

Abstract
Endothelial cell migration is a fundamental process during angiogenesis and, therefore, a point of intervention for therapeutic strategies aimed at controlling pathologies involving blood vessel growth. We sought to determine the role of the gap junction protein connexin 43 (Cx43) in key features of angiogenesis in the central nervous system. We used an in vitro model to test the hypothesis that a complex of interacting proteins, including Cx43 and zonula occludens-1 (ZO-1), regulates the migratory behavior of cerebral endothelium. With knockdown and overexpression experiments, we demonstrate that the rate of healing following scrape-wounding of endothelium is regulated by the level of Cx43 protein expression. The effects on cell motility and proliferation were independent of gap junction communication as cells were sensitive to altered Cx43 expression in single plated cells. Coupling of Cx43/ZO-1 critically regulates this process as demonstrated with the use of a Cx43 α-carboxy terminus 1 peptide mimetic (αCT1) and overexpression of a mutant ZO-1 with the Cx43-binding PDZ2 domain deleted. Disrupting the Cx43/ZO-1 complex with these treatments resulted in collapse of the organized F-actin cytoskeleton and the appearance of actin nodes. Preincubation with the myosin 2 inhibitors blebbistatin or Y-27632 disrupted the Cx43/ZO-1 complex and inhibited cell spreading at the leading edge of migration. Cells studied individually in time-lapse open field locomotion assays wandered less when Cx43/ZO-1 interaction was disrupted without significant change in speed, suggesting that faster wound healing is a product of linearized migration. In contrast to the breakdown of F-actin architecture, microtubule architecture was not obviously affected by treatments. This study provides new insight into the fundamental regulatory mechanisms of cerebral endothelial cell locomotion. Cx43 tethers the F-actin cytoskeleton through a ZO-1 linker and supports cell spreading and exploration during locomotion. Here, we demonstrate that releasing this actin-coupled tether shifts the balance of directional migration control to a more linear movement that enhances the rate of wound healing.
Keywords: connexin43, wound healing, ZO-1, cytoskeleton, angiogenesis
the central nervous system requires an uninterrupted supply of nutrients, which is provided by the microcirculation. When vascular supply is compromised, such as in stroke or disease, angiogenesis is necessary to increase blood flow to tissue. Endothelial cell migration is fundamental to the angiogenic processes. While many of the pathways and molecular interactions underlying these processes have been established, new paradigms are emerging. For example, the ubiquitous gap junction protein connexin 43 (Cx43) has been reported to have significant influence over cell locomotion and proliferation (4, 16, 20).
We recently found that a neurovascular disease risk factor, homocysteine, impairs cerebral endothelial wound healing, while increasing Cx43 levels (3). Our observation of this correlation led us to question the extent to which Cx43 modulates endothelial migration. We reasoned that regulation of cytoskeletal dynamics may be involved through a complex with zonula occludens-1 (ZO-1). The carboxyl tail of Cx43 (Cx43-CT) binds the second postsynaptic density/disc-large/ZO-1 (PDZ2) domain of ZO-1 (11, 14, 29, 31, 32), which is an actin cytoskeleton binding protein (5, 6, 30, 33). Previous studies have concluded that disabling the Cx43/ZO-1 interaction with a peptide mimetic of the binding region results in faster skin (10) and corneal wound healing (21, 22).
In the present study, we hypothesized that Cx43 regulates cellular actin architecture through its interaction with ZO-1 and that this complex is responsible for modulating cell migration in a model of cerebral endothelial wound repair. Our results suggest that non–channel functions of Cx43 exert significant control over cell behavior through an F-actin cytoskeletal tethering function that is necessary for cell spreading and migratory exploration.
MATERIALS AND METHODS
All procedures and safety measures were approved by the Institutional Animal Care and Use Committee of Idaho State University and performed in accord with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All chemicals were purchased from Fisher Scientific (Waltham, MA) unless indicated otherwise.
Cell culture and pharmaceutical treatment.
Mouse brain microvascular endothelial cells (bEnd.3; catalog no. CRL-2299, American Type Culture Collection, Manassas, VA) were grown to confluence in 8-well chambered slides or 96-well plates in complete media comprising Dulbecco's modified Eagle's medium high glucose (DMEM-H; catalog no. SH30243.02, Hyclone, Logan, UT) supplemented with 10% bovine calf serum (vol/vol) and penicillin-streptomycin. Cells were maintained in a humidified incubator at 37°C with a 5% CO2 in air atmosphere. Experiments began at 3 days postconfluence. The Cx43-CT peptide α-connexin carboxyl terminal 1 (αCT1) was dissolved in sterile pure water as a stock solution (30 mM) and stored at −20°C until dilution to 150 μM just prior to use on the day of experiments. The αCT1 peptide is a binary construct with an amino terminal (NT) antennapedia cell penetration domain (RQPKIWFPNRRKPWKK) fused to the carboxyl terminal-most 9 amino acids of Cx43 (RPRPDDELI) (14). In experiments to study the role of myosin 2, cells were treated with either dimethyl sulfoxide (DMSO) as a vehicle control (−)−blebbistatin (Cayman Chemicals, Ann Arbor, MI), or Y-27632-dihydrochloride (Alexis Biochemicals, San Diego, CA).
Transient knockdown and overexpression of Cx43.
One day before transfection, bEnd.3 cells were incubated in Opti-MEM (catalog no. 51985-034, Invitrogen, Carlsbad, CA). Transfection procedures were performed with 0.5 μl Lipofectin reagent (catalog no. 18292, Invitrogen) in 100 μl Opti-MEM according to the manufacturer's instructions. Small interfering RNA (siRNA; 250 nM) targeting the mouse Cx43 gene Gja 1 (sequence: 5′-UGUUCUAUGUGAUGAGAAATT-3′) (catalog no. 4390771, Invitrogen) was used to knock down levels of Cx43 and the nontargeting siRNA (catalog no. 4390846, Invitrogen) was used as a negative control. To overexpress Cx43, 2 μg of DNA was used to transfect the cells. The largest effective period for Cx43 knockdown and overexpression was between 1–3 and 2–4 days posttransfection, respectively. Transfection experiments were timed so that cells were studied during these windows.
Transfection of ZO-1.
ZO-1 was overexpressed with the PDZ2 domain intact (ZO-1/PDZ2int) and deleted (ZO-1/PDZ2del), respectively (15). The protocol was similar to that described above, except the dose and time course. A total of 1.2 μg of DNA was used to transfect the cells, and the optimal time window was at 5–7 days.
In-cell ELISA.
In-cell ELISAs were performed in 96-well plates as previously described (1, 2, 19). Anti-Cx43 primary antibody (catalog no. C6219, Sigma-Aldrich, St. Louis, MO) was used at a dilution of 1:5,000 at 4°C overnight. The absorbance of blue phosphatase substrate (KPL, Gaithersburg, MO) was quantified at a wavelength of 620 nm using a plate reader (Synergy HT, BioTek Instruments, Winooski, VT).
Immunocytochemistry.
Cells were fixed in 4% paraformaldehyde (PFA) at room temperature for 15 min. Immunodetection followed standard procedures. Nuclei were stained with 10 ng/ml Hoechst 33258 (catalog no. 861405, Sigma-Aldrich) at room temperature for 5 min. Anti-Cx43 primary antibody was used at a dilution of 1:2,000. Anti-β tubulin primary antibody was used at a dilution of 1:1,000 (catalog no. T8328, Sigma-Aldrich). Phalloidin (catalog no. A12381, Molecular Probes, Grand Island, NY) was used at a dilution of 1:1,000 at 4°C overnight. Donkey anti-rabbit secondary antibody conjugated to AlexaFluor-488 (catalog no. A21206, Invitrogen, Eugene, OR) was used at a dilution of 1:500 at 4°C overnight. Images were acquired using a confocal microscope and Fluoview 4.0 software (FV100, Olympus Imaging America, Center Valley, PA).
Wound repair assays.
Wound repair assays were performed using an in vitro scrape model. The cells were seeded in 96-well plates and studied 3 days postconfluence. Cells were removed (“scraped”) using a sterile 200 μl pipette tip by drawing the tip across the middle of each well through the center. Cells were rinsed with warm minimal essential medium (MEM; catalog no. SH30235.02, Hyclone) three times to remove scraped and loosened cells followed by incubation in DMEM-H with control and αCT1 treatments. The DMEM-H with treatments were replaced every 24 h. Three days after wounding, the cells were fixed by 4% PFA at room temperature and wounds were scored 0 (open) or 1 (closed) at 5 evenly spaced regions along each wound and averaged as a single measurement for each well.
For time-lapse confocal microscopy of closing wounds, the cells were seeded in 8-well chambered coverslips. Cells were scraped using a sterile 10 μl pipette tip. Subsequent steps were performed as described above. Twenty-four hours after wounding, the slides were plated in a stage-top incubator (Tokai Hit, Japan) on the confocal microscope and differential interference contrast (DIC) images were recorded every 15 min for 16 h at 3 different fields of view of leading edges per well. The image capture was controlled by the Fluoview software (version 4.0). The wound closure rate was quantified from the growth area, using ImageJ software (28), divided by the respective time period.
Locomotion assays.
Cell locomotion was assessed by plating cells at 10% confluence in 8-well chambered slides in the late afternoon. The following morning, transfection procedures were carried out as described above. For time-lapse microscopy, the slides were placed in the stage-top incubator as described above and DIC images were recorded every 20 min for 16 h at 6 different fields of view per well. The time-lapse DIC images were analyzed to track each single cell using MetaMorph software. Average speed was defined as the tracking distance divided by the time period. Net speed was defined as the distance between initial and terminal positions divided by the time period. Directionality was then calculated by net speed divided by average speed; a value of one means that the cell moved in a straight line.
Cell proliferation assays.
Cell proliferation was assessed by plating cells at 25% confluence in 96-well plates in the late afternoon. The following morning, designated as time zero, any nonviable and nonadherent cells were removed with a media change and a random subset of wells were fixed (4% PFA) to define the baseline number of viable cells in the assay. After 72 h, cells in all conditions were fixed. Nuclei were stained with Hoechst 33258. Cell number was quantified by counting the number of cells in 5 microscope fields of view per well (×10 objective) and averaged as a single measurement for each well. Proliferation was defined as the increase in the number of cells: mean count minus mean count at day zero.
Statistical analyses.
Alpha was set at 0.05 for statistical significance. One-way or two-way ANOVA (Tukey post hoc test) was used to compare groups using computer software programs (SPSS, IBM, Armonk, NY; SAS, Cary, NC). Data are presented as means ± standard error of the mean.
RESULTS
Manipulating levels of Cx43.
As shown in Fig. 1, Cx43 levels were effectively decreased and increased by transfection procedures using Lipofectin and siRNA, mock, or DNA as described in materials and methods. The result was cell populations with a range of ∼60% to ∼120% Cx43 levels compared with mock-transfected cells (P < 0.05). This was quantified by ELISA (Fig. 1C) and is illustrated by immunocytochemistry (Fig. 1B).
Fig. 1.

Manipulating levels of connexin 43 (Cx43). Cx43 expression was knocked down (Cx43low) or increased (Cx43high) relative to mock transfection (Cx43ctrl; control). A: density of immunohistochemistry for Cx43 in the cells at the leading edge of the wound in respective groups (*P < 0.05 vs. Cx43ctrl, n = 6). B: representative results of immunocytochemistry for Cx43 at wound edges (dashed lines). C: in-cell enzyme-linked immunosorbent assay (ELISA) of Cx43 expression in respective groups. The ELISA quantifies expression throughout the full well of the culture and is dominated by the monolayer of cells behind the wound edge (*P < 0.05 vs. Cx43ctrl, n = 8).
Cx43 regulates wound healing through ZO-1 binding.
Wound repair rate was inversely related to the level of Cx43 (Fig. 2). Disrupting the interaction between Cx43 and ZO-1, using the peptide αCT1, enhanced wound healing in control (mock transfected) and overexpressing cells.
Fig. 2.
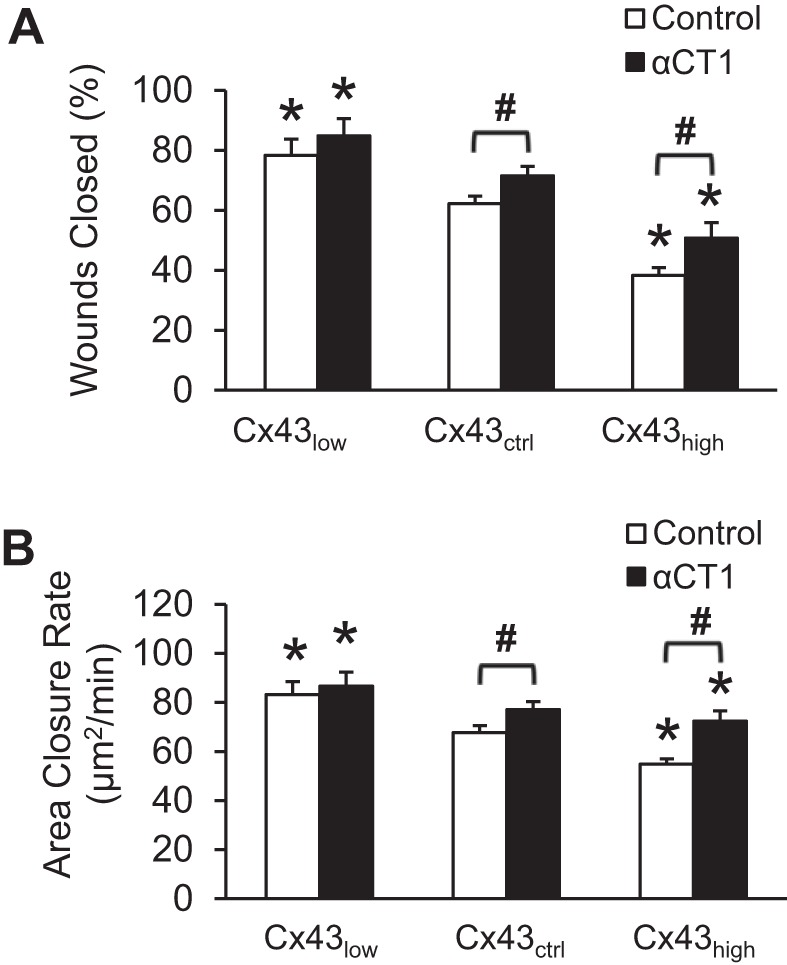
Cx43 regulates wound healing through ZO-1 binding. A: relative number of wounds closed 72 h after wounding cerebral endothelial monolayer. B: area of wound repaired per minute measured from time-lapse confocal microscopy. *P < 0.05 vs. Cx43ctrl within each group; #P < 0.05 for Control vs. α-connexin carboxyl terminal 1 (αCT1); n = 12–18 per mean.
Cx43 reduces cell locomotion and directionality through ZO-1 binding.
Locomotion of dispersed cells was studied to better understand the wound healing process. Given that wound healing requires efficient directional movement into the wound area, and may involve gap junction-based cell-cell communication to coordinate motility, we questioned the extent to which the regulatory complex of Cx43 and ZO-1 operated on the single cell level, absent cell-cell contact. We quantified cell movement as average speed (total distance moved per unit time), net speed (net distance moved per unit time), and directionality (linearity of movement). A cell that moved quickly in one direction will have high values for all three whereas a cell moving in circles may have any average speed but will have low net speed and low directionality. Figure 3A shows that overexpression of Cx43 reduced the average speed and that disrupting Cx43/ZO-1 interaction fully rescued the impairment. Net speed (Fig. 3B) and directionality (Fig. 3C) were improved by siRNA knockdown of Cx43 and impaired by progressively higher levels of Cx43. Disrupting Cx43/ZO-1 enhanced net speed and directionality of control cells (mock transfection) and of cells overexpressing Cx43.
Fig. 3.
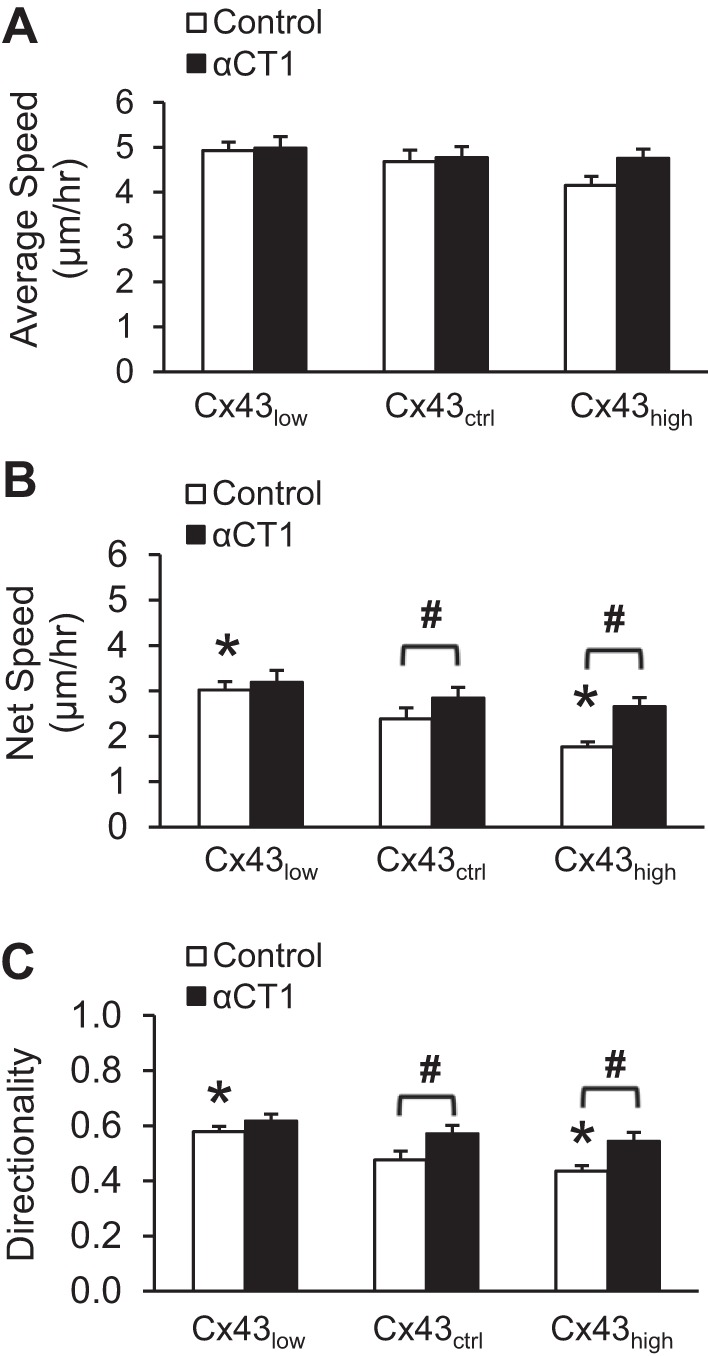
Cx43 reduces cell locomotion and directionality through ZO-1 binding. Locomotor behavior of cerebral endothelial cells was measured from time-lapse confocal microscopy of single cells dispersed in culture. A: average speed of cells is the total distance traveled per hour. B: net speed is the displacement from start to end of recording, regardless of the path traveled, per hour. Cells that travel tortuous paths or move slowly will have a lower net speed. C: average speed divided by net speed quantifies the directionality of movement. Cells that move quickly along a straight line will have a value of one. *P < 0.05 vs. Cx43ctrl; #P < 0.05 for Control vs. αCT1; n = 151 per mean.
Cx43 regulates proliferation through ZO-1 binding.
In addition to motility, wound healing requires cell proliferation. Next, we tested the hypothesis that Cx43 contributes to the regulation of endothelial cell proliferation via interaction with ZO-1. As shown in Fig. 4, the extent of proliferation was a function of the level of Cx43. Higher Cx43 levels produced less proliferation and vice versa. Moreover, disrupting the Cx43/ZO-1 complex enhanced proliferation at all levels of Cx43.
Fig. 4.
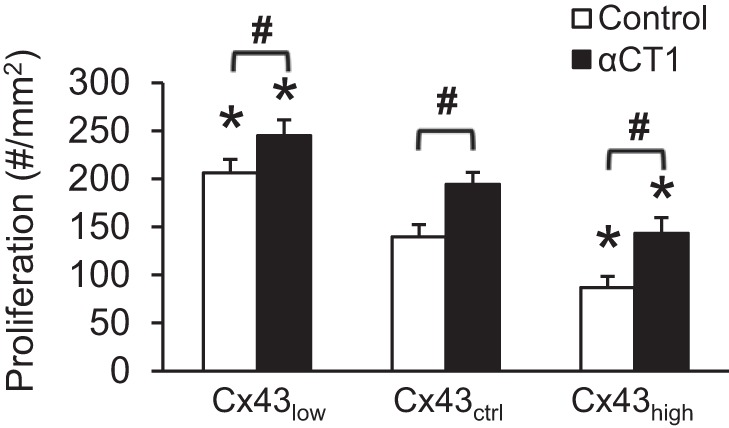
Cx43 regulates proliferation through ZO-1. Cells were plated at 25% confluence. Proliferation is the increase in the number of cells after 72 h of incubation. *P < 0.05 vs. Cx43ctrl; #P < 0.05 for Control vs. αCT1; n = 6 per mean.
Cx43 modulates F-actin architecture through ZO-1 binding.
ZO-1 links Cx43 to the actin cytoskeleton. Next, we addressed whether this link provides a mechanism for controlling F-actin architecture. Figure 5 shows representative wound edges labeled with fluorescent phalloidin in each of the conditions studied. The density of F-actin staining was inversely proportional to the level of Cx43. In addition, the density of F-actin punctae or large nodes in cells of the leading edge also declined with increasing levels of Cx43. Disrupting the Cx43/ZO-1 complex resulted in loss of the F-actin skeleton independent of Cx43 level, leaving behind F-actin-deficient pockets. Consistent with these relations, Cx43/ZO-1 complexes were localized along F-actin strands but not at nodes (Fig. 6A). We did not observe gross alterations of microtubule structure after disrupting the Cx43/ZO-1 complex (Fig. 6B).
Fig. 5.

Cx43 modulates F-actin architecture through ZO-1 binding. A: representative images of cells at wound edges, delineated with dashed lines. Wound space is to the left. The density of cytoskeletal F-actin staining with phalloidin (red) was inversely proportional to the level of Cx43. Disrupting the Cx43/ZO-1 complex with αCT1 caused a distinct change in F-actin architecture characterized by a high density of nodes in all conditions. B and C: quantification of the density of F-actin (B) and F-actin nodes (C) in cells at wound edges using ImageJ. *P < 0.05 vs. Cx43ctrl; #P < 0.05 for Control vs. αCT1; n = 6 per mean.
Fig. 6.

Microtubule architecture is not grossly altered by Cx43/ZO-1 uncoupling. A: Cx43 (red) and zonula occludens-1 (ZO-1; green) are colocalized (arrows) along continuous strands of F-actin (purple). F-actin nodes (arrowheads) occur only in the absence of Cx43/ZO-1 colocalization. B: representative images of microtubules (β-tubulin) in cells at wound edges with varying levels of Cx43; wound space is to the left, delineated with dashed lines. The architecture of cytoskeletal microtubule labeling with an antibody to tubulin (green) under control conditions and following disruption of the Cx43/ZO-1 complex with αCT1. Nuclei are shown in blue.
Myosin 2 contraction participates in F-actin architectural integrity and partially masks the relative importance of Cx43/ZO-1/F-actin tethering. When the contraction of myosin 2 was inhibited with either blebbistatin or Y-27632, there was a moderate decrement of structural normalcy in F-actin morphology (Fig. 7). But, when these treatments were combined with Cx43/ZO-1 uncoupling, cells lost protrusive competency with collapse of the F-actin cytoskeletal structure and cell-cell separation throughout the leading edge cells (Fig. 7).
Fig. 7.

Inhibition of myosin 2 contraction further unmasks stabilizing role of Cx43/ZO-1 on F-actin and cell protrusions in migrating cells. A–C: baseline F-actin architecture (red) under respective treatment conditions: vehicle control (A), blebbistatin inhibition (B) of myosin 2 contraction (blebbistatin slows phosphate release from myosin 2), and Y-27632 inhibition (C) of myosin 2 contraction (Y-27632 inhibits phosphorylation of myosin light chain by Rho-kinase). D–F: effect of Cx43/ZO-1 uncoupling with αCT1 in the context of respective baseline conditions. Arrowheads indicate examples of cell separation. Nuclei are shown in blue.
Binding of Cx43 at the PDZ2 domain of ZO-1 regulates wound healing.
αCT1 is a class II PDZ binding motif, exhibiting a consensus CT amino acid sequence of Hydrophobic-X-Hydrophobic (14, 24). It thus cannot be excluded that αCT1 may bind PDZ domains on proteins other than that on ZO-1 that have affinity for PDZ binding ligands of this class. To address the issue of whether the PDZ2 domain of ZO-1 was specifically determining our results, we directly overexpressed a previously characterized mutant ZO-1 in which the Cx43-binding PDZ2 domain is deleted (15) in our endothelial scratch-wound models and compared the cell behaviors to cells transfected with overexpressed full-length ZO-1. Results from the experiments with the ZO-1 PDZ2 deletion mutant confirmed those testing αCT1 (Fig. 8). When the nonbinding form of ZO-1 was overexpressed, wound healing was accelerated compared with overexpression of a ZO-1 with an intact PDZ2 domain. These results are consistent with both knockdown of Cx43 expression (siRNA) and with the application of αCT1.
Fig. 8.
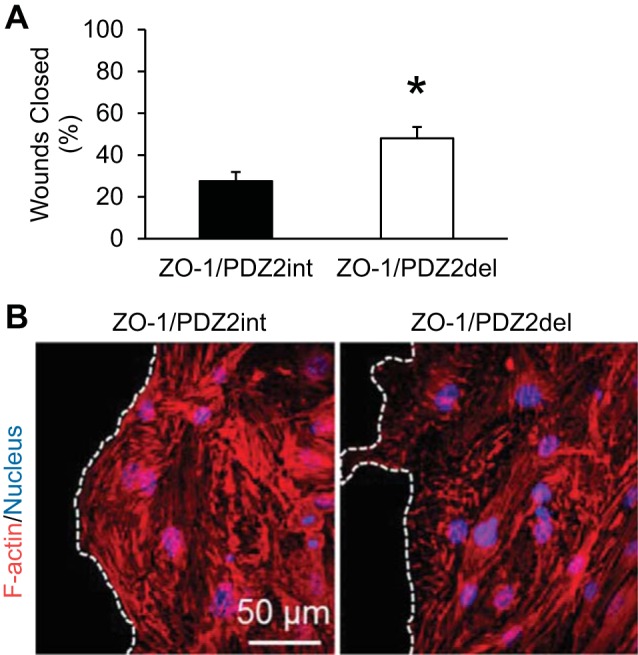
Confirmation that the Cx43-binding PDZ2 domain of ZO-1 is the critical element in regulating wound healing. A: overexpression of ZO-1 with deletion of the PDZ2 domain resulted in faster wound repair of cerebral endothelium compared with overexpression of ZO-1 with the PDZ2 domain intact. *P < 0.05; n = 16 per mean. B: representative images of cells at the wound edge in respective conditions with dashed lines delineating the wound edge; wound space to left of lines. Similar to the effects of αCT1, deletion of the ZO-1 PDZ2 domain increased the detection of F-actin nodes at the leading edge of the wound.
DISCUSSION
The gap junction protein Cx43 is one of the most ubiquitously expressed connexins. The Cx43/ZO-1 complex regulates the size, structure, and stability of gap junctions (26). Here, we report our discovery of another essential function of the Cx43/ZO-1 complex, regulation of F-actin cytoskeletal architecture and cellular migration in wounded endothelium. This study provides the first evidence that cerebral endothelial cell wound healing (Fig. 2), directionality of locomotion (Fig. 3), and proliferation (Fig. 4) are regulated, in part, through interactions between Cx43 and ZO-1. From the results of experiments in which we blocked Cx43/ZO-1 interaction with a mimetic peptide (Figs. 5–7) and additional experiments (Fig. 8) overexpressing a mutant ZO-1 that lacks the Cx43-binding PDZ2 domain, we concluded that this complex was specific and necessary for the regulation of wound healing of cerebral endothelium in our in vitro assay system.
Actin microfilaments are essential in forming protrusive structures such as lamellipodia and filopodia in migrating cells. When the Cx43/ZO-1 complex was disrupted, either directly by using the peptide αCT1 or by overexpressing a mutant form of ZO-1 that does not bind Cx43, much of the F-actin structure was lost and became dominated by large nodes (Figs. 5 and 8). These nodes are reminiscent of the actin “spots” first described as dynamic actin assemblies of the cortical actin cytoskeleton (27). More recently, the Bershadsky lab has identified actin nodes that are revealed when cells are treated with actin depolymerizing agents and the nodes move and enlarge through the contraction of myosin 2 (18). In endothelial cells, myosin 2 plays several roles that are relevant to our results. For example, myosin 2 is important for cell-cell adhesion through adherens junctions (13). Inhibiting myosin 2 activity resulted in some contact disruption and primed the cells by placing them in a compromised state. Disconnecting ZO-1 from Cx43 then resulted in gross absence of spreading cell fronts of migration, which is consistent with the roles of both myosin 2 and F-actin in cell spreading during migration. These nodes are not sights of Cx43/ZO-1 tethering (Fig. 6) and remain to be fully characterized.
Channel-dependent effects of Cx43 require the formation of gap junctions or functional hemichannels. Gap junction intercellular communication (GJIC) may play an important role in the collective migration of cells as a cohesive group during embryonic morphogenesis, wound healing, and cancer invasion (7). For example, cultured adrenocortical cells transfected with Cx43-green fluorescent protein were shown to use GJIC for collective migration in a wound healing assay (7). Additionally, heterocellular coupling of gap junctions via Cx43 between glioma cells and astrocytes regulated glioma cell invasion and migration in brain parenchymal tissue (25). In time-lapse experiments, we studied individual cells that had no contact sites with other cells, and therefore no GJIC. Doing so, we were able to demonstrate that GJIC was not required for control of cellular migration by Cx43 (Fig. 3). In light of our previous results showing an elevation in Cx43 levels (3) and a disruption of endothelial F-actin orientation (19a), after treatment with the neurovascular disease risk factor homocysteine, the Cx43/ZO-1 complex may be involved in the impaired angiogenesis in hyperhomocysteinemia (23). Moreover, the results of our experiments indicate a dominant role for the Cx43/ZO-1 complex in dictating whether cell locomotion will be more linear vs. exploratory. Specifically, αCT1 enhanced the linearity, and thus the net speed, of migration that likely contributed to the faster wound healing we observed in scrape-wounded monolayers when Cx43 binding to ZO-1 was inhibited. When Cx43 levels were low, αCT1 did not significantly affect average cell speed (Fig. 3A).
Microtubules are 1) necessary for directed movement of large cells like fibroblasts and endothelial cells (12), 2) important for maximal migration speeds (17), and 3) responsible for linearizing the direction of cell movement (8). We did not, however, find significant alterations in the gross architecture of the microtubule cytoskeleton in these studies (Fig. 6B). Moreover, interaction between microtubules and F-actin is critical for turning of neurites during outgrowth such that disrupting their interaction inhibits turning of growth cones (reviewed in ref. 9). Consistent with this background, our results provide evidence that αCT1 untethers the F-actin cytoskeleton from a Cx43 anchor, resulting in reduced spreading and exploration of the cell migratory front that leads to linearized movement based, most likely, on the underlying microtubule network polarity.
Wound healing requires both cell migration and proliferation. The rate of cell proliferation was inversely proportional to levels of Cx43, which is consistent with previous findings in corneal endothelium [7]. The Cx43/ZO-1 complex plays a significant role in proliferation of cerebral endothelial cells because disrupting this complex rescued proliferation at any level of Cx43 expression tested. The present study focused on cytoskeletal elements during cell migration. It could be hypothesized that Cx43/ZO-1 altered the mechanical kinetics of cell division at the level of the cytoskeleton. Our results do not preclude a role for Cx43/ZO-1 on cell cycle progression by modulating the relevant signaling pathways (e.g., cyclin-coupled pathways).
Collectively, these results demonstrate that a level of intra- and intercellular cytoskeletal cohesion exists for which the Cx43/ZO-1 complex is necessary. When we push the system by knocking down another important level of cell-cell coupling and cell edge spreading cohesion, inhibiting myosin 2 contractile function, the cell edges collapse away from cell-cell junctions and fail to develop spreading migration fronts. On some level this appears to be useful for the endothelium because αCT1 improves wound healing. Our data show that these endothelial cells migrate in a more linear pattern under the influence of αCT1 and that αCT1 has a negligible effect on locomotor speed per se because single cells without αCT1 treatment migrate in more tortuous or even circular patterns relative to straighter lines of travel when treated with αCT1. So, there is some link between this cytoskeletal cohesiveness and locomotor direction that our data support. We might next hypothesize that the cells are exploratory when wounded, but loss of cellular cohesive structures causes a default to reliance on unidirectional migration that is more resistant to change, probably established by the underlying cell polarity and microtubule dynamics. Collectively, these results support the idea that Cx43/ZO-1 is a key tethering protein complex for the F-actin cytoskeleton that is required for coordinated protrusive spreading, turning, and exploratory migration.
GRANTS
This work was supported in part by National Institutes of Health Grants R15 HL106548 (S. E. Bearden), R01 HL56728-10A2 (R. G. Gourdie), R01-1DE019355-01 (R. G. Gourdie; subcontract), and ISUGSRSC S11-28U (C.-H. Chen).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
C.-H.C., J.N.M., R.G.G., S.R.J., and S.E.B. conceived of and designed research; C.-H.C., J.N.M., and S.E.B. performed experiments; C.-H.C. analyzed data; C.-H.C., J.N.M., R.G.G., S.R.J., B.E.I., and S.E.B. interpreted results of experiments; C.-H.C. prepared figures; C.-H.C. and S.E.B. drafted manuscript; C.-H.C., J.N.M., R.G.G., S.R.J., B.E.I., and S.E.B. edited and revised manuscript; C.-H.C., J.N.M., R.G.G., S.R.J., B.E.I., and S.E.B. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the technical contribution of Jane Jourdan.
REFERENCES
- 1.Beard RS Jr, Reynolds JJ, Bearden SE. Hyperhomocysteinemia increases permeability of the blood-brain barrier by NMDA receptor-dependent regulation of adherens and tight junctions. Blood 118: 2007–2014, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beard RS Jr, Reynolds JJ, Bearden SE. Metabotropic glutamate receptor 5 mediates phosphorylation of vascular endothelial cadherin and nuclear localization of beta-catenin in response to homocysteine. Vascul Pharmacol 56: 159–167, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen CH, Beard RS, Bearden SE. Homocysteine impairs endothelial wound healing by activating metabotropic glutamate receptor 5. Microcirculation 19: 285–295, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cina C, Maass K, Theis M, Willecke K, Bechberger JF, Naus CC. Involvement of the cytoplasmic C-terminal domain of connexin43 in neuronal migration. J Neurosci 29: 2009–2021, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem 273: 29745–29753, 1998. [DOI] [PubMed] [Google Scholar]
- 6.Fanning AS, Ma TY, Anderson JM. Isolation and functional characterization of the actin binding region in the tight junction protein ZO-1. FASEB J 16: 1835–1837, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 10: 445–457, 2009. [DOI] [PubMed] [Google Scholar]
- 8.Ganguly A, Yang H, Sharma R, Patel KD, Cabral F. The role of microtubules and their dynamics in cell migration. J Biol Chem 287: 43359–43369, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geraldo S, Gordon-Weeks PR. Cytoskeletal dynamics in growth-cone steering. J Cell Sci 122: 3595–3604, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghatnekar GS, O'Quinn MP, Jourdan LJ, Gurjarpadhye AA, Draughn RL, Gourdie RG. Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen Med 4: 205–223, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol 8: 931–934, 1998. [DOI] [PubMed] [Google Scholar]
- 12.Gotlieb AI, Subrahmanyan L, Kalnins VI. Microtubule-organizing centers and cell migration: effect of inhibition of migration and microtubule disruption in endothelial cells. J Cell Biol 96: 1266–1272, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoelzle MK, Svitkina T. The cytoskeletal mechanisms of cell-cell junction formation in endothelial cells. Mol Biol Cell 23: 310–323, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell 16: 5686–5698, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunter AW, Gourdie RG. The second PDZ domain of zonula occludens-1 is dispensable for targeting to connexin 43 gap junctions. Cell Commun Adhes 15: 55–63, 2008. [DOI] [PubMed] [Google Scholar]
- 16.Kwak BR, Pepper MS, Gros DB, Meda P. Inhibition of endothelial wound repair by dominant negative connexin inhibitors. Mol Biol Cell 12: 831–845, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao G, Nagasaki T, Gundersen GG. Low concentrations of nocodazole interfere with fibroblast locomotion without significantly affecting microtubule level: implications for the role of dynamic microtubules in cell locomotion. J Cell Sci 108: 3473–3483, 1995. [DOI] [PubMed] [Google Scholar]
- 18.Luo W, Yu CH, Lieu ZZ, Allard J, Mogilner A, Sheetz MP, Bershadsky AD. Analysis of the local organization and dynamics of cellular actin networks. J Cell Biol 202: 1057–1073, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayo JN, Beard RS Jr, Price TO, Chen CH, Erickson MA, Ercal N, Banks WA, Bearden SE. Nitrative stress in cerebral endothelium is mediated by mGluR5 in hyperhomocysteinemia. J Cereb Blood Flow Metab 32: 825–834, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19a.Mayo JN, Chen CH, Liao FF, Bearden SE. Homocysteine disrupts outgrowth of microvascular endothelium by an iNOS-dependent mechanism. Microcirculation 21: 541–550, 2014. doi: 10.1111/micc.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendoza-Naranjo A, Cormie P, Serrano AE, Wang CM, Thrasivoulou C, Sutcliffe JE, Gilmartin DJ, Tsui J, Serena TE, Phillips AR, Becker DL. Overexpression of the gap junction protein Cx43 as found in diabetic foot ulcers can retard fibroblast migration. Cell Biol Int 36: 661–667, 2012. [DOI] [PubMed] [Google Scholar]
- 21.Moore K, Bryant ZJ, Ghatnekar G, Singh UP, Gourdie RG, Potts JD. A synthetic connexin 43 mimetic peptide augments corneal wound healing. Exp Eye Res 115: 178–188, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore K, Ghatnekar G, Gourdie RG, Potts JD. Impact of the controlled release of a connexin 43 peptide on corneal wound closure in an STZ model of type I diabetes. PloS One 9: e86570, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagai Y, Tasaki H, Takatsu H, Nihei S, Yamashita K, Toyokawa T, Nakashima Y. Homocysteine inhibits angiogenesis in vitro and in vivo. Biochem Biophys Res Commun 281: 726–731, 2001. [DOI] [PubMed] [Google Scholar]
- 24.O'Quinn MP, Palatinus JA, Harris BS, Hewett KW, Gourdie RG. A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ Res 108: 704–715, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oliveira R, Christov C, Guillamo JS, de Bouard S, Palfi S, Venance L, Tardy M, Peschanski M. Contribution of gap junctional communication between tumor cells and astroglia to the invasion of the brain parenchyma by human glioblastomas. BMC Cell Biol 6: 7, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhett JM, Gourdie RG. The perinexus: a new feature of Cx43 gap junction organization. Heart 9: 619–623, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schafer DA, Welch MD, Machesky LM, Bridgman PC, Meyer SM, Cooper JA. Visualization and molecular analysis of actin assembly in living cells. J Cell Biol 143: 1919–1930, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh D, Solan JL, Taffet SM, Javier R, Lampe PD. Connexin 43 interacts with zona occludens-1 and -2 proteins in a cell cycle stage-specific manner. J Biol Chem 280: 30416–30421, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sonsino J, Gong H, Wu P, Freddo TF. Co-localization of junction-associated proteins of the human blood–aqueous barrier: occludin, ZO-1 and F-actin. Exp Eye Res 74: 123–129, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Sorgen PL, Duffy HS, Sahoo P, Coombs W, Delmar M, Spray DC. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J Biol Chem 279: 54695–54701, 2004. [DOI] [PubMed] [Google Scholar]
- 32.Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M, Tada M. Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem 273: 12725–12731, 1998. [DOI] [PubMed] [Google Scholar]
- 33.Wittchen ES, Haskins J, Stevenson BR. Protein interactions at the tight junction. Actin has multiple binding partners, and ZO-1 forms independent complexes with ZO-2 and ZO-3. J Biol Chem 274: 35179–35185, 1999. [DOI] [PubMed] [Google Scholar]