

Abstract
Cadmium (Cd) is a carcinogenic metal which is implicated in breast cancer by epidemiological studies. It is reported to promote breast cancer cell growth in vitro through membrane receptors. The study described here examined Cd-mediated growth of non-metastatic human breast cancer derived cells that lack receptors for estrogen, progesterone, and HER2. Treatment of triple-negative HCC 1937 cells with 0.1–0.5 μM Cd increased cell growth by activation of AKT and ERK. Accelerated cell cycle progression was achieved by increasing the levels of cyclins A, B, and E, as well as those of CDKs 1 and 2. Although triple negative cells lack estrogen receptor, they express high levels of EGFR. Therefore, further studies on HCC 1937 and another triple-negative cell line, HCC 38, were conducted using specific siRNA and an inhibitor of EGFR to determine whether EGFR was responsible for mediating the effect of Cd. The results revealed that in both cell types EGFR was not only activated upon Cd treatment, but was also essential for the downstream activation of AKT and ERK. Based on these observations, it is concluded that, in breast cancer cells lacking estrogen receptor, sub-micromolar concentration of Cd can promote cells proliferation. Furthermore, that EGFR plays a critical role in this process.
Keywords: Cadmium, Triple-negative breast cancer cells, Cell proliferation, Cell cycle, EGFR
Introduction
Cadmium (Cd) is a toxic metal which is widely distributed in the environment. The general population is exposed to this element from fuel combustion, waste burning, and cigarette smoking, as well as through dietary intake from food and polluted water (Satarug et al., 2010). Besides its acute toxicity to kidney and bone, Cd is an established Group 1 carcinogen because it causes lung cancer (Stayner et al., 1992). Retrospective and prospective epidemiology studies indicate that dietary Cd intake is also associated with increased breast cancer incidence (Julin et al., 2012; Itoh et al., 2014). Also, bioaccumulation of Cd in breast tissue of breast cancer patients is higher than in normal subjects (Romanowicz-Makowska et al., 2011; Strumylaite et al., 2011). In studies with rats, Cd was found to be a highly potent endocrine disruptor because it promoted growth of mammary gland and uterus after a single 5 μg/kg ip injection (Johnson et al., 2003).
The mechanism of breast cancer cell growth by Cd has been explored by a number of investigators. Garcia-Morales et al. (1994) reported that Cd stimulated growth of MCF-7 cells by activating estrogen receptor alpha (ERα) and inducing the expression of ERα target genes involved in cell growth. Cd was shown to bind to the ligand-binding domain of ERα in a noncompetitive manner (Stoica et al., 2000). Several other studies have also reported the proliferation of Cd in ERα-positive MCF7 and T47D cells (Martin et al., 2003; Zang et al., 2009). However, Silva et al. (2006) were unable to observe the estrogenicity of Cd in MCF7 cells by E-Screen assay. Similarly, Benbrahim-Tallaa et al. (2009) reported Cd induced malignant transformation of non-tumorigenic breast epithelial MCF10A cells by an ER-independent mechanism. Furthermore, in ERα-negative breast cancer SKBR3 cells, Yu et al. (2010) reported that Cd-induced cell growth via G protein coupled receptor 30 (GPR30). Thus, the role of ERα in facilitating the estrogenic effects of Cd in breast cancer cells is controversial. Lack of involvement of ERα in other types of cells has also been demonstrated. For example, in leiomyoma cancer ht-UtLM cells, Cd was reported to neither bind to ERα or β, or stimulate ER-induced transcriptional activity (Gao et al., 2015). Moreover, in a transgenic estrogen reporter mouse model, Cd did not induce estrogen-like effect via classical ER signaling (Ali et al., 2010).
There is a general agreement that Cd activates the mitogen signaling pathways such as mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) in breast cancer cells (Choe et al., 2003; Liu et al., 2008; Zang et al., 2009). These pathways converge signaling from various membrane receptors, including ER, GPR30, receptor tyrosine kinases (RTKs), and result in activation of genes involved in cell cycle regulation, cell proliferation and cell survival (Martin et al., 2000). Breast cancer is classified into different subtypes according to the expression of ER, progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2). Epidermal growth factor receptor (EGFR) is one of the RTKs which plays a pivotal roles in integrating hormone- and growth factor-mediated stimulation and subsequent activation of MAPK and PI3K pathways (Hoadley et al., 2007).
Although triple-negative breast cancer cells lack ER, PR, and HER2, the level of EGFR (or HER1) is amplified in this type of cancer as compared to the other breast cancer subtypes (Kao et al., 2009). Recently, our laboratory reported that even though Cd did not phosphorylate EGFR or ERα in breast cancer-derived MCF7 cells, both of these receptors were essential for the activation of ERK (Song et al., 2015). It is interesting to note that while the MCF7 cells are positive for ER, PR, and HER2, these cells have only low level of EGFR. Indeed, in recent years Cd has been implicated in EGFR-mediated MAPK activation in liver, lung and uterine cancer cells (Kundu et al., 2011; Ali et al., 2015; Gao et al., 2015). Therefore, the present study was designed to further delineate the role of EGFR in Cd-induced cell signaling and cell growth. We chose non-metastasized human ductal breast cancer cells that were triple-negative, but were known to be EGFR-positive. Thus these cells rendered themselves a good model to study the tumorigenic potential of Cd in the absence of ER.
Materials and methods
Reagents and supplies
PD 184161, Wortmannin and AG 1478 were purchased from Cayman Chemical (Ann Arbor, MI). Cd chloride, RIPA buffer, trypan blue, MTT, BSA, DMSO, PMSF and protease inhibitor were from Sigma-Aldrich (Dallas, TX). Apoptosis Detection Kit, containing propidium iodide (PI) and annexin V-FITC staining solutions was from BD Bioscience (San Jose, CA). Phosphatase inhibitor cocktail was from Roche (Indianapolis, IN). Primary antibodies for phospho-ERK (pERK; Thr 202/Tyr 204), ERK, phospho-AKT (pAKT; Ser 473), and AKT were from Cell Signaling Technology (Danvers, MA). Antibodies for phospho-EGFR (pEGFR; Tyr 845), EGFR (1005), cyclin A (H-432), cyclin B1 (M-20), cyclin D1 (H-295), cyclin E (M-20), CDK 1 (p34) and CDK 2 (H-298) were from Santa Cruz Biotechnology (Dallas, TX). Goat anti-rabbit/mouse secondary antibodies were from Li-Cor (Lincoln, NE). EGFR siRNA (siGENOME Human EGFR 1956 – SMARTpool, catalog #M-003114-03-0005), nonsense siRNA (catalog #D-001206-01-05), transfection reagent, and RNase were from Thermo Scientific (Pittsburgh, PA). RPMI 1640 medium and supplements were from Life Technologies (Grand Island, NY). Fetal bovine serum (FBS) was from Atlanta Biologicals (Flowery Branch, GA). Human breast cancer cells HCC 1937 and HCC 38 were from ATCC (Manassas, VA). Cell counting slides were from Nexcelom Bioscience (Lawrence, MA).
Cell proliferation
The HCC 1937 cells were seeded into 12-well plates at a density of 1×105 cells/well and cultured in phenol red-free RPMI 1640 medium supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and non-essential amino acids at 37°C and 5% CO2 for 24 h. To deplete the growth factors, the medium was changed to phenol red-free RPMI 1640 supplemented with 0.2% BSA instead of FBS and the cells were cultured for 48 h. The serum-starved cells were treated with 0.05 – 1.0 μM CdCl2 or 1 ng/mL EGF in 0.2 % BSA-supplemented phenol-red free medium, with a change of medium after 2 days. At the end of 4 days, the cells were harvested and stained with 10% trypan blue solution. The stained cells were counted using the Cellometer (Nexcelom Biosciences, Lawrence, MA).
Cell proliferation was also determined by the MTT assay. In this case, the HCC 1937 cells were seeded into 24-well plates at a density of 8×103 cells/well and cultured, serum-starved and treated with Cd and EGF as described above. After 4 days of treatment, the cells were incubated with 10% MTT solution (5 mg/mL). The solution was aspirated after 3 h and the insoluble formazan formed in the cells was dissolved by adding DMSO. Cell proliferation was determined by measuring the absorbance at 570 nm and subtracting the background absorbance at 690 nm.
Apoptosis analysis
The HCC 1937 cells were serum-starved to deplete growth factors and treated with 0.1 or 0.5 μM CdCl2 for 48 h as mentioned above. Cells incubated with BSA alone or with FBS served as negative and positive controls, respectively. After completion of incubation, the medium was aspirated and saved and the cells were harvested by using trypsin. Once harvested, the cells were returned to the aspirated medium to stop the activity of trypsin and centrifuged. The cell pellet was re-suspended and stained with annexin V-FITC and PI for 15 min. The stained cells were analyzed for apoptosis using a flow cytometer (FACSVerse, BD Biosciences, CA).
Cell cycle analysis
Serum-starved HCC1937 cells were retreated with 0.5 μM CdCl2 for up to 48 h. Cells cultured in BSA alone or FBS were the negative and positive controls, respectively. At the end of 12, 24, 36, and 48 h treatment, the cells were harvested and fixed overnight in 70% ethanol. For measuring DNA content by flow cytometry, the cells were washed with phosphate-buffered saline (PBS) and incubated with a solution of 18 μg/mL PI and 40 μg/mL RNase A for 40 min.
Transfection with siRNA
The HCC 1937 and HCC 38 cells were cultured in RPMI 1640 medium containing 10% FBS and transfected with 2 μL EGFR siRNA (20 μM) and 2 μL transfection reagent for 48 h. Cells transfected with nonsense siRNA were used as negative control. The transfected cells were serum-starved in 0.2% BSA-supplemented phenol red-free medium for 12 h before treatment with Cd or EGF.
Western blot analysis
To determine the expression of cyclins and CDKs, serum-starved cells were treated with CdCl2 for 48 h in 0.2 % BSA-supplemented phenol red-free RPMI 1640 medium. For determination of ERK and AKT phosphorylation, the serum-starved cells were treated with CdCl2 in serum-free medium for 15 min or 6 h, respectively. Phosphorylation of EGFR was also determined after 15 min incubation with Cd. The cells were washed with PBS before harvest and lysed in RIPA buffer supplemented with protease inhibitor, 2 mM PMSF, and phosphatase inhibitor. The cell lysates were centrifuged at 14,000 × g for 10 min and proteins in the supernatant were separated by electrophoresis. Protein bands were transferred from the gel to the nitrocellulose membrane in an SD Semi-dry Transfer Cell (Bio-Rad, Hercules, CA) at 24 V for 1 h. The membrane was blocked in 5% non-fat milk for 1 h and incubated with corresponding primary antibody at 4°C. After overnight incubation, the membrane was washed three times with 0.1% PBS-Tween 20 for 10 min each and incubated, with the goat anti-rabbit/mouse secondary antibody (1:10,000 dilution). The membrane was washed again three times for 10 min each with PBS-Tween and scanned in an Odyssey Infrared Imager (Li-Cor, Lincoln, NE).
Data analysis
All experiments were repeated at least three times. Flow cytometry data were analyzed by ModFit LT Software. Western blot data were analyzed and quantified by the Odyssey Infrared Imaging Software. One-way ANOVA and Tukey’s post hoc test were performed using SPSS and p < 0.05 was considered statistically significant.
Results
Cd treatment promotes HCC 1937 cell growth
In order to establish that Cd promoted the growth of ERα-negative breast cancer cells, the HCC 1937 cells, derived from a triple-negative human breast cancer, were used in this study. Since Cd is not a potent mitogen, its effect on cell growth could be masked by other more potent growth factors. Thus the cells were serum-starved for 48 h prior to Cd and EGF treatment in order to deplete potential growth promoters. As shown in Fig. 1A, there were 2.37×105 live cells when the cells were cultured with BSA alone for 4 days following the initial serum starvation. Cd treatment, similar to EGF, had a growth promoter effect on the cells and at 0.5 μM Cd concentration the cell number increased by about 50% to 3.57×105. In comparison, cells cultured in the presence of FBS had 82% more cells than in the presence of BSA (data not shown).
Fig. 1.

Effect of Cd treatment on growth of HCC 1937 cells. The cells were serum-starved for 48 h before being treated with CdCl2 or 1 ng/mL EGF for 4 days. (A) The harvested cells were stained with trypan blue and live cells were counted. Counted cell number from three independent experiments are plotted as mean ± SD (n=3). (B) Cell growth was measured by the MTT assay and cell proliferation relative to the BSA control was plotted. Data from three independent experiments are plotted as mean ± SD (n=3). *Significantly different from the untreated control group (p < 0.05).
Result obtained from the MTT assay had similar trend and validated the observation that at low levels Cd treatment resulted in cell proliferation in a concentration-dependent manner. As depicted in Fig. 1B, the MTT assay showed that at 0.5 μM Cd concentration, there was 36% increase in cell proliferation over the BSA control. Higher concentration of Cd (1.0 μM) did not promote cell growth and was potentially cytotoxic. Thus, these results demonstrated that, in triple-negative HCC 1937 breast cancer cells, sub-micromolar concentrations of Cd promote cell growth even in the absence of ERα. Based on these results, most of the studies described below were carried out at 0.5 μM Cd.
Cd treatment is not anti-apoptotic
To evaluate whether the increase in cell number observed upon low level Cd treatment could be due to a reduction in apoptosis, the serum-starved cells were cultured in the presence of 10% FBS or 0.2% BSA with or without Cd. After 4 days the cells were harvested and stained with annexin V-FITC and PI and analyzed for apoptosis by flow cytometry (Fig. 2A and B). Based on the staining characteristics, live cells appeared in the lower left quadrant and cells undergoing apoptosis appeared in the lower right quadrant (Fig. 2A). As expected, cells cultured in the presence of FBS had the least number of apoptotic cells (5%). In comparison, 24% of the cells cultured in the presence of BSA were apoptotic. The proportion of cells undergoing apoptosis remained unchanged when Cd was also present (Fig. 2B). Thus, cell proliferation in response to Cd treatment was not due to prevention of apoptosis.
Fig. 2.

Quantification of apoptosis in HCC 1937 cells by flow cytometry. The cells were serum-starved for 48 h before being treated with CdCl2 for 4 days. Cells cultured in FBS and BSA were positive and negative controls, respectively. Cells were harvested and stained with Annexin V-FITC/PI. (A) Representative contour plots of the distribution of cells according to PI staining (y-axis) versus annexin V-FITC staining (x-axis). Live cells were concentrated in the lower left quadrant and cells undergoing apoptosis appeared in the lower right quadrant. (B) Apoptotic cell population following various treatments. Data from three independent experiments are plotted as mean ± SD (n=3). *Significantly different from the FBS positive control group (p < 0.05).
Cd promotes cell cycle progression
To further establish that Cd promoted the growth of HCC 1937 cells through cell cycle progression, the cells were first synchronized by 48 h serum starvation and then treated with 0.5 μM Cd for up to 48 h. Cells grown in the presence of 10% FBS or 0.2% BSA served as positive and negative controls, respectively. As shown in Fig. 3A and B, whereas cells grown in the presence of FBS continued to progress through the cell cycle, those cultured in the presence of BSA remained static. However, with the addition of Cd, the cell cycle was stimulated in cells cultured in BSA to about the same extent as in cells cultured with FBS. Over the 48 h period, Cd treatment resulted in a significant decrease in the cell population that was in S phase, with a concomitant increase in the population in G2/M phase. At 48 h, 52% more cells were in the G2/M phase in the Cd-treated group than in the BSA control group. This marked change in cell distribution from the S to G2/M phase of the cell cycle indicated that Cd treatment caused the progression of cells through the cell cycle.
Fig. 3.

Effect of Cd treatment on progression of cell cycle in HCC 1937 cells. The cells were serum-starved before being treated with 0.5 μM CdCl2 in 0.2% BSA. After incubation for 12, 24, 36 or 48 h, the cells were harvest and DNA was stained with PI. Untreated cells and cells incubated with FBS served as negative and positive controls, respectively. (A) Representative plot of relative distribution of cells in various phases of cell cycle showing PI staining (y-axis) versus DNA content (x-axis). (B) Relative distribution of cells in various phases of cell cycle at different times after treatment is plotted as mean ± SD (n = 6). *Significantly different from the untreated control group (p < 0.05).
Cd increases expression of cyclins and CDKs
Cell cycle progression is precisely regulated by periodic expression of phase-specific heterodimeric protein kinases consisting of cyclins (regulatory subunit) and CDKs (catalytic subunit). Cyclin D/CDK 4 and 6 complex prompts cells to enter cell cycle and drive cells going through G1 phase. Cyclin E/CDK 2 complex is responsible for G1/S transition, and cyclin A/CDK 2 and cyclin B/CDK 1 drive the cells through S and G2 phases, respectively. Therefore, to investigate the mechanism of Cd-induced cell cycle progression, the expression of various cyclins and CDKs was examined in serum-starved cells treated with 0.05–1.0 μM Cd for 48 h. The Western blot results summarized in Fig. 4A and B show that Cd treatment significantly increased the expression of cyclins A, B, and E, but not that of cyclin D. Also, whereas the expression of CDK 1 was significantly elevated at 0.1–1.0 μM Cd, CDK 2 expression was significantly elevated only at 0.5 μM Cd. These observations agreed with the cell cycle data presented in Fig. 3A and B which showed that Cd treatment accelerated the progression of HCC 1937 cells into G2/M phase.
Fig. 4.

Effect of Cd treatment on the expression of cyclins and CDKs in HCC 1937 cells. The cells were serum-starved and treated with 0.05–1.0 μM CdCl2 in 0.2% BSA for 48 h. Untreated cells and cells grown in the presence of FBS served as negative and positive controls, respectively. (A) Representative Western blot of the expression of cyclins A, B, D, and E and CDKs 1 and 2. (B) Density relative to the untreated control group after various treatments is plotted as mean ± SD (n = 3). *Significantly different from the untreated control group (p < 0.05).
Cd induces cell cycle progression through EGFR
Cd-induced mitogenic effect has been reported previously in ERα-positive breast cancer cells. The HCC 1937 cells used in the present study were ERα-negative, but EGFR positive. To examine whether EGFR alone played a role in Cd-induced cell cycle progression in the triple-negative cells, the serum-starved cells were treated for 48 h with 0.5 μM Cd in 0.2% BSA, with or without the EGFR inhibitor AG 1478. Cells treated with EGF served as positive controls. As depicted in Fig. 5A and B, both Cd and EGF treatments significantly increased the cell population in G2/M phase (56 and 96%, respectively). However, in the presence of EGF inhibitor, Cd had no significant effect on cell cycle progression. The importance of EGFR in Cd-induced expression of various cyclins and CDKs was also studied (Fig. 6A and B). As with the cell cycle, AG 1478 also blocked the Cd-induced increase in the expression of cyclins A, B, and E, as well as CDKs 1 and 2. These results demonstrate that in the triple-negative breast cancer cells EGFR is critical in mediating the effect of Cd on cell cycle progression.
Fig. 5.

Effect of inhibition of EGFR on progression of cell cycle in HCC 1937 cells. The cells were serum-starved and treated with 0.5 μM CdCl2 in 0.2% BSA, with or without AG 1478 for 48 h and DNA was stained with PI. Untreated cells and cells treated with EGF served as negative and positive controls, respectively. (A) Representative plot of relative distribution of cells in various phases of cell cycle showing PI staining (y-axis) versus DNA content (x-axis). (B) Relative distribution of cells in G2/M phase after various treatments is plotted as mean ± SD (n = 3). *Significantly different from the untreated control group (p<0.05).
Fig. 6.

Effect of inhibition of EGFR on the expression of cyclins and CDKs in HCC 1937 cells. The cells were serum-starved and treated with 0.5 μM CdCl2 in 0.2% BSA, with or without AG 1478 for 48 h. Untreated cells and cells treated with EGF served as negative and positive controls, respectively. (A) Representative Western blot of the expression of cyclins A, B, and E and CDKs 1 and 2. (B) Density relative to the untreated control group is plotted as mean ± SD (n =3). *Significantly different from the untreated control group (p < 0.05).
Cd-induces cell cycle progression through MAPK and PI3K
MAPK and PI3K signaling pathways are downstream to EGFR and regulate cell cycle progression and cell proliferation. In Cd-treated breast cancer cells, both ERK 1/2 and AKT are phosphorylated (Liu et al., 2008). To test whether inhibition of these kinases would block the Cd-induced cell cycle progression, inhibitors of MEK (PD 184161) and PI3K (Wortmannin) were utilized. As shown in Fig. 7A and B, both inhibitors prevented the Cd-induced increases in the expression of cyclins A, B, and E, and CDKs 1 and 2. These results indicated that the activation of both MAPK and PI3K pathways was a required intermediary step in signal transduction for Cd-induced cell cycle progression.
Fig. 7.

Effect of inhibition of ERK and AKT on the expression of cyclins and CDKs in HCC 1937 cells. The serum-starved cells were treated with 0.5 μM CdCl2 in 0.2% BSA, in the presence or absence of PD 184161 or Wortmannin for 48 h. (A) Representative Western blot of the expression of cyclins A, B, and E and CDKs 1 and 2. (B) Density relative to the untreated control group is plotted as mean ± SD (n =3). *Significantly different from the untreated control group (p < 0.05).
Cd-induces activation of MAPK and PI3K through EGFR
EGFR is a key membrane receptor in signal transduction through the MAPK and PI3K pathways. To identify the contribution of EGFR in Cd-induced MAPK and PI3K activation, the EGFR inhibitor AG 1478 was utilized. The time course of ERK 1/2 and AKT phosphorylation by Cd (data not shown) revealed that maximum activation of these transcription factors in the HCC 1937 cells was at 15 min and 6 h, respectively.
Besides lacking ER, the HCC 1937 cells have a mutation in BRCA1 gene. Wild type BRCA1 reportedly inhibits estradiol- as well as EGF-induced activation of ERK and cell proliferation, but not the activation of AKT (Razandi et al., 2004). To examine whether the BRCA1 mutation had any effect on the Cd-induced activation of ERK and AKT, HCC 38 cells containing wild type BRCA1 were compared with the HCC 1937 cells. The cells were serum-starved and treated for either 15 min or 6 h with 0.5 μM Cd or 1 ng/mL EGF in the presence or absence of 2 μM AG 1478 for 48 h. As depicted in Fig. 8A and B, in both cell lines Cd as well as EGF increased phosphorylation of ERK and AKT and this effect was blocked by inhibition of EGFR. Thus, these results show that BRCA1 mutation in the HCC 1937 cells does not affect EGFR-mediated activation of ERK and AKT by Cd.
Fig. 8.
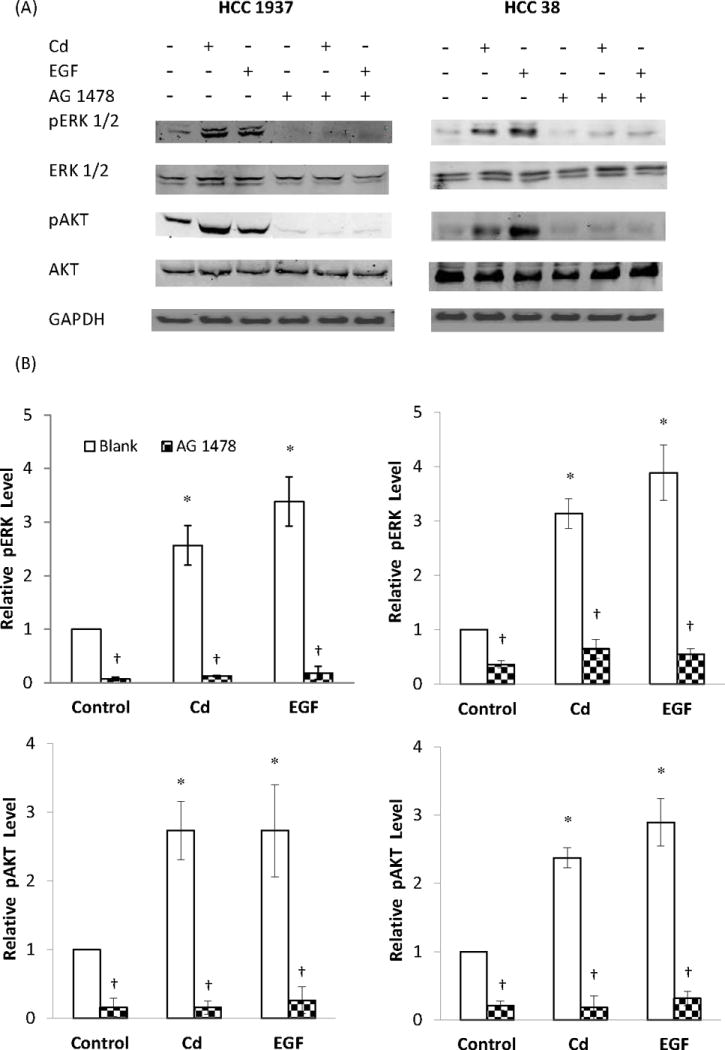
Effect of inhibition of EGFR on ERK and AKT phosphorylation in HCC 1937 and HCC 38 cells. The serum-starved cells were treated with 0.5 μM CdCl2 in 0.2% BSA in the presence or absence of AG 1478 for either 15 min or 6 h. Untreated cells and cells treated with EGF served as negative and positive controls, respectively. (A) Representative Western blot of the phosphorylated ERK and AKT. (B) Density relative to the untreated control is plotted as mean ± SD (n = 3). *Significantly different from the untreated control group (p < 0.05). †Significantly different from the respective non-AG 1478 treated blank group (p < 0.05).
EGFR’s role in Cd-induced ERK 1/2 phosphorylation was further studied in both HCC 1937 and HCC 38 cells by knocking down EGFR expression with siRNA as well as by inhibition of EGFR with AG 1478. The cells were serum-starved and treated with 0.5 or 1.0 μM Cd, or 1 ng/mL EGF for 15 min. As shown in Fig. 9A and B, in both cell types, when the expression of EGFR was knocked down by transfection with siRNA, or its activity was blocked by the inhibitor, not only the Cd-induced phosphorylation of EGFR disappeared, but also that of ERK. These results further confirm that phosphorylation of EGFR is a prerequisite for Cd-induced activation of MAPK.
Fig. 9.
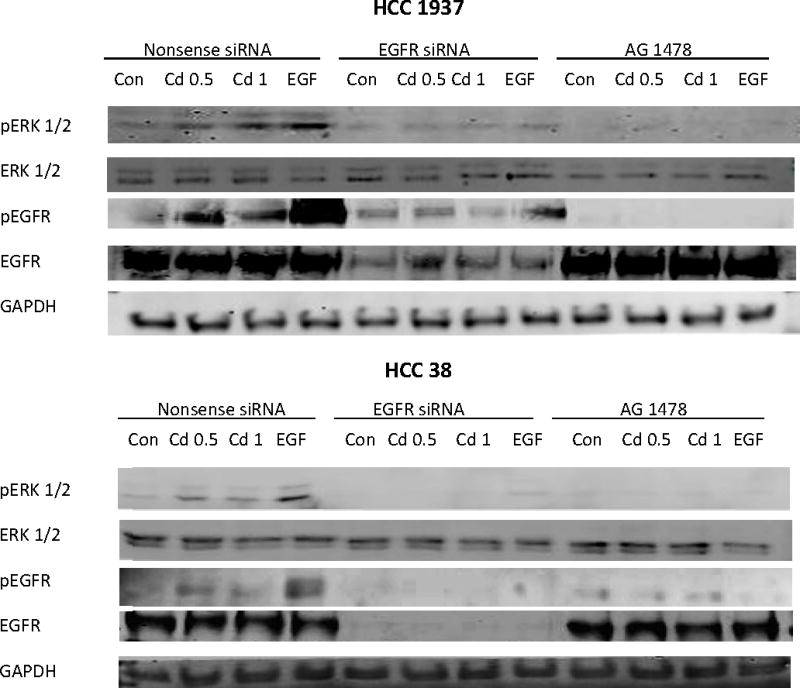
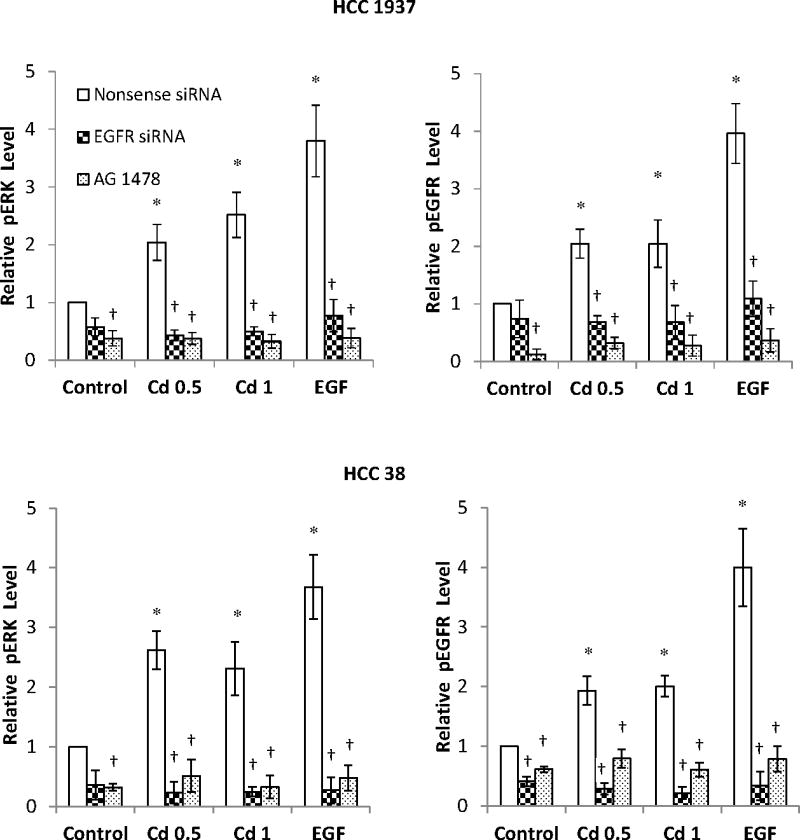
Effect of knock down of EGFR expression by siRNA on the phosphorylation of EGFR and ERK in HCC 1937 and HCC 38 cells. The cells were transfected with EGFR siRNA for 48 h and serum-starved for 12 h prior to treatment with 0.5 or 1.0 μM CdCl2 in 0.2% BSA for 15 min. Cells transfected with either nonsense siRNA or EGFR siRNA alone, or treated with AG 1478 alone served as the respective negative controls. Cells treated with EGF served as positive controls. (A) Representative Western blot of the phosphorylated EGFR and ERK. (B) Density relative to the nonsense siRNA control is plotted as mean ± SD (n = 3). *Significantly different from the nonsense siRNA control group (p < 0.05). †Significantly different from the respective nonsense siRNA group (p < 0.05).
Discussion
Cd treatment produces cell cycle arrest at 10 μM and higher concentrations (Yang et al., 2004; Xie and Shaikh, 2006). However, more recent studies in prostate and lung epithelial cells, mesangial cells, and breast cancer MCF-7 cells show that lower concentrations of Cd actually promote cell growth (Bakshi et al., 2008; Kundu et al., 2011). Similar to these reports, the present study showed that sub-micromolar concentration of Cd increased the growth of breast cancer HCC 1937 cells. Furthermore, Cd did so by regulating MAPK and P13K activation, and increasing expression of cyclins and CDKs. Inhibition of MAPK by PD 184161, and PI3K by Wortmannin, suppressed the expression cyclins and CDKs. This study further examined the involvement of EGFR in Cd-induced signal transduction and cell cycle progression by the use of an EGFR inhibitor and by siRNA knockdown. Both treatments abolished the Cd-induced activation of MAPK and PI3K pathways not only in the HCC 1937 cells, but also in HCC 38 cells, thus establishing the importance of EGFR in this process.
A number of studies report that metals such as chromium, manganese, nickel, arsenic and lead activate Ras/MAPK pathways (Tessier and Pascal, 2006; Andrew et al., 2009; Chan et al., 2015). Cd-induced activation of MAPK and PI3K has also been reported in the ERα-positive cells and this effect is attributed to the xenoestrogenic effect of Cd (Brama et al., 2007; Ponce et al., 2013). The present study is the first to report the proliferative effect of low level Cd treatment on triple-negative breast cancer cells which are ERα negative. About 20% of the breast cancer patients have triple-negative receptor phenotype (ER−, PR−, HER2−). These patients are resistant to hormone therapy and have the highest mortality rate and worst outcome. EGFR is an important biomarker in clinical therapy for this group of patients (Chen and Russo, 2011). In preclinical studies, suppressing triple-negative breast cancer cell growth by inhibition of EGFR is observed in both monoclonal antibody and small molecule inhibitor treatments (Oliveras-Ferraros et al., 2008). Non-metastasized ductal carcinoma with triple-negative receptor phenotype tends to proceed to invasive breast cancer (Bryan et al., 2006). HCC cell lines are epithelial cells derived from ductal carcinoma of breast tissue. The HCC 1937 and HCC 38 cells used in the present study were derived from two patients with different stage IIB, grade 3 breast cancer. Both cell lines are reported to respond about equally to EGF-induced phosphorylation of ERK and AKT (Niepel, et al., 2014). The results presented here show that, in addition to EGF, these cells are also responsive to Cd-induced activation of ERK and AKT. This finding is important in that it establishes a strong association between EGFR and Cd exposure in promoting the growth of triple-negative breast cancer cells. By linking EGFR and its downstream signaling to Cd-induced cell cycle promotion, this study further strengthens the necessity of managing Cd exposure in triple-negative breast cancer patients.
Progression into G2/M phase requires precise control of cell cycle machinery, which is regulated by kinases involved in signal transduction through a series of steps. It is well accepted that major cell cycle regulatory pathways are well conserved (Klein and Assoian, 2008; Ma et al., 2010). Activation of MAPK pathway leads to enhanced activity of transcription factors such as Myc, AP-1, E2F1 and STAT3 (Lo et al., 2005; Hanada et al., 2006). These transcription factors promote expression of cyclins and CDKs. Cyclin A/CDK 2 and cyclin B/CDK 1 complexes sequentially phosphorylate FoxM1 to translocate into the nucleus and to transcribe executor of mitosis, such as cyclin B, and centromere protein F (Major et al., 2004; Laoukili et al., 2008; Chen et al., 2009). Activation of PI3K is commonly regarded as an anti-apoptotic signal because AKT phosphorylation induces anti-apoptotic protein B-cell lymphoma 2 (Bcl-2) and survivin expression (Siddiqa et al., 2008). The sustained AKT activation contributes to cell survival by directly inactivating caspase family (Longo et al., 2008). The activation of PI3K/AKT pathway causes the cell to progress into late G2 phase, but the mechanism is not fully understood (Ornelas et al., 2013).
The results obtained in the present study suggest that Cd is an endocrine disruptor in the triple-negative breast cancer cells since it activated EGFR and downstream pathways which promoted cell cycle progression and cell proliferation. Recent reports in other cell types support our finding that Cd-induced cell proliferation is mediated through EGFR, however, the underlying mechanisms of Cd-activated EGFR downstream pathways seem to differ between various cell types. For example, the human lung and airway epithelial cells respond to Cd independent of endocrine-relevant pathways (Kundu et al., 2011). In these cells, Cd activates EGFR and promotes an initial inflammatory response. This leads to the up-regulation of NFκB, activation of AKT and STAT3, and increased expression of cyclin D1. The end result is cell cycle progression and cell proliferation. Also in human airway epithelial cells, another study reported that Cd treatment caused reactive oxygen species generation, upregulating HIF-1 and VEGF expression which is involved in tumorigenesis and angiogenesis (Jing et al., 2012). Growth factor receptors such as EGFR, HGFR, and VEGFR-R1, but not ERα, were responsible for Cd-induced MAPK activation in human uterine cancer leiomyoma cells (Gao et al., 2015). Activation of EGFR and downstream phosphorylation of Src, ERK and AKT has also been reported in rat mesangial cells upon Cd treatment (Xiao et al., 2009).
EGFR is a member of the HER family. When a ligand binds to EGFR (HER 1), it either undergoes homodimerization or heterodimerizes with other members of the HER family. Dimerization triggers autophosphorylation in the C-terminal residues and serves as a docking site for adaptor proteins. Although Cd is not a specific ligand for EGFR, it has the potential to interact with a number of amino acid residues in the receptor. In so doing, it is likely that Cd alters the conformation of EGFR and promotes the receptor-receptor contacts which result in dimerization. In the ectodomain crystals of human EGFR (PDB: 1MOX), presence of 11 Cd2+ ions has been reported by Garrett et al. (2002), suggesting that Cd has ample binding sites on EGFR. Further study is needed to delineate the nature of these interactions.
In summary, the present study demonstrates that sub-micromolar concentrations of Cd cause proliferation of triple-negative breast cancer cells which lack ERα, and it does so by regulating the cell cycle machinery through EGFR mediated cell signaling.
Highlights.
Sub-micromolar concentrations of Cd promote cell growth in breast cancer cells that lack ER, PR, and HER2.
The increase in cell number is not due to reduction in apoptosis.
Growth promotion involves AKT and ERK signaling and downstream stimulation of cell cycle progression.
Initiation of cell growth by Cd occurs at the cell membrane and requires the activation of EGFR.
Acknowledgments
The authors acknowledge partial salary support for ZW and the use of Centralized Research Core Facility of the RI Institutional Development Award (IDeA) Network of Biomedical Research Excellence (RI-INBRE) Program supported by grant number P20GM103430 from the National Institute of General Medical Sciences at the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Zhengxi Wei, Email: weizhengxi@gmail.com.
Zahir A. Shaikh, Email: zshaikh@uri.edu.
References
- Ali I, Damdimopoulou P, Stenius U, Halldin K. Cadmium at nanomolar concentrations activates Raf-MEK-ERK1/2 MAPKs signaling via EGFR in human cancer cell lines. Chem Biol Interact. 2015;231:44–52. doi: 10.1016/j.cbi.2015.02.014. [DOI] [PubMed] [Google Scholar]
- Ali I, Penttinen-Damdimopoulou PE, Makela SI, Berglund M, Stenius U, Akesson A, Hakansson H, Halldin K. Estrogen-like effects of cadmium in vivo do not appear to be mediated via the classical estrogen receptor transcriptional pathway. Environ Health Perspect. 2010;118:1389–1394. doi: 10.1289/ehp.1001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew AS, Mason RA, Memoli V, Duell EJ. Arsenic activates EGFR pathway signaling in the lung. Toxicol Sci. 2009;109:350–357. doi: 10.1093/toxsci/kfp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakshi S, Zhang X, Godoy-Tundidor S, Cheng RY, Sartor MA, Medvedovic M, Ho SM. Transcriptome analyses in normal prostate epithelial cells exposed to low-dose cadmium: oncogenic and immunomodulations involving the action of tumor necrosis factor. Environ Health Perspect. 2008;116:769–776. doi: 10.1289/ehp.11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Tokar EJ, Diwan BA, Dill AL, Coppin JF, Waalkes MP. Cadmium malignantly transforms normal human breast epithelial cells into a basal-like phenotype. Environ Health Perspect. 2009;117:1847–1852. doi: 10.1289/ehp.0900999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brama M, Gnessi L, Basciani S, Cerulli N, Politi L, Spera G, Mariani S, Cherubini S, d’Abusco AS, Scandurra R, Migliaccio S. Cadmium induces mitogenic signaling in breast cancer cell by an ERalpha-dependent mechanism. Mol Cell Endocrinol. 2007;264:102–108. doi: 10.1016/j.mce.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Bryan BB, Schnitt SJ, Collins LC. Ductal carcinoma in situ with basal-like phenotype: a possible precursor to invasive basal-like breast cancer. Mod Pathol. 2006;19:617–621. doi: 10.1038/modpathol.3800570. [DOI] [PubMed] [Google Scholar]
- Chan CP, Tsai YT, Chen YL, Hsu YW, Tseng JT, Chuang HY, Shiurba R, Lee MH, Wang JY, Chang WC. Pb2+ induces gastrin gene expression by extracellular signal-regulated kinases 1/2 and transcription factor activator protein 1 in human gastric carcinoma cells. Environ Toxicol. 2015;30:129–136. doi: 10.1002/tox.21878. [DOI] [PubMed] [Google Scholar]
- Chen JQ, Russo J. Potential Roles of ERb, GPR-30/EGFR, and ERR in -pathogenesis of ERa-negative and triple-negative breast cancer. Eur J Clin Med Oncol. 2011;3:50–62. [Google Scholar]
- Chen YJ, Dominguez-Brauer C, Wang Z, Asara JM, Costa RH, Tyner AL, Lau LF, Raychaudhuri P. A conserved phosphorylation site within the forkhead domain of FoxM1B is required for its activation by cyclin-CDK1. J Biol Chem. 2009;284:30695–30707. doi: 10.1074/jbc.M109.007997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe SY, Kim SJ, Kim HG, Lee JH, Choi Y, Lee H, Kim Y. Evaluation of estrogenicity of major heavy metals. Sci Total Environ. 2003;312:15–21. doi: 10.1016/S0048-9697(03)00190-6. [DOI] [PubMed] [Google Scholar]
- Gao X, Yu L, Moore AB, Kissling GE, Waalkes MP, Dixon D. Cadmium and proliferation in human uterine leiomyoma cells: evidence of a role for EGFR/MAPK pathways but not classical estrogen receptor pathways. Environ Health Perspect. 2015;123:331–336. doi: 10.1289/ehp.1408234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Morales P, Saceda M, Kenney N, Kim N, Salomon DS, Gottardis MM, Solomon HB, Sholler PF, Jordan VC, Martin MB. Effect of cadmium on estrogen receptor levels and estrogen-induced responses in human breast cancer cells. J Biol Chem. 1994;269:16896–16901. [PubMed] [Google Scholar]
- Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, Zhu HJ, Walker F, Frenkel MJ, Hoyne PA, Jorissen RN, Nice EC, Burgess AW, Ward CW. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell. 2002;110:763–773. doi: 10.1016/s0092-8674(02)00940-6. [DOI] [PubMed] [Google Scholar]
- Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y, Hung MC. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol Carcinog. 2006;45:10–17. doi: 10.1002/mc.20147. [DOI] [PubMed] [Google Scholar]
- Hoadley KA, Weigman VJ, Fan C, Sawyer LR, He X, Troester MA, Sartor CI, Rieger-House T, Bernard PS, Carey LA, Perou CM. EGFR associated expression profiles vary with breast tumor subtype. BMC Genomics. 2007;8:258. doi: 10.1186/1471-2164-8-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh H, Iwasaki M, Sawada N, Takachi R, Kasuga Y, Yokoyama S, Onuma H, Nishimura H, Kusama R, Yokoyama K, Tsugane S. Dietary cadmium intake and breast cancer risk in Japanese women: a case-control study. Int J Hyg Environ Health. 2014;217:70–77. doi: 10.1016/j.ijheh.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Jing Y, Liu LZ, Jiang Y, Zhu Y, Guo NL, Barnett J, Rojanasakul Y, Agani F, Jiang BH. Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol Sci. 2012;125:10–19. doi: 10.1093/toxsci/kfr256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MD, Kenney N, Stoica A, Hilakivi-Clarke L, Singh B, Chepko G, Clarke R, Sholler PF, Lirio AA, Foss C, Reiter R, Trock B, Paik S, Martin MB. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nat Med. 2003;9:1081–1084. doi: 10.1038/nm902. [DOI] [PubMed] [Google Scholar]
- Julin B, Wolk A, Bergkvist L, Bottai M, Akesson A. Dietary cadmium exposure and risk of postmenopausal breast cancer: a population-based prospective cohort study. Cancer Res. 2012;72:1459–1466. doi: 10.1158/0008-5472.CAN-11-0735. [DOI] [PubMed] [Google Scholar]
- Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J, Kwei KA, Hernandez-Boussard T, Wang P, Gazdar AF, Minna JD, Pollack JR. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One. 2009;4:e6146. doi: 10.1371/journal.pone.0006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein EA, Assoian RK. Transcriptional regulation of the cyclin D1 gene at a glance. J Cell Sci. 2008;121:3853–3857. doi: 10.1242/jcs.039131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu S, Sengupta S, Bhattacharyya A. EGFR upregulates inflammatory and proliferative responses in human lung adenocarcinoma cell line (A549), induced by lower dose of cadmium chloride. Inhal Toxicol. 2011;23:339–348. doi: 10.3109/08958378.2011.572931. [DOI] [PubMed] [Google Scholar]
- Laoukili J, Alvarez M, Meijer LA, Stahl M, Mohammed S, Kleij L, Heck AJ, Medema RH. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol Cell Biol. 2008;28:3076–3087. doi: 10.1128/MCB.01710-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Yu X, Shaikh ZA. Rapid activation of ERK1/2 and AKT in human breast cancer cells by cadmium. Toxicol Appl Pharmacol. 2008;228:286–294. doi: 10.1016/j.taap.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y, Bartholomeusz G, Shih JY, Hung MC. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Longo PG, Laurenti L, Gobessi S, Sica S, Leone G, Efremov DG. The Akt/Mcl-1 pathway plays a prominent role in mediating antiapoptotic signals downstream of the B-cell receptor in chronic lymphocytic leukemia B cells. Blood. 2008;111:846–855. doi: 10.1182/blood-2007-05-089037. [DOI] [PubMed] [Google Scholar]
- Ma RY, Tong TH, Leung WY, Yao KM. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1. Methods Mol Biol. 2010;647:113–123. doi: 10.1007/978-1-60761-738-9_6. [DOI] [PubMed] [Google Scholar]
- Major ML, Lepe R, Costa RH. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol Cell Biol. 2004;24:2649–2661. doi: 10.1128/MCB.24.7.2649-2661.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MB, Franke TF, Stoica GE, Chambon P, Katzenellenbogen BS, Stoica BA, McLemore MS, Olivo SE, Stoica A. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology. 2000;141:4503–4511. doi: 10.1210/endo.141.12.7836. [DOI] [PubMed] [Google Scholar]
- Martin MB, Reiter R, Pham T, Avellanet YR, Camara J, Lahm M, Pentecost E, Pratap K, Gilmore BA, Divekar S, Dagata RS, Bull JL, Stoica A. Estrogen-like activity of metals in MCF-7 breast cancer cells. Endocrinology. 2003;144:2425–2436. doi: 10.1210/en.2002-221054. [DOI] [PubMed] [Google Scholar]
- Niepel M, Hafner M, Pace EP, Chung M, Chai DH, Zhou L, Muhlich JL, Schoeberl B, Sorger PK. Analysis of growth factor signaling in genetically diverse breast cancer cell lines. BMC Biology. 2014;12:20. doi: 10.1186/1741-7007-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveras-Ferraros C, Vazquez-Martin A, Lopez-Bonet E, Martin-Castillo B, Del Barco S, Brunet J, Menendez JA. Growth and molecular interactions of the anti-EGFR antibody cetuximab and the DNA cross-linking agent cisplatin in gefitinib-resistant MDA-MB-468 cells: new prospects in the treatment of triple-negative/basal-like breast cancer. Int J Oncol. 2008;33:1165–1176. [PubMed] [Google Scholar]
- Ornelas IM, Silva TM, Fragel-Madeira L, Ventura AL. Inhibition of PI3K/Akt pathway impairs G2/M transition of cell cycle in late developing progenitors of the avian embryo retina. PLoS One. 2013;8:e53517. doi: 10.1371/journal.pone.0053517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce E, Aquino NB, Louie MC. Chronic cadmium exposure stimulates SDF-1 expression in an ERalpha dependent manner. PLoS One. 2013;8:e72639. doi: 10.1371/journal.pone.0072639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razandi M, Pedram A, Rosen EM, Levin ER. BRCA1 inhibits membrane estrogen and growth factor receptor signaling to cell proliferation in breast cancer. Mol Cell Biol. 2004;24:5900–5913. doi: 10.1128/MCB.24.13.5900-5913.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanowicz-Makowska H, Forma E, Brys M, Krajewska WM, Smolarz B. Concentration of cadmium, nickel and aluminium in female breast cancer. Pol J Pathol. 2011;62:257–261. [PubMed] [Google Scholar]
- Satarug S, Garrett SH, Sens MA, Sens DA. Cadmium, environmental exposure, and health outcomes. Environ Health Perspect. 2010;118:182–190. doi: 10.1289/ehp.0901234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqa A, Long LM, Li L, Marciniak RA, Kazhdan I. Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic proteins Survivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K) signalling pathways. BMC Cancer. 2008;8:129. doi: 10.1186/1471-2407-8-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva E, Lopez-Espinosa MJ, Molina-Molina JM, Fernandez M, Olea N, Kortenkamp A. Lack of activity of cadmium in in vitro estrogenicity assays. Toxicol Appl Pharmacol. 2006;216:20–28. doi: 10.1016/j.taap.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Song X, Wei Z, Shaikh ZA. Requirement of ERα and basal acticvities of EGRF and Src kinase in Cd-induced activation of MAPK/ERK pathway in human breast cancer MCF-7 cells. Toxicol Appl Pharmacol. 2015;287:26–34. doi: 10.1016/j.taap.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stayner L, Smith R, Thun M, Schnorr T, Lemen R. A quantitative assessment of lung cancer risk and occupational cadmium exposure. IARC Sci Publ. 1992:447–455. [PubMed] [Google Scholar]
- Stoica A, Katzenellenbogen BS, Martin MB. Activation of estrogen receptor-alpha by the heavy metal cadmium. Mol Endocrinol. 2000;14:545–553. doi: 10.1210/mend.14.4.0441. [DOI] [PubMed] [Google Scholar]
- Strumylaite L, Bogusevicius A, Abdrachmanovas O, Baranauskiene D, Kregzdyte R, Pranys D, Poskiene L. Cadmium concentration in biological media of breast cancer patients. Breast Cancer Res Treat. 2011;125:511–517. doi: 10.1007/s10549-010-1007-8. [DOI] [PubMed] [Google Scholar]
- Tessier DM, Pascal LE. Activation of MAP kinases by hexavalent chromium, manganese and nickel in human lung epithelial cells. Toxicol Lett. 2006;167:114–121. doi: 10.1016/j.toxlet.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Xiao W, Liu Y, Templeton DM. Pleiotropic effects of cadmium in mesangial cells. Toxicol Appl Pharmacol. 2009;238:315–326. doi: 10.1016/j.taap.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Xie J, Shaikh ZA. Cadmium induces cell cycle arrest in rat kidney epithelial cells in G2/M phase. Toxicology. 2006;224:56–65. doi: 10.1016/j.tox.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Yang PM, Chiu SJ, Lin KA, Lin LY. Effect of cadmium on cell cycle progression in Chinese hamster ovary cells. Chem Biol Interact. 2004;149:125–136. doi: 10.1016/j.cbi.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Yu X, Filardo EJ, Shaikh ZA. The membrane estrogen receptor GPR30 mediates cadmium-induced proliferation of breast cancer cells. Toxicol Appl Pharmacol. 2010;245:83–90. doi: 10.1016/j.taap.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Zang Y, Odwin-Dacosta S, Yager JD. Effects of cadmium on estrogen receptor mediated signaling and estrogen induced DNA synthesis in T47D human breast cancer cells. Toxicol Lett. 2009;184:134–138. doi: 10.1016/j.toxlet.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]