 8k. Despite this limitation, as speculated earlier, the better binding energy for the allyl analog, 5, derives mainly from an improved lipophilic term (-27.5 and -26.0 kJ/mol for compounds 5 and 8g, respectively, for the hydrophobic interactions). Hence, replacement of the C1 allyl substituent with a more polar nitrile group leads to a reduction in favorable contacts in this hydrophobic region of the binding pocket.
Figure 4.
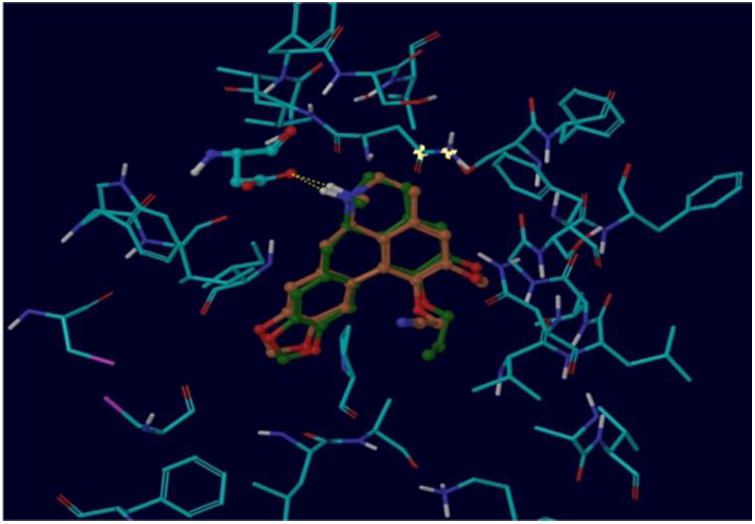
Superimposed binding poses for compounds 5 (C atoms in green) and 8g (C atoms in brown). The key H-bonding interaction between the ligand quaternary N and Asp155 is depicted by the dashed line.
In summary, modifications at the C1 position of 5 with various allyl isosteres has resulted in the identification of compound 8k, the most potent h5-HT2A receptor antagonist with an aporphine core reported thus far. Our SAR study indicates that hydrophobic, steric and electronic effects are important to the 5-HT2A receptor affinity of the analogues; the relative contribution of each parameter depends on the particular C1 substituent. Substituted allyl, alkynyl, nitrile and ester functionalities are not well tolerated at C1 for high 5-HT2A affinity. In the case of p-substituted benzyl analogues, hydrophobicity and larger steric size favors, while electron-withdrawing ability of the p-substituent disfavors 5-HT2A receptor affinity. For compounds 8i – 8k, the 5-HT2A receptor model does not clearly differentiate compounds with high affinity from compounds with low affinity when absolute binding energies are considered. Nevertheless, the binding energies for 8i – 8k were consistent with the trend in ligand affinities observed and this was also paralleled in comparing 5 and 8g. The extent to which the SAR trends observed above may be generalized as well as the capabilities and limitations of the 5-HT2A receptor model for prospective ranking, will benefit from evaluations of larger libraries of analogues. The results obtained herein form the basis for such future extrapolations.
Supplementary Material
Acknowledgments
This publication was made possible by Grant Numbers 1SC-1GM092282 and G12RR003037 from the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or its divisions. Ki determinations, and receptor binding profiles were generously provided by the National Institute of Mental Health's Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. For experimental details please refer to the PDSP website http://pdsp.med.unc.edu/ and click on “Binding Assay” or “Functional Assay” on the menu bar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.McCorvy JD, Roth BL. Pharmacol Ther. 2015;150:129. doi: 10.1016/j.pharmthera.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanger DJ, Soubrane C, Scatton B. Ann Pharm Fr. 2007;65:268. doi: 10.1016/s0003-4509(07)90046-2. [DOI] [PubMed] [Google Scholar]
- 3.Monti JM. Drugs Today (Barc) 2010;46:18. doi: 10.1358/dot.2010.46.3.1437247. [DOI] [PubMed] [Google Scholar]
- 4.Griebel G, Beeske S, Jacquet A, Laufrais C, Alonso R, Decobert M, Avenet P, Francon D. Neuropharmacology. 2013;70:19. doi: 10.1016/j.neuropharm.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Teegarden BR, Al Shamma H, Xiong Y. Curr Top Med Chem. 2008;8:969. doi: 10.2174/156802608784936700. [DOI] [PubMed] [Google Scholar]
- 6.Zhang A, Zhang Y, Branfman AR, Baldessarini RJ, Neumeyer JL. J Med Chem. 2007;50:171. doi: 10.1021/jm060959i. [DOI] [PubMed] [Google Scholar]
- 7.Zhao R, Lu W, Fang X, Guo L, Yang Z, Ye N, Zhao J, Liu Z, Jia J, Zheng L, Zhao B, Zhang A, Zhen X. Pharmacol Biochem Behav. 2014;124:204. doi: 10.1016/j.pbb.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 8.Munusamy V, Yap BK, Buckle MJ, Doughty SW, Chung LY. Chem Biol Drug Des. 2013;81:250. doi: 10.1111/cbdd.12069. [DOI] [PubMed] [Google Scholar]
- 9.Liu Z, Zhang H, Ye N, Zhang J, Wu Q, Sun P, Li L, Zhen X, Zhang A. J Med Chem. 2010;53:1319. doi: 10.1021/jm9015763. [DOI] [PubMed] [Google Scholar]
- 10.Pecic S, Makkar P, Chaudhary S, Reddy BV, Navarro HA, Harding WW. Bioorg Med Chem. 2010;18:5562. doi: 10.1016/j.bmc.2010.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaudhary S, Pecic S, Legendre O, Navarro HA, Harding WW. Bioorg Med Chem Lett. 2009;19:2530. doi: 10.1016/j.bmcl.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ponnala S, Gonzales J, Kapadia N, Navarro HA, Harding WW. Bioorg Med Chem Lett. 2014;24:1664. doi: 10.1016/j.bmcl.2014.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ponnala S, Kapadia N, Navarro HA, Harding WW. Chem Biol Drug Des. 2014;84:558. doi: 10.1111/cbdd.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaudhary S, Ponnala S, Legendre O, Gonzales JA, Navarro HA, Harding WW. Bioorg Med Chem. 2011;19:5861. doi: 10.1016/j.bmc.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaudhary S, Pecic S, Legendre O, Harding WW. Tetrahedron Lett. 2009;50:2437. doi: 10.1016/j.tetlet.2009.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higgins DG, Sharp PM. Gene. 1988;73:237. doi: 10.1016/0378-1119(88)90330-7. [DOI] [PubMed] [Google Scholar]
- 17.Abagyan RA, Totrov MM, Kuznetsov DA. J Comput Chem. 1994;15:488. [Google Scholar]
- 18.Baldwin JM, Schertler GF, Unger VM. J Mol Biol. 1997;272:144. doi: 10.1006/jmbi.1997.1240. [DOI] [PubMed] [Google Scholar]
- 19.Alberts IL, Todorov NP, Dean PM. J Med Chem. 2005;48:6585. doi: 10.1021/jm050196j. [DOI] [PubMed] [Google Scholar]
- 20.Alberts IL, Todorov NP, Källblad P, Dean PM. QSAR Comb Sci. 2005;24:503. [Google Scholar]
- 21.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Proteins. 2003;52:609. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.