

Abstract
Over 200 types of human papillomaviruses (HPV) have been identified that infect epithelial cells at different anatomic locations. HPVs are grouped into five genra with the alpha and beta viruses being the most commonly studied. Members of the alpha HPV genus infect genital epithelia and are the causative agents of many anogenital cancers. Beta HPVs infect cutaneous epithelia and have been suggested as co-factors in the development of non-melanoma skin cancers. Recent studies have shown that activation of DNA damage pathways is important for the productive life cycle of the alpha HPVs while the beta viruses suppress their activation. These differences likely contribute to the varying types of lesions and malignancies that are associated with these viruses.
Introduction
Human papillomaviruses (HPVs) are associated with a spectrum of manifestations ranging from unapparent infections to malignant neoplasias. HPVs are grouped phylogenetically into five genera, alpha, beta, gamma, mu and nu, and are further divided into species[1]. The alpha-papillomaviruses contain viruses that infect mucosal epithelium, some of which are considered high-risk (HR) and others low-risk (LR) based on their association with cancers. The LR-HPVs can cause cutaneous lesions, and other alpha papillomaviruses only cause benign cutaneous infections. HR-HPVs have been conclusively linked to the etiology of anogenital and oropharyngeal cancers[2]. Infection with HR-HPVs is common in young sexually active women and in men, and is generally resolved by an immune response, though latent HPV infection may remain[3,4]. In some cases HR-HPV infections persist causing genetic instability that can progress to invasive cancer if untreated [5,6]. In these cancers the HPV genome persists, generally integrated into host chromosomes, with expression of the E6 and E7 genes [7,8].
Genus beta HPVs commonly infect the skin[9]. Unlike the genital tract HPVs, serologic evidence indicates that beta HPV infections occur early in life [10]. Several of the genus beta HPVs were initially detected in squamous cell skin cancers (SCSC) from patients with the rare disease epidermodysplasia verruciformis [11], leading to the hypothesis that beta HPVs were causal for SCSC, just as HR-HPVs are causal for anogenital cancers. However integration of beta HPVs and continued expression of the E6 and E7 genes in SCSC has not been reported, indicating that these HPVs are not required for maintenance of the tumor. Similarly, epidemiologic studies have not supplied strong support for a causal association of the beta HPVs with SCSC. Both mechanistic studies on the beta HPV E6 and E7 proteins, and transgenic mouse models are consistent with the beta HPVs being able to act as cofactors with UV damage to promote carcinogenesis [12–15].
Life cycle of human papillomaviruses
The life cycle of human papillomaviruses is dependent upon differentiation of the host-infected cell and cellular replication proteins (Figure 1) [16]. Human papillomaviruses have small double strand genomes of approximately 8 kilobases in length that encode between 6 and 8 genes. None of these genes except for the E1 and E2 encode polymerases, or other replication factors. E1 encodes an origin recognition and helicase protein while E2 facilitates the assembly of E1 complexes on viral DNAs. This makes viral replication dependent largely upon cellular proteins for both the stable maintenance of viral genomes in undifferentiated cells as well as productive replication or amplification in differentiated cells. HPVs infect stratified squamous epithelia and enter cells in the basal layer that become exposed following trauma or wounding. In these cells, viral genomes are maintained as extrachromosomal elements that replicate in S phase in synchrony with cellular replication. Following entry into basal cells, viral genome copy numbers rapidly increase to about 50 to 100 copies per cell on average and this copy number is maintained at similar levels throughout the course of productive infections. Upon differentiation of suprabasal cells, high level replication of viral genomes is induced in a process referred to as amplification and this occurs concurrently with synthesis of the capsid proteins followed by virion assembly and release.
Figure 1.
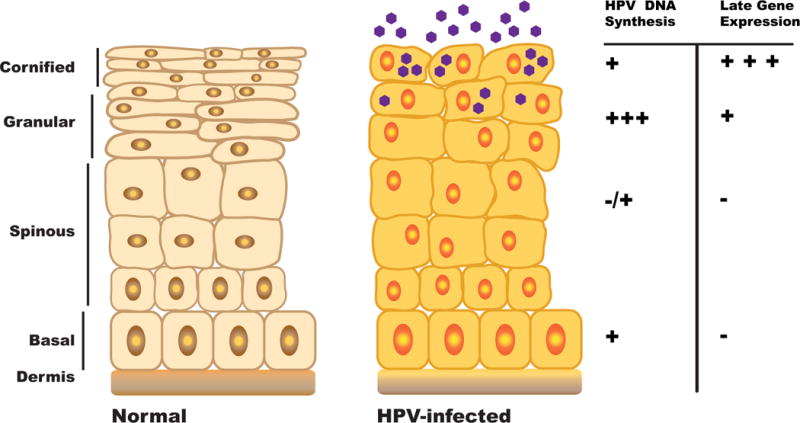
Differentiation-dependent life cycle of human papillomaviruses. HPVs establish persistent infections in basal epithelial cells where viral genomes are maintained as low copy episomes. Productive replication or amplification occurs upon differentiation in suprabasal layers.
Amplification of HPV genomes requires differentiating cells to remain active in the cell cycle. For the high-risk alpha viruses, amplification also requires the activation of the ataxia telangiectasia (ATM) pathway[17]. In normal cells, the ATM pathway functions to repair double strand DNA breaks, while in HPV positive cells ATM factors such as pCHK2, NBS1 and pSMC1 are constitutively activated and recruited to viral genomes in both undifferentiated and differentiated cells[18–20]. Interestingly, ATM activation is not required for the stable maintenance of episomes in basal cells but is critical for genome amplification in differentiated cells[17]. The levels of a small number of ATM factors that are recruited to HPV genomes including ⅕-H2AX, pCHK2 and pNBS1 increase upon differentiation and contribute to regulation of amplification. Members of the homologous recombination repair arm of the ATM pathway such as RAD51 and SMC1 are specifically required for amplification that occurs in G2 and this is when normal recombination repair also occurs[18,19]. Interestingly, activation of the DNA damage pathway is required for productive replication of the polyomavirus SV40 where it acts as a quality control mechanism to prevent default to a rolling circle mechanism of replication in favor of theta replication[21]. Whether a similar activity is important for HPV genome amplification remains to be determined.
The requirement for ATM activation does not, however, appear to be shared with the beta papillomaviruses and may be restricted to viruses from high-risk alpha subgroup. In fact, beta HPV E6 protein expression results in reduced expression of ATM and ATR, as well as BRCA1/BRCA2[22]. It has been difficult to study the virus lifecycle of LR-HPVs or beta HPVs because it has not yet been possible to develop experimental systems to study replication. However some differences have been noticed by careful examination of infected tissues. Whereas high risk HPVs stimulate proliferation in the basal and parabasal cells, presumably through degradation of pRb and p107[23–25], LR HPVs only stimulate proliferation in the mid-to upper epithelial layers, because their E7 proteins only degrade p130[26]. The E5 protein of alpha HPVs promotes genome amplification, at least in part through stabilization of the EGF receptor[27,28], but this protein is missing in beta HPVs.
Regulation of HPV Gene Expression
Two major viral promoters along with several minor ones regulate viral gene expression during the differentiation-dependent life cycle. The early promoter of high-risk HPVs is located in the Upstream Regulatory Region (URR) and directs initiation of transcription at sites upstream of the E6 open reading frame. HPV transcripts are all polycistronic encoding between 2 and 4 open reading frames and share a common polyadenylation site at the end of the early region. Alternative splicing and leaky scanning translation initiation regulate the differential expression and synthesis of early viral proteins[29]. The HPV E2 proteins are negative regulators of early promoter expression and this is part of a copy number control mechanism that acts by autoregulating expression of the replication proteins E2 and E1[30]. One characteristic of many HPV-positive cancers is that viral genomes are found integrated into host chromosomes so as to retain expression of only the E6 and E7 genes. Integration leads to the loss of E2 expression and high-level transcription of E6 and E7 viral oncoproteins that contributes to malignant progression. Upon differentiation, the late viral promoter is activated by cellular factors and directs initiation of late transcripts from a series of start sites located in the middle of the E7 open reading frame. The late promoter is not a target of E2 repression and its activation induces high levels of expression of the replication proteins E1 and E2 as well as E1ˆE4, E5 L1 and L2. Differentiation-dependent changes in polyadenylation site usage as well as alternate splicing regulate expression of the capsid proteins L1 and L2[31]. Expression of viral proteins is also regulated by microRNAs (miRNAs), however, HPVs do not encode their own miRNAs but rather modulate the expression of cellular miRNAs[32]. These cellular miRNAs regulate a variety of pathways involving proliferation, differentiation and innate immune surveillance. A large number of cellular miRNAs are altered by HPV proteins suggesting that the mechanisms regulating their expression are highly complex. Interestingly, only one miRNA, miR-145, has been shown to directly regulate viral transcription and to be critical for effects on the viral life cycle[33].
The role of high-risk E6 and E7 in viral oncogenesis
The high-risk HPVs are the causative agents of anogenital as well as some oral cancers and the expression of two viral proteins, E6 and E7, is essential for their development[34]. HPVs often integrate into host chromosomes during malignant progression retaining expression only of E6 and E7. The E6 protein interacts with a number of cellular pathways such as those regulating DNA damage repair through its interactions with p53. In normal cells, p53 acts to arrest cells following DNA damage and allow for repair. In HR-HPV infected cells, E6 recruits the cellular ubiquitin ligase UBE3A (E6AP) into a trimeric complex with p53. This results in the ubiquitination of p53 leading to its rapid turnover[34,35]. E6 proteins from the HPVs from the high-risk species also contain PDZ binding domains at their C-terminus that mediate interactions with a number of PDZ domain containing factors that control differentiation and cell polarity. These interactions are regulated by phosphorylation of the E6 PDZ domain and lead to their rapid degradation[36]. The high-risk HPVs E6 proteins bind and degrade the MAGI (membrane-associated guanylate kinase) as well as DLG and Scribble PDZ-domain containing factors implicating these factors as potentially important for oncogenesis. Other binding partners of the high-risk E6 proteins include the acetyl-transferases p300 and CREB[37], which leads to inhibition of p53 acetylation[38]. Finally, E6 proteins activate expression of the catalytic subunit of telomerase, hTert by degrading the transcriptional repressor, NFX1-191[35]. Activation of hTert is found in most human cancers and this activity appears largely restricted to the alpha viruses.
The E7 oncoprotein is co-expressed with E6 in HPV-induced cancers and the two act synergistically to promote oncogenesis. During the productive viral life cycle, these two factors are responsible for maintaining differentiating cells active in the cell cycle but also act to promote malignant progression. E7 proteins bind members of the Rb family of proteins with high-affinity which leads to the their degradation and constitutive activation of E2F transcription factors[24,25]. High-risk E7 proteins also activate the ATM DNA damage pathway, which occurs through the release of E2Fs from Rb complexes[17]. Interestingly, overexpression of E1 can also activate the DNA damage response in the absence of viral replication and acts by inducing replication from cellular pseudo-origins resulting in stalled replication forks[39]. E7 proteins also bind Cullen 2 ubiquitin ligases as well as class I histone deacetylases (HDACs) and both activities are important for the viral life cycle[25,40]. In addition to E6 and E7, the E5 proteins have been shown to induce tumors in transgenic mice suggesting they may play some role in enhancing the oncogenic activity of E6 and E7 in human cancers. This activity appears to be associated with E5’s interactions with EGF receptor[41].
E6 and E7 of the Beta HPVs
The E6 and E7 proteins of the beta HPVs share some similarities with their homologous counterparts in the HR-alpha HPVs, but also have differences. E6 proteins from both genera can target the proapoptotic protein, BAK for degradation and inhibit the apoptotic response to DNA damage[12,42]. Similarly E6 proteins from both genera can bind the transcriptional regulator and histone acetylase, p300[43,44]. However beta E6 proteins bind p300 very tightly, blocking a stabilizing phosphorylation of p300 by AKT, leading to reduced levels of p300 in beta E6 expressing cells[43]. Unlike HR HPV E6 the beta HPV E6 proteins do not target p53 for degradation. Degradation of p53 by E6 is mediated by its binding to the ubiquitin ligase, E6AP, through a LXXLL motif on E6AP[45]. The beta E6s proteins either bind E6AP very weakly or not at all. Instead the beta E6 proteins bind MAML1, which has a similar LXXLL motif[37,46]. MAML1 is a transcriptional coactivator of Notch regulated genes. The binding of beta E6 proteins to MAML1 blocks Notch signaling. One function of Notch is to promote keratinocyte differentiation and the beta E6 proteins have been reported to perturb differentiation[47].
The alpha and beta E7 proteins all contain an LXCXE motif and bind Rb and the related pocket proteins, p107 and p130[48,49], however their ability to target Rb for degradation is restricted to the HR alpha E7 proteins and an occasional beta E7 protein, such as HPV 38 E7[50]. To varying degrees the Rb/E7 interaction promotes entry into S phase. All of the E7 proteins bind to the ubiquitin ligases UBR4 and KCMF1, though the significance of these interactions for the HPV lifecycle is not known[51].
The beta E6 and E7 proteins have some activities that can promote oncogenesis. Some studies have shown that these genes either singly or in combination can extend the lifespan of keratinocytes[52]. Though most do so only weakly, HPV 38 E6/E7 was shown to immortalize cells [53,54]. Two beta types, HPVs 8 and 38, were shown to promote tumorigenesis in transgenic mice[15,55]. When either the HPV 8 early region, HPV 8 E6 or HPV 38 E6/E7 have been expressed using a K14 promoter they induced epidermal hyperplasia and at low frequency squamous cell carcinomas. Interestingly, UV greatly increased the frequency of SCSC at doses that did not cause tumors in nontransgenic littermates.
UV exposure is the major risk factor for SCSCs leading to the hypothesis that the beta HPVs might inhibit the response to UV damage, allowing cells with mutations to persist. Indeed, blocking apoptosis following UV damage has been shown for several beta HPV types [42,56]. Several beta E6 proteins have been shown to inhibit the repair of UV damage. The ability of HPV 5 and 8 to bind p300 delays the induction of ATR, ATM, their downstream targets CHK1 and CHK2, and BRCA1 and BRCA2[13,22,43]. Although DNA repair foci form they are often delayed and persist longer than it takes to resolve DNA damage in control cells.
Conclusion
Human papillomaviruses are important pathogens that are responsible for a range of effects in humans ranging from unapparent infections to cancers. All HPVs target epithelia tissues and link their productive life cycles to differentiation of the infected host cell. HPVs from different genra have distinct effects on cellular pathways such as those regulating DNA damage and cell cycle control that contribute to the types of lesions as well as cancers that they induce (Table 1). The mechanisms regulating persistent infection and progression to malignancy are fertile areas for future study.
Table 1.
Differential activities of high-risk alpha and beta papillomaviruses
| Beta HPVs | High risk Alpha HPVs |
|---|---|
| Infect cutaneous epithelia | Infects genital and oral epithelia |
| Possible association with non-melanoma cancers | Causative agents of cervical cancers |
| Differentiation-dependent life cycle | Differentiation-dependent life cycle |
| E6 does not target p53 for degradation | E6 induces p53 degradation through UBE3A |
| E7 does not affect RB levels | E7 induces degradation of Rb |
| E6 reduces ATM/ATR signaling | E7 and E1 activate ATM pathway |
| E6 fails to bind PDZ proteins | E6 binds and degrades PDZ proteins |
Highlights.
HPVs infect epithelia cells and link their life cycles to differentiation
HPVs are grouped into five genra with the alpha and beta viruses being the most commonly studied.
Alpha HPVs are the causative agents of anogenital cancers while beta HPVs have been implicated as co-factors in some skin cancers
Activation of DNA damage pathways is important for the productive life cycle of the alpha HPVs while the beta viruses suppress their activation
Acknowledgments
DAG and LAL were supported by grants from the NCI and NIAID. We thank Kristina Simanis for her artwork.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers EM. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology. 2010;401:70–79. doi: 10.1016/j.virol.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.zur Hausen H. Papillomaviruses in the causation of human cancers - a brief historical account. Virology. 2009;384:260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 3.Koutsky LA, Galloway DA, Holmes KK. Epidemiology of genital human papillomavirus infection. Epidemiol Rev. 1988;10:122–163. doi: 10.1093/oxfordjournals.epirev.a036020. [DOI] [PubMed] [Google Scholar]
- 4.Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med. 1998;338:423–428. doi: 10.1056/NEJM199802123380703. [DOI] [PubMed] [Google Scholar]
- 5.Ho GY, Burk RD, Klein S, Kadish AS, Chang CJ, Palan P, Basu J, Tachezy R, Lewis R, Romney S. Persistent genital human papillomavirus infection as a risk factor for persistent cervical dysplasia. J Natl Cancer Inst. 1995;87:1365–1371. doi: 10.1093/jnci/87.18.1365. [DOI] [PubMed] [Google Scholar]
- 6.Herrero R, Hildesheim A, Bratti C, Sherman ME, Hutchinson M, Morales J, Balmaceda I, Greenberg MD, Alfaro M, Burk RD, et al. Population-based study of human papillomavirus infection and cervical neoplasia in rural Costa Rica. J Natl Cancer Inst. 2000;92:464–474. doi: 10.1093/jnci/92.6.464. [DOI] [PubMed] [Google Scholar]
- 7.Schwarz E, Durst M, Demankowski C, Lattermann O, Zech R, Wolfsperger E, Suhai S, zur Hausen H. DNA sequence and genome organization of genital human papillomavirus type 6b. EMBO J. 1983;2:2341–2348. doi: 10.1002/j.1460-2075.1983.tb01744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Tine BA, Kappes JC, Banerjee NS, Knops J, Lai L, Steenbergen RD, Meijer CL, Snijders PJ, Chatis P, Broker TR, et al. Clonal selection for transcriptionally active viral oncogenes during progression to cancer. J Virol. 2004;78:11172–11186. doi: 10.1128/JVI.78.20.11172-11186.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9**.Howley PM, Pfister HJ. Beta genus papillomaviruses and skin cancer. Virology. 2015;479–480:290–296. doi: 10.1016/j.virol.2015.02.004. This review article summarizes the epidemiology of genus beta HPV infection of skin and describes potential oncogenic activity of the E6 and E7 proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michael KM, Waterboer T, Pfister H, Gariglio M, Majewski S, Favre M, Pawlita M. Seroreactivity of 38 human papillomavirus types in epidermodysplasia verruciformis patients, relatives, and controls. J Invest Dermatol. 2010;130:841–848. doi: 10.1038/jid.2009.356. [DOI] [PubMed] [Google Scholar]
- 11.Lutzner MA, Orth G, Dutronquay V, Ducasse MF, Kreis H, Crosnier J. Detection of human papillomavirus type 5 DNA in skin cancers of an immunosuppressed renal allograft recipient. Lancet. 1983;2:422–424. doi: 10.1016/s0140-6736(83)90389-6. [DOI] [PubMed] [Google Scholar]
- 12.Jackson S, Harwood C, Thomas M, Banks L, Storey A. Role of Bak in UV-induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes Dev. 2000;14:3065–3073. doi: 10.1101/gad.182100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallace NA, Robinson K, Howie HL, Galloway DA. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog. 2012;8:e1002807. doi: 10.1371/journal.ppat.1002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marcuzzi GP, Hufbauer M, Kasper HU, Weissenborn SJ, Smola S, Pfister H. Spontaneous tumour development in human papillomavirus type 8 E6 transgenic mice and rapid induction by UV-light exposure and wounding. J Gen Virol. 2009;90:2855–2864. doi: 10.1099/vir.0.012872-0. [DOI] [PubMed] [Google Scholar]
- 15.Viarisio D, Mueller-Decker K, Kloz U, Aengeneyndt B, Kopp-Schneider A, Grone HJ, Gheit T, Flechtenmacher C, Gissmann L, Tommasino M. E6 and E7 from beta HPV38 cooperate with ultraviolet light in the development of actinic keratosis-like lesions and squamous cell carcinoma in mice. PLoS Pathog. 2011;7:e1002125. doi: 10.1371/journal.ppat.1002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2011;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 17.Moody CA, Laimins LA. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009;5:e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18**.Gillespie KA, Mehta KP, Laimins LA, Moody CA. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J Virol. 2013;86:9520–9526. doi: 10.1128/JVI.00247-12. The homologous recombination arm of the ATM pathway is shown to be activated by HPV proteins during differentiation-dependent productive replication of alpha HPVs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19**.Mehta K, Gunasekharan V, Satsuka A, Laimins LA. Human papillomaviruses activate and recruit SMC1 cohesin proteins for the differentiation-dependent life cycle through association with CTCF insulators. PLoS Pathog. 2015;11:e1004763. doi: 10.1371/journal.ppat.1004763. The cohesion protein, SMC1, is implicated as critical for productive replication of alpha HPVs by contributing to recruitment of ATM factors as well as CTCF to viral genomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anacker DC, Gautam D, Gillespie KA, Chappell WH, Moody CA. Productive replication of human papillomavirus 31 requires DNA repair factor Nbs1. J Virol. 2014;88:8528–8544. doi: 10.1128/JVI.00517-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sowd GA, Mody D, Eggold J, Cortez D, Friedman KL, Fanning E. SV40 utilizes ATM kinase activity to prevent non-homologous end joining of broken viral DNA replication products. PLoS Pathog. 2014;10:e1004536. doi: 10.1371/journal.ppat.1004536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22**.Wallace NA, Robinson K, Howie HL, Galloway DA. beta-HPV 5 and 8 E6 disrupt homology dependent double strand break repair by attenuating BRCA1 and BRCA2 expression and foci formation. PLoS Pathog. 2015;11:e1004687. doi: 10.1371/journal.ppat.1004687. Expression of a number of proteins in the homologous recombination pathway are repressed by genus beta HPV E6 proteins, reducing the repair of double strand breaks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munger K, Jones DL. Human papillomavirus carcinogenesis: an identity crisis in the retinoblastoma tumor suppressor pathway. J Virol. 2015;89:4708–4711. doi: 10.1128/JVI.03486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helt AM, Galloway DA. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J Virol. 2001;75:6737–6747. doi: 10.1128/JVI.75.15.6737-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huh K, Zhou X, Hayakawa H, Cho JY, Libermann TA, Jin J, Harper JW, Munger K. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J Virol. 2007;81:9737–9747. doi: 10.1128/JVI.00881-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang B, Chen W, Roman A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc Natl Acad Sci U S A. 2006;103:437–442. doi: 10.1073/pnas.0510012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fehrman FK D, Laimins L. The E5 protein of HPV 31 acts to augment cell prolfilertion and activatin of differntiation-edependent late viral functions. J Virol. 2003 [Google Scholar]
- 28.Genther SS S, Duensing S, Munger K, Sattler C, Lambert P, Lambert P. Quantitaive role of HPV 16 E5 gene during the productive stage of viral life cycle. J Virol. 2003;77:2832–2842. doi: 10.1128/JVI.77.5.2832-2842.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong S, Laimins LA. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol. 2013;8:1547–1557. doi: 10.2217/fmb.13.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McBride AA. The papillomavirus E2 proteins. Virology. 2013;445:57–79. doi: 10.1016/j.virol.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz S. Papillomavirus transcripts and posttranscriptional regulation. Virology. 2013;445:187–196. doi: 10.1016/j.virol.2013.04.034. [DOI] [PubMed] [Google Scholar]
- 32.Cai X, Li G, Laimins LA, Cullen BR. Human papillomavirus genotype 31 does not express detectable microRNA levels during latent or productive virus replication. J Virol. 2006;80:10890–10893. doi: 10.1128/JVI.01175-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gunasekharan V, Laimins LA. Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J Virol. 2013;87:6037–6043. doi: 10.1128/JVI.00153-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mesri EA, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe. 2014;15:266–282. doi: 10.1016/j.chom.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katzenellenbogen RA, Vliet-Gregg P, Xu M, Galloway DA. NFX1-123 increases hTERT expression and telomerase activity posttranscriptionally in human papillomavirus type 16 E6 keratinocytes. J Virol. 2009;83:6446–6456. doi: 10.1128/JVI.02556-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ganti K, Broniarczyk J, Manoubi W, Massimi P, Mittal S, Pim D, Szalmas A, Thatte J, Thomas M, Tomaic V, et al. The Human Papillomavirus E6 PDZ Binding Motif: From Life Cycle to Malignancy. Viruses. 2015;7:3530–3551. doi: 10.3390/v7072785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White EA, Kramer RE, Tan MJ, Hayes SD, Harper JW, Howley PM. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J Virol. 2012;86:13174–13186. doi: 10.1128/JVI.02172-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev Med Virol. 2006;16:83–97. doi: 10.1002/rmv.488. [DOI] [PubMed] [Google Scholar]
- 39.McKinney CC, Hussmann KL, McBride AA. The Role of the DNA Damage Response throughout the Papillomavirus Life Cycle. Viruses. 2015;7:2450–2469. doi: 10.3390/v7052450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Longworth MS, Laimins LA. The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J Virol. 2004;78:3533–3541. doi: 10.1128/JVI.78.7.3533-3541.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maufort JP, Shai A, Pitot HC, Lambert PF. A role for HPV16 E5 in cervical carcinogenesis. Cancer Res. 2010;70:2924–2931. doi: 10.1158/0008-5472.CAN-09-3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Underbrink MP, Howie HL, Bedard KM, Koop JI, Galloway DA. E6 proteins from multiple human betapapillomavirus types degrade Bak and protect keratinocytes from apoptosis after UVB irradiation. J Virol. 2008;82:10408–10417. doi: 10.1128/JVI.00902-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Howie HL, Koop JI, Weese J, Robinson K, Wipf G, Kim L, Galloway DA. Beta-HPV 5 and 8 E6 promote p300 degradation by blocking AKT/p300 association. PLoS Pathog. 2011;7:e1002211. doi: 10.1371/journal.ppat.1002211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patel D, Huang SM, Baglia LA, McCance DJ. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999;18:5061–5072. doi: 10.1093/emboj/18.18.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huibregtse JM, Scheffner M, Howley PM. Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol Cell Biol. 1993;13:775–784. doi: 10.1128/mcb.13.2.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brimer N, Lyons C, Wallberg AE, Vande Pol SB. Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene. 2012;31:4639–4646. doi: 10.1038/onc.2011.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boxman IL, Mulder LH, Noya F, de Waard V, Gibbs S, Broker TR, ten Kate F, Chow LT, ter Schegget J. Transduction of the E6 and E7 genes of epidermodysplasia-verruciformis-associated human papillomaviruses alters human keratinocyte growth and differentiation in organotypic cultures. J Invest Dermatol. 2001;117:1397–1404. doi: 10.1046/j.0022-202x.2001.01602.x. [DOI] [PubMed] [Google Scholar]
- 48.Munger K, Werness B, Dyson N, Phelps W, Harlow E, Howley P. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barbosa MS, Vass WC, Lowy DR, Schiller JT. In Vitro biological activities of the E6 and E7 genes vary among human papillomaviruses of different oncogenic potential. Journal of Virology. 1991;65:292–298. doi: 10.1128/jvi.65.1.292-298.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caldeira S, Zehbe I, Accardi R, Malanchi I, Dong W, Giarre M, de Villiers EM, Filotico R, Boukamp P, Tommasino M. The E6 and E7 proteins of the cutaneous human papillomavirus type 38 display transforming properties. J Virol. 2003;77:2195–2206. doi: 10.1128/JVI.77.3.2195-2206.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.White EA, Sowa ME, Tan MJ, Jeudy S, Hayes SD, Santha S, Munger K, Harper JW, Howley PM. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc Natl Acad Sci U S A. 2012;109:E260–267. doi: 10.1073/pnas.1116776109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bedard KM, Underbrink MP, Howie HL, Galloway DA. The E6 oncoproteins from human betapapillomaviruses differentially activate telomerase through an E6AP-dependent mechanism and prolong the lifespan of primary keratinocytes. J Virol. 2008;82:3894–3902. doi: 10.1128/JVI.01818-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Massimi P, Zori P, Roberts S, Banks L. Differential regulation of cell-cell contact, invasion and anoikis by hScrib and hDlg in keratinocytes. PLoS One. 2012;7:e40279. doi: 10.1371/journal.pone.0040279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cornet I, Bouvard V, Campo MS, Thomas M, Banks L, Gissmann L, Lamartine J, Sylla BS, Accardi R, Tommasino M. Comparative analysis of transforming properties of E6 and E7 from different beta human papillomavirus types. J Virol. 2012;86:2366–2370. doi: 10.1128/JVI.06579-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schaper ID, Marcuzzi GP, Weissenborn SJ, Kasper HU, Dries V, Smyth N, Fuchs P, Pfister H. Development of skin tumors in mice transgenic for early genes of human papillomavirus type 8. Cancer Res. 2005;65:1394–1400. doi: 10.1158/0008-5472.CAN-04-3263. [DOI] [PubMed] [Google Scholar]
- 56.Jackson S, Storey A. E6 proteins from diverse cutaneous HPV types inhibit apoptosis in response to UV damage. Oncogene. 2000;19:592–598. doi: 10.1038/sj.onc.1203339. [DOI] [PubMed] [Google Scholar]