

Abstract
Vitamin D deficiency affects more that 1 billion people worldwide. Although thought to increase risk of bacterial infections, the importance of vitamin D on host defense against intestinal bacterial pathogens is currently unclear since injection of the active form of vitamin D, 1,25(OH)2D3, increased susceptibility to the enteric bacterial pathogen Citrobacter rodentium by suppressing key immune/inflammatory factors. To further characterize the role of vitamin D during bacteria-induced colitis, we fed weanling mice either vitamin D3-deficient or vitamin D3-sufficient diets for 5 wk and then challenged them with C. rodentium. Vitamin D3-deficient mice lost significantly more body weight, carried higher C. rodentium burdens, and developed worsened histological damage. Vitamin D3-deficient mice also suffered greater bacterial translocation to extra-intestinal tissues, including mesenteric lymph nodes, spleen, and liver. Intestinal tissues of infected vitamin D3-deficient mice displayed increased inflammatory cell infiltrates as well as significantly higher gene transcript levels of inflammatory mediators TNF-α, IL-1β, IL-6, TGF-β, IL-17A, and IL-17F as well as the antimicrobial peptide REG3γ. Notably, these exaggerated inflammatory responses accelerated the loss of commensal microbes and were associated with an impaired ability to detoxify bacterial lipopolysaccharide. Overall, these studies show that dietary-induced vitamin D deficiency exacerbates intestinal inflammatory responses to infection, also impairing host defense.
Keywords: vitamin D, Citrobacter rodentium, inflammation, colitis, segmented filamentous bacteria
it is being increasingly recognized that vitamin D plays an important role in host defense against pathogenic microbes. Vitamin D deficiency affects more than 1 billion people worldwide and is associated with an increased risk of respiratory infections, including Mycobacterium tuberculosis (18, 53) and Pseudomonas aeruginosa (8, 48). Recently, vitamin D status was also shown to influence the risk and severity of Clostridium difficile infection, which is the most common cause of hospital-acquired infectious diarrhea (2, 51). Furthermore, vitamin D deficiency is associated with an increased risk of systemic bacterial infection leading to sepsis (31). Animal models of vitamin D deficiency, including the vitamin D receptor (VDR)−/− mice, also have increased susceptibility to infection and carry higher bacterial burdens during challenge with Salmonella typhimurium (56). Surprisingly, despite its broad linkage to host defense against predominantly mucosal bacterial pathogens, the mechanisms and pathways through which vitamin D affects a host's susceptibility to these infections is poorly understood.
Vitamin D refers to a group of fat-soluble prohormones, including vitamin D3 or cholecalciferol, which is produced endogenously in the skin during exposure to UVB light and is also naturally present in certain foods such as fatty fish (23). Studies of people in Northern latitudes, such as Canada, suggest that endogenous synthesis of vitamin D is minimal from October to March due to limited UVB exposure (22). Furthermore, recent efforts to limit sun exposure and increased usage of sunscreen have dramatically reduced the levels of vitamin D3 produced in the skin of people worldwide (22). Dietary and supplemental forms of vitamin D3 are thus becoming more important sources of this vitamin. Most dietary vitamin D3 is absorbed via passive diffusion in the small intestine, together with dietary fats and bile salts in chylomicrons. Classically, vitamin D3 enters the systemic circulation bound to the D-binding protein and is transported to the liver and kidney, where it is metabolized by various enzymes, including CYP27B1 to yield 1,25(OH)2D3, the active hormonal form of vitamin D, also known as calcitriol (23). 1,25(OH)2D3 signals through the VDR, a nuclear receptor located in most cells in the body. Studies in mice have shown the VDR is highly expressed throughout the small intestine and colon, with the highest expression found in the cecum and proximal colon (29, 56). Ligand binding activates the VDR to heterodimerize with the retinoid X receptor (RXR), and the VDR-RXR complex binds to specific DNA sequences, known as vitamin D-responsive elements (VDRE) to induce transcription (6). 1,25(OH)2D3 directly or indirectly regulates many different genes through the VDR and is involved in diverse cellular functions including calcium metabolism, cell proliferation, and immune modulation (6), all of which could impact intestinal host defense.
Another important group of mucosal bacterial pathogens comprise the attaching and effacing (A/E) family of pathogenic Escherichia coli including enterohemorrhagic E. coli (EHEC) and enteropathogenic E. coli (EPEC). Both microbes are important causes of infectious diarrhea, with EPEC causing as many as 1 million infant deaths per year in developing nations (37). Since these clinically important pathogens do not colonize mice, these infections have been modeled using the related mouse-specific A/E bacterial pathogen Citrobacter rodentium. Following infection, C. rodentium intimately attaches to epithelial cells lining the cecum and colon, resulting in barrier disruption, crypt hyperplasia, and goblet cell depletion as well as immune and inflammatory cell infiltration of the intestinal mucosa (14). More specifically, Toll-like receptor (TLR)-based recognition of C. rodentium products, such as peptidoglycan and lipopolysaccharide (LPS), drives the inflammatory response during infection (19, 32). Ultimately the host immune/inflammatory response not only helps control these infections, but it also causes significant pathophysiology changes. Similarly while the induction of antimicrobial genes at the mucosal surface helps to eventually clear C. rodentium from the host, these responses also remove competing commensal microbes and reduce intestinal colonization resistance (45).
At present, the impact of vitamin D on host defense against C. rodentium is poorly understood. Previous research from our group has shown that treatment with the active form of vitamin D, 1,25(OH)2D3 can increase the susceptibility of mice to C. rodentium by suppressing key inflammatory factors required for bacterial clearance (41). This is in keeping with several studies showing that 1,25(OH)2D3 can inhibit inflammatory responses in the host through a number of mechanisms, including suppressing TLR expression (42), targeting MAPK phosphatase-1 (58), blocking NF-κB signaling (57), modulating dendritic cell and macrophage behavior (15), and skewing T-cell responses toward a regulatory phenotype (26, 33). Moreover 1,25(OH)2D3 acts directly on T cells to inhibit proliferation and production of inflammatory cytokines, including IL-2, IFN-γ, TNF-α, and IL-17 (7). As such, 1,25(OH)2D3 can be considered a potent immunosuppressive agent and may inadvertently impair host defenses against enteric microbes.
Correspondingly, studies suggest that loss of vitamin D3 signaling promotes exaggerated intestinal inflammatory responses. Mice fed a vitamin D3-deficient diet and mice deficient in the VDR gene have both been shown to exhibit a higher baseline colonic inflammatory tone, as well as elevated serum levels of IL-6 (3, 56). Published studies that have examined the impact of vitamin D3 deficiency during C. rodentium infection have shown an exaggerated colitic phenotype (3, 9). Chen et al. (9) showed that VDR−/− mice are resistant to colonization with C. rodentium, but their resistance appeared to depend on a dysbiotic microbiota rather than a direct effect of VDR deficiency. Assa et al. (3) did not directly examine pathogen burdens but focused on the impact of a vitamin D3-deficient diet on epithelial barrier function. To complement these studies, we sought to test whether a vitamin D-deficient diet would be beneficial or detrimental to intestinal host defense, as well as host-specific inflammatory/antimicrobial factors that during C. rodentium infection are regulated by vitamin D.
We fed C57Bl/6 mice either a vitamin D3-deficient or -sufficient diet for 5 wk and challenged them with C. rodentium. Interestingly, despite having a higher baseline inflammatory tone in their intestines, vitamin D3-deficient mice carried significantly higher pathogen burdens in the ceca and in the mesenteric lymph nodes (MLN), spleen, and liver at day 10 postinfection (pi), indicating greater susceptibility to pathogen translocation. In accordance with their heightened immune responses, intestinal tissues of infected vitamin D3-deficient mice showed significantly higher gene transcript levels of the inflammatory mediators TNF-α, IL-1β, IL-6, TGF-β, IL-17A, and IL-17F as well as the antimicrobial peptide REG3γ. Vitamin D3-deficient mice also carried significantly more segmented filamentous bacteria (SFB) in their stool, compared with vitamin D3-sufficient mice. Moreover, they showed defects in the ability to detoxify bacterial LPS and carried higher serum levels of cluster of differentiation 14 (CD14). Overall, these findings show that dietary-induced vitamin D deficiency alters host mucosal defense and increases susceptibility to an enteric bacterial pathogen.
MATERIALS AND METHODS
Mice and experimental diets.
Weanling (3-wk-old) female C57BL/6 mice were obtained from Charles River Laboratories (St. Constant, QC, Canada). Mice were fed either a vitamin D3-deficient diet (0 IU) or a vitamin D3-sufficient diet (1,000 IU) for 6 wk, as previously described (35). All diets were procured from Research Diets (New Brunswick, NJ). Mice were maintained in sterilized, filter-topped cages, handled in tissue culture hoods, and given free-access to water under specific pathogen-free conditions in the animal facility at the Child and Family Research Institute. Sentinel animals were routinely tested for common pathogens. The protocols used were approved by the University of British Columbia's Animal Care Committee and in direct accordance with guidelines drafted by the Canadian Council on the Use of Laboratory Animals.
Bacterial strains and infection of mice.
Mice were infected by oral gavage with 0.1 ml of an overnight culture of Luria broth (LB) containing ∼2.5 × 108 colony-forming units of streptomycin-resistant C. rodentium (formerly C. freundii biotype 4280, strain DBS100). Mice were weighed daily and monitored for signs of illness or distress.
FITC-dextran intestinal permeability assay.
The FITC-dextran assay was performed as previously described (4). Briefly, mice were orally gavaged with 150 μl of 80 mg/ml 4-kDa FITC-dextran (Sigma; FD4) in PBS 4 h before death. In subsequent studies, mice were given 100 μl of 80 mg/ml 4-kDa FITC-dextran intrarectally 2 h before death. Mice were anaesthetized, and blood was collected by cardiac puncture. Blood was then immediately added to a final concentration of 3% acid-citrate dextrose (20 mM citric acid, 100 nM sodium citrate, and 5 mM dextrose; H. Schulze, Shivdasani Laboratory, Dana-Farber Cancer Institute). Plasma was collected and fluorescence was quantified using a Wallac Victor (Perkin-Elmer Life Sciences, Boston, MA) at excitation 485 nm and emission 530 nm for 0.1 s.
Tissue and serum collection.
Mice were anesthetized with halothane, and blood was collected by cardiac puncture. Blood was allowed to clot naturally at room temperature, the cells were removed by centrifugation, and then serum was collected and stored at −80°C until analysis. Anesthetized mice were euthanized by cervical dislocation, and cecal and colonic tissues were collected and immediately placed in 10% neutral buffered formalin (Fisher; 48 h, 4°C) for histological studies or placed in RNAlater (Qiagen) and stored at −80 °C for subsequent RNA extraction.
Serum calcium analysis.
Serum samples were analyzed with the calcium colorimetric assay kit (cat no. K380-250; BioVision Research, Mountain View, CA). This assay utilizes the chromogenic complex, which forms between calcium ions and 0-cresolphthalein. Samples were quantified at an optical density of 575 nm using a Wallac Victor (Perkin-Elmer Life Sciences) and compared with the calcium standard provided with the kit.
Serum 25(OH)D3 analysis.
Serum samples were processed and 25(OH)D3 was assessed using ultra-high-performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) as previously described (12).
C. rodentium enumeration.
For enumeration of C. rodentium, tissues were prepared as previously described (4). Briefly, tissues were collected, weighed, and homogenized in a MixerMill 301 bead miller (Retche). Sample homogenates were serially diluted in PBS and plated onto LB agar plates containing 100 mg/ml strep and incubated overnight at 37°C, and C. rodentium colonies were enumerated the following day, normalizing them to the tissue or stool weight (per gram). For fecal bacterial burden analysis, stool pellets were collected from live mice at different time points pi and processed as described above. For cecal and colonic samples, tissues were opened longitudinally, luminal contents were collected, and then tissues were gently cleaned three times in PBS rinse before collection.
Assessment of total microbes via DAPI staining.
Enumerating total microbes was performed as previously described (45). Briefly, two fecal pellets were collected from each animal. After homogenization, samples were placed in 10% neutral buffered formalin diluted to a final concentration of 3%. Samples were further diluted 1:10 in PBS, vortexed briefly, and stored at 4°C. Next, 2–5 ml of the 1:10 diluted sample were further diluted in 1 ml PBS and filtered onto Anodisc 25 filters (Whatman International) with a pore size of 0.2 mM and a 2.5-cm diameter. The samples were thoroughly dried and then mounted on glass slides with ProLong Gold Antifade reagent containing DAPI (Molecular Probes), and sections were viewed on a Zeiss AxioImager microscope and images taken using an AxioCam HRm camera operating through AxioVision software. The mean number of DAPI-positive microbes was counted in three to six randomly chosen fields per disc (×1,000). The total number of commensal microbes was calculated based on the mean numbers of all the counted fields and the dilution factor. The total number of commensal microbes was presented as the percentage of uninfected controls.
Commensal microbe analysis.
Microbial composition analysis was performed by quantitative PCR (qPCR) as previously described (5). DNA was extracted from at least two fecal pellets from each animal using the Qiagen DNA stool extraction kit. Extracted DNA with 50 ng/reaction was used for qPCR. Group-specific primers for 16S rRNA were used to determine the relative abundance of the selected bacterial phyla: Actinobacteria (forward: 5′-TAC GGC CGC AAG GCTA-3′; reverse: 5′-CGT CAT CCC CAC CTT CCT CCG-3′); Bifidobacterium (forward: 5′-GGG TGG TAA TGC CGG ATG-3′; reverse: 5′-CCA CCG TTA CAC CGG GAA-3′); Lactobacillus (forward: 5′-AGC AGT AGG GAA TCT TCC A-3′; reverse: 5′-CAC CGC TAC ACA TGG AG-3′); SFB (Candidatus savagella) (forward: 5′-CGG AGC ATG TGG TTT AAT TC; reverse: 5′-GCT GTC TCG CTA AAG TGC TC-3′); and α-Proteobacteria (forward: 5′-CTA GTG TAG AGG TGA AATT-3′; reverse: 5′-CCC CGT CAA TTC CTT TGA GTT-3′). Primer sequences for Bacteroidetes, Firmicutes, and γ-Proteobacteria have previously been described (5). Universal Eubacteria primers (5′-ACTCCT ACG GGA GGC AGC AGT-3′ and 5′-ATT ACC GCG GCT GCT GGC-3′) were used to determine total bacterial 16S rRNA in each sample, and the relative abundance of each taxonomic group was determined by calculating the average threshold cycle (CT) value relative to this number, normalized to each primer's determined efficiency.
Histopathological scoring.
To assess tissue pathology, paraffin-embedded cecal and colonic tissue sections (5 μm) were stained with hematoxylin and eosin and were examined by two blinded observers. Tissue sections were assessed for submucosal edema (0 = no change; 1 = mild; 2 = moderate; and 3 = profound), epithelial hyperplasia (scored based on percentage above the height of the control where 0 = no change; 1 = 1–50%; 2 = 51–100%; and 3 = >100%), epithelial integrity (0 = no change; 1 = <10 epithelial cells shedding per lesion; 2 = 11–20 epithelial cells shedding per lesion; 3 = epithelial ulceration; and 4 = epithelial ulceration with severe crypt destruction) and neutrophil and mononuclear cell infiltration (0 = none; 1 = mild; 2 = moderate; and 3 = severe), as previously described (4). The maximum score possible was 15 points.
Immunofluorescence staining.
Immunofluorescence staining of cecal tissues for Ki67, F4/80, and Ly6G was performed as previously described (4, 19, 49). Briefly, paraffin-embedded sections were deparaffinized and then rehydrated, followed by antigen retrieval using 0.1 M citric acid monohydrate (Sigma) with 0.05% Tween 20 (pH 6.0) and steam for 45 min. Slides were blocked in PBS with 2% normal goat serum, 1% BSA, 0.1% Triton X-100, and 0.05% Tween 20. Primary antibodies used were rabbit antisera generated against Ki67 (1:200; Abcam), F4/80 (1:8K; Serotec), or Ly6G (Abcam). This was followed by secondary Alexa568-conjugated goat anti-rabbit or anti-rat IgG antibodies (Molecular Probes) and Prolong Gold antifade reagent containing 4′,6′-diamidino-2-phenylindole (DAPI; Invitrogen). Sections were viewed at 350 and 594 nm on a Zeiss AxioImager microscope. Images were obtained using a Zeiss AxioImager microscope equipped with an AxioCam HRm camera operating through AxioVision software (Version 4.4).
RNA extraction and quantitative RT-PCR.
Cecal tissues were collected and stored in RNAlater (Qiagen) at −80°C with total RNA extracted using the Qiagen RNeasy kit, as previously described (4). Total RNA was quantified using a NanoDrop Spectrophotometer (ND1000). RNA was reverse transcribed using a Qiagen Omniscript RT kit (Qiagen), according to the manufacturer's instructions. Quantitative PCR was carried out using a Bio-Rad Miniopticon or Opticon2, as previously described (4). Melting point analysis confirmed the specificity for each of the PCR reactions. Quantitation was performed using GeneEx Macro OM 3.0 software. Primer sequences and annealing temperatures were as follows: IL-10 (forward: 5′-GTT GCC AAG CCT TAT CGG AA-3′; reverse: 5′-CCA GGG AAT TCA AAT GCT CCT-3′; annealing 55°C); TGF-β (forward: 5′-GAC TCT CCA CCT GCA AGA CCA T′; reverse: 5′-GGG ACT GGC GAG CCT TAG TT; annealing 59°C); β-defensin 1 (forward: 5′-TCC TCT CTG CAC TCT GGA CC′; reverse: 5′-ATC GCT CGT CCT TTA TGT CC; annealing 72°C); and β -defensin 3 (forward: 5′-CTC CAC CTG CAG CTT TTA GC′; reverse: 5′ - GCT AGG GAG CAC TTG TTT GC; annealing 72°C). The primer sequences and reaction conditions for β-actin, TNF-α, IL-6, IL-1β, IL-17A, IL-17F, mcramp, and Reg3γ have previously been described (4).
Serum CD14 measurement.
Serum samples were collected and prepared as described above. CD14 was measured using the Quantikine ELISA Mouse CD14 Immunoassay Kit (R&D Systems, Minneapolis, MN), and the assays were performed according to directions provided by manufacturer. This assay employs the quantitative sandwich enzyme immunoassay technique. Samples were quantified at an optical density of 450 nm using a Wallac Victor (Perkin-Elmer Life Sciences) and compared with the standard provided with the kit.
Lipopolysaccharide-dephosphorylation assay.
Lipopolysaccharide (LPS)-dephosphorylating activity was measured by the malachite green assay, which measures free phosphate release, as previously described (17). In brief, intestinal tissues were homogenized in 500 ml of homogenization buffer and then centrifuged at 11,000 rpm for 3 min to remove insoluble material. To determine the protein concentrations, a Bradford assay (Bio-Rad) was performed on the tissue samples according to the manufacturers instructions. A standard curve was created using a stock solution of 1 mg/ml BSA (Sigma) in triplicates with multiple concentrations. Forty milliliters of a 5 mg/ml solution of E. coli 055:B5 LPS (Sigma L2880) were then added to 15 ml lysate and left for 2 h at room temperature. Forty milliliters of a solution composed of 0.01% malachite green (Sigma), 16% sulfuric acid (Fisher), 1.5% ammonium molybdate (Sigma), and 0.18% Tween-20 (Sigma) were incubated with the lysate for 10 min. LPS- dephosphorylating activity was determined from colorimetric measurements taken at an absorbance of 620 nm.
Statistical analysis.
Statistical significance was determined using either a two-tailed Student's t-test or the Mann-Whitney test unless otherwise indicated, with assistance from GraphPad Prism Software Version 4.00 (GraphPad Software, San Diego, CA; www.graphpad.com). P ≤ 0.05 was considered significant. The results are expressed as the means ± SE.
RESULTS
Vitamin D3-deficient mice are more susceptible to C. rodentium infection, carrying higher cecal and extraintestinal pathogen burdens.
To determine the role of vitamin D3 during enteric infection, we first fed weanling mice either a vitamin D3-deficient (0 IU) or vitamin D3-sufficient (1,000 IU) diets for 5 wk. There was no difference in food intake or body weight between the two groups during the 5-wk feeding trial (data not shown). Dietary vitamin D3 is converted in the liver into 25(OH)D3, which is the major circulating form of vitamin D in the body and used to assess vitamin D status. After 5 wk, vitamin D3-deficient mice had significantly lower levels of serum 25(OH)D3, compared with vitamin D3-sufficient mice (Fig. 1A), in agreement with previous studies (3, 35). Vitamin D also plays an important role in regulating calcium metabolism in the body, and supplemental dietary calcium has previously been shown to protect against C. rodentium infection (40, 50), but we found no difference in serum calcium between the diet groups (Fig. 1B), similar to findings by Lagishetty et al. (35), whose feeding protocol (including diet manufacturer) we replicated.
Fig. 1.

Vitamin D3-deficient mice lose more body weight during infection with Citrobacter rodentium and have thickened colon and shrunken ceca at day 10 postinfection (pi). Weanling (3-wk-old) female C57Bl/6 mice were fed vitamin D3-deficient (0 IU) or vitamin D3-sufficient (1,000 IU) diets for 5 wk and then orally infected with C. rodentium for 10 days. A: serum 25(OH)D3. Results are representative of 3 independent experiments; n = 5–6 per group; *P < 0.05 by Mann-Whitney test. B: serum calcium. Results are representative of 3 independent experiments; n = 6–8 per group. C: body weight. Each data point represents the average body weight pooled from 8 mice and is expressed as the percentage of the initial body weight with SE. Results are representative of 3 independent experiments; n = 8 per group; Student's t-test was conducted at each time point; *P < 0.05 by Mann-Whitney test. D: Macroscopic images of lower gastrointestinal tract (cecum + colon) taken at day 10 postinfection (pi) are representative of group phenotype. VD3, vitamin D3; Def, deficient; Suff, sufficient; CR, C. rodentium; D10 = day 10.
On challenge with C. rodentium, vitamin D3-deficient mice lost 5–8% of their body weight by day 2 pi, significantly greater than vitamin D3-sufficient mice, with this greater weight loss maintained until the mice were euthanized at day 10 pi (Fig. 1C). Vitamin D3-deficient mice had thicker colons and shrunken ceca, which were often devoid of stool contents compared with infected vitamin D3-sufficient mice (Fig. 1D). To determine pathogen burdens, tissues were homogenized and plated to quantify C. rodentium. While no significant differences were found regarding pathogen burdens in the colon between the groups at day 10 pi, vitamin D3-deficient mice were found to carry 5- to 10-fold higher C. rodentium burdens in their ceca (cecal tissue + contents) than the vitamin D3-sufficient mice (Fig. 2A). Interestingly, vitamin D3-deficient mice also carried significantly more culturable C. rodentium from extraintestinal tissues, including MLN, spleen, and liver, compared with vitamin D3-sufficient mice at day 10 pi, indicating greater bacterial translocation to these systemic sites with vitamin D3 deficiency (Fig. 2B).
Fig. 2.

Vitamin D3-deficient mice are more susceptible to C. rodentium infection and carry higher bacteria burdens in cecum and extra-intestinal tissues at day 10 pi. Weanling (3-wk-old) female C57Bl/6 mice were fed vitamin D3-deficient (0 IU) or vitamin D3-sufficient (1,000 IU) diets for 5 wk and then orally infected with C. rodentium for 10 days. Whole tissues were homogenized and plated on LB/strep-treated plates to enumerate C. rodentium burdens. A: cecum and colon at day 10 pi. CFU, colony-forming units. Results are representative of 3 independent experiments; n = 7–8 per group; **P < 0.01 by Mann-Whitney test. B: spleen, liver, and mesenteric lymph nodes (MLN) at day 10 pi. Results are representative of 3 independent experiments; n = 8 per group; *P < 0.05 by Mann-Whitney test. C: to assess intestinal barrier integrity, FITC/dextran was administered orally or intrarectally and plasma was assessed for levels of translocated FITC/dextran at day 10 pi. Results are representative of 2 independent experiments; n = 7–9 per group. NS, nonsignificant.
To determine if the exaggerated pathogen translocation in vitamin D deficiency reflected increased disruption of intestinal barrier integrity, mice were administered FD4 through oral or intrarectal routes as measures of proximal or distal intestine, respectively, and their serum was collected and assessed for translocated FD4. We found no significant differences in translocated FD4 levels between vitamin D3-deficient and -sufficient mice at day 6 pi (data not shown) or day 10 pi (Fig. 2C), indicating the exaggerated bacterial translocation may not reflect an overt epithelial barrier defect. Overall these findings indicate that vitamin D3-deficient mice are more susceptible to infection with C. rodentium.
Vitamin D3-deficient mice carry heavier C. rodentium burdens in their cecal contents at day 10 pi.
To explore the basis for the greater C. rodentium burdens seen in vitamin D3-deficient mice, we carefully separated colon and cecal tissues from their luminal contents and quantified the pathogen burdens. While no differences were found in C. rodentium numbers in the colon tissue or colon contents between diet groups at day 10 pi (Fig. 3, A and B), vitamin D3-deficient mice carried significantly more C. rodentium in their cecal contents, whereas there was no significant difference in cecal tissue burdens between groups. These findings indicated that the increased C. rodentium burdens seen in the ceca of vitamin D3 mice were not adherent to the tissue but rather residing in either cecal crypts or in the lumen (Fig. 3A). To determine if vitamin D3-deficient mice showed any impairment in clearing C. rodentium infection, we assessed bacterial burdens at later time points. At day 18 pi, vitamin D3-deficient mice showed a trend for higher C. rodentium burdens in both the colon and cecum compared with vitamin D3-sufficient mice (Fig. 3C); however, the burdens in both groups were still threefold lower than those seen at day 10 pi (height of infection), suggesting that pathogen clearance was still occurring.
Fig. 3.

Vitamin D3-deficient mice carry higher C. rodentium burdens in the cecal contents at day 10 pi and show delayed clearance of pathogen. Weanling (3-wk-old) female C57Bl/6 mice were fed vitamin D3-deficient (0 IU) or vitamin D3-sufficient (1,000 IU) diets for 5 wk and then orally infected with C. rodentium. A: cecal tissues were separated from their contents, homogenized, and plated on Luria broth (LB)/strep-treated plates to enumerate C. rodentium burdens at day 10 pi. Results are representative of 3 independent experiments; n = 7–9 per group; *P < 0.05 by Mann-Whitney test. B: colonic tissues were separated from their contents, homogenized and plated on LB/strep-treated plates to enumerate C. rodentium burdens at day 10 pi. Results are representative of 3 independent experiments; n = 7–9 per group. C: whole cecal and colonic tissues were homogenized and plated on LB/strep-treated plates to enumerate C. rodentium burdens at day 18 pi. Results are from 1 experiment; n = 4 per group.
Vitamin D3-deficient mice display exaggerated cecitis during C. rodentium infection.
During infection, C. rodentium initially colonizes the cecum and then spreads to the distal colon of wild-type mice, resulting in characteristic histological damage to these regions, including goblet cell depletion, crypt hyperplasia, loss of epithelial integrity, and inflammatory cell infiltration. At day 10 pi, the ceca of vitamin D3-deficient mice displayed worsened histological damage, with significantly more submucosal edema and crypt hyperplasia compared with vitamin D3-sufficient mice, whose ceca had modest damage, as typical for a wild-type C56Bl/6 mouse at day 10 pi (Fig. 4). We also examined Swiss-rolled sections of the colon for histological damage. Although we found a trend for worsened C. rodentium-induced damage in the distal colon of vitamin D3-deficient mice at day 10 pi, the differences between dietary groups did not reach statistical significance (data not shown). Furthermore, there was no difference in histological scores between dietary groups under uninfected conditions in the colon or cecum (data not shown). Since we found higher pathogen burdens and greater tissue damage in the ceca of vitamin D3-deficient mice, we focused our additional analysis on cecal tissues. To further characterize the mucosal pathology and responses to infection, epithelial cell proliferation in the cecum was determined by Ki67 staining. At day 10 pi, vitamin D3 deficiency mice displayed significantly more Ki67+ve intestinal epithelial cells compared with vitamin D3-sufficient infected mice (Fig. 4, C and D). These results are in agreement with previous work showing that active vitamin D [1,25(OH)2D3] can inhibit cell proliferation in human colonic epithelial cells in vitro (43). However, these changes were only seen during infection, as there were no overt differences in intestinal epithelial cell proliferation between uninfected groups.
Fig. 4.

Vitamin D3-deficient mice suffer worsened histological damage with increased cell proliferation in the ceca at day 10 pi with C. rodentium. Weanling (3-wk-old) female C57Bl/6 mice were fed vitamin D3-deficient (0 IU) or vitamin D3-sufficient (1,000 IU) diets for 5 wk and then orally infected with C. rodentium. A: representative image of cross section of the cecum at day 10 pi. Original magnification = ×50 for top and ×200 for bottom. Scale bar = 200 μm. B: cecum was assessed for histological damage by scoring system for C. rodentium described in materials and methods. GC, goblet cell. Results are representative of 3 independent experiments; n = 8 per group; *P < 0.05 by Mann-Whitney test. C: representative images of formalin fixed cross section of cecum at day 10 pi. Immunofluorescence stained: blue, DAPI; red, Ki67. Original magnification = ×50; scale bar = 200 μm. D: cecum was assessed for number of Ki67+ve cells in the lumen (percent of +ve Ki67 per +ve DAPI per area measured). Results are representative of 3 independent experiments; n = 4 per group (uninfected) and n = 11 per group at day 10 pi; **P < 0.01 by Mann-Whitney test.
Vitamin D3-deficient mice develop an elevated inflammatory tone in the cecum under both uninfected and infected conditions.
To determine if the increased susceptibility of vitamin D3-deficient mice to C. rodentium infection could reflect an altered host immune response, we assessed cecal tissues for inflammatory cells and mediators. While no differences in macrophage/neutrophil numbers were noted in the cecum of vitamin D3-deficient or -sufficient groups under uninfected conditions, at day 10 pi, vitamin D3-deficient mice showed more infiltrating macrophages and neutrophils in their cecal tissues, particularly in the submucosal regions, compared with vitamin D3-sufficient mice (data not shown).
We next assessed cecal tissues for the transcription of genes encoding cytokines that influence susceptibility to C. rodentium infection. Interestingly, vitamin D3-deficient mice showed higher transcript levels for the acute inflammatory cytokines TNF-α, IL-1β, and IL-6 under both uninfected and day 10 pi conditions (Fig. 5), indicating a higher inflammatory tone. As expected, infection also induced an increase in transcript levels for Th17-related cytokines (IL-17A and IL-17F) in the ceca, similar to findings previously described in the infected colon (10, 60). However, the infection induced increase in IL-17A and IL-17F transcripts was significantly greater in vitamin D3-deficient mice compared with vitamin D3-sufficient mice (Fig. 6A). Interestingly, the vitamin D3-deficient mice also showed higher transcript levels of the anti-inflammatory cytokines IL-10 and TGF-β (Fig. 6B), suggesting an attempt to counteract the increased inflammatory tone.
Fig. 5.
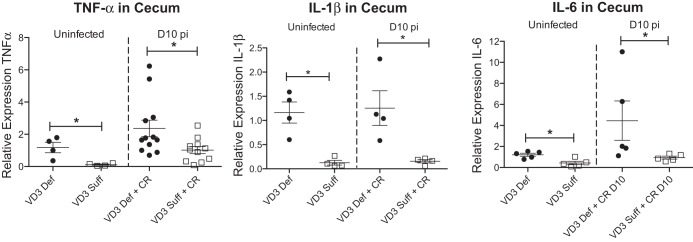
Vitamin D3-deficient mice have higher cecal expression of TNF-α, IL-1β, and IL-6. Weanling (3-wk-old) female C57Bl/6 mice were fed vitamin D3-deficient (0 IU) or vitamin D3-sufficient (1,000 IU) diets for 5 wk and then orally infected with C. rodentium. Expression of TNF-α, IL-1β, and IL-6 in cecum during uninfected conditions and day 10 pi as assessed by quantitative (q)RT-PCR. Results are representative of 3 independent experiments; n = 4–12 per group; *P < 0.05 by Mann-Whitney test.
Fig. 6.

Vitamin D3-deficient mice have higher cecal expression of IL-17A, IL-17F, and TGF-β. Weanling (3-wk-old) female C57Bl/6 mice were fed vitamin D3-deficient (0 IU) or vitamin D3-sufficient (1,000 IU) diets for 5 wk and then orally infected with C. rodentium. Expression of IL-17A, IL-17F, IL-10, and TGF-β in cecum during uninfected conditions and day 10 pi as assessed by qRT-PCR. Results are representative of 3 independent experiments; n = 4–10 per group; *P < 0.05 by Mann-Whitney test.
Vitamin D3 deficiency alters commensal bacteria at baseline and during infection with C. rodentium.
Considering the higher luminal and systemic pathogen burdens carried by infected vitamin D3-deficient mice, we next examined whether their increased susceptibility could be attributed to differences in their commensal intestinal microbiota. It has recently been shown that vitamin D3-deficient mice have an altered fecal microbiome composition (3), while mice with altered vitamin D signaling, including VDR−/− mice and Cyp27b1−/− mice, have been described as suffering intestinal microbial dysbiosis (39). Assessing the microbiota within the stool (using Sybr Green stain) of our two dietary groups under uninfected and infected conditions, we found no difference in the number of commensal bacteria/gram between the dietary groups under uninfected conditions (Fig. 7A). Interestingly, vitamin D3-deficient mice carried significantly more SFB in their stool at day 0, compared with vitamin D3-sufficient mice (Fig. 7B), as determined by qPCR. In contrast, we found no significant differences in Bacteroidetes or γ-Proteobacteria in the stool between dietary groups during uninfected conditions (Fig. 7B), and although there was a trend (P = 0.0850) for vitamin D3-deficient mice to carry fewer Firmicutes, it did not reach statistical significance.
Fig. 7.

Vitamin D-deficient mice have more segmented filamentous bacteria in stool at baseline conditions and have a more dramatic drop in commensal bacteria at day 6 pi with C. rodentium. Weanling (3-wk-old) female C57Bl/6 mice were fed vitamin D3-deficient (0 IU) or vitamin D3-sufficient (1,000 IU) diets for 5 wk and then orally infected with C. rodentium. A: total commensals were assessed in stool during uninfected conditions by DAPI stain. Results are representative of 2 independent experiments; n = 8 per group. B: microbial composition of stool was analyzed by qPCR. SFB, segmented filamentous bacteria. Results are representative of 2 independent experiments; n = 6–7 per group; *P < 0.05 by Mann-Whitney test. C: %commensal bacteria in stool relative to baseline levels were assessed at day 2 pi and day 6 pi. Results are representative of 2 independent experiments; n = 6–7 per group; ***P < 0.0001 by Mann-Whitney test. D: makeup of cecal microbiota at day 10 pi was assessed by qPCR. Each bar represents 1 sample. E: makeup of cecal microbiota at day 10 pi was assessed by qPCR. Results are representative of 2 independent experiments; n = 5–9 per group; *P < 0.05, Mann-Whitney test.
Previous studies have shown that C. rodentium infection is associated with a host-driven depletion of commensal microbes that potentially aids pathogen colonization by reducing colonization resistance (45). We assessed commensal microbe numbers over the course of infection and found a modest but significant acceleration in commensal loss in the vitamin D3-deficient mice by day 6 pi, in keeping with the increased inflammatory response seen in these mice (Fig. 7C). The makeup of the cecal microbiota at day 10 pi was also assessed (Fig. 7D), with vitamin D3-deficient mice found to carry significantly higher levels of Actinobacteria and Bifidobacteria, compared with vitamin D3-sufficient mice (Fig. 7E). There was also a trend for higher levels of γ-proteobacteria and SFB in the cecal contents of vitamin D3-deficient mice at day 10 pi (Fig. 7E), but no significant differences in the major bacteria phyla- Firmicutes or Bacteroides were found between dietary groups at day 10 pi (Fig. 7, D and E).
Vitamin D3-deficient mice upregulate expression of the antimicrobial peptide REG3γ during infection with C. rodentium.
To understand a potential basis for the altered commensal microbes seen in the vitamin D3-deficient group, as well as explore why higher numbers of C. rodentium would be only found within their cecal lumen as opposed to being tissue adherent, we assessed gene transcripts for several antimicrobial factors including REG3γ, which has been shown to play a role in mucosal repair and host defense during infection with C. rodentium (60). Moreover in our previous studies, supplementation with the active form of vitamin D, 1,25(OH)2D3, suppressed transcription of REG3γ, thereby impacting on the susceptibility of mice to C. rodentium infection (41). Notably, REG3γ transcription was dramatically elevated in vitamin D3-deficient mice (Fig. 8), supporting the belief that it is regulated by vitamin D3 and suggesting it may be protecting the cecal mucosa from exaggerated C. rodentium colonization. In contrast, no difference between diet groups was detected for transcript levels of other antimicrobial genes that encode β-defensin 1, β-defensin 3, or mcramp at day 10 pi (data not shown).
Fig. 8.
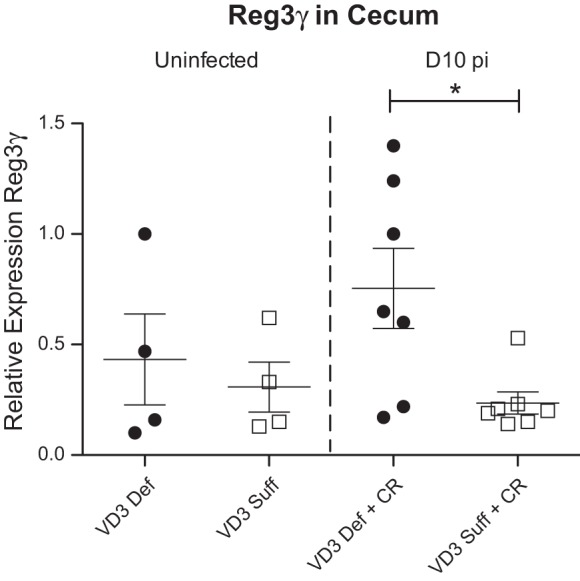
Vitamin D3-deficient mice have higher cecal expression of Reg3γ at day 10 pi with C. rodentium. Weanling (3-wk-old) female C57Bl/6 mice were fed vitamin D3-deficient (0 IU) or vitamin D3-sufficient (1,000 IU) diets for 5 wk and then orally infected with C. rodentium. Expression of Reg3γ in cecum during uninfected conditions and day 10 pi as assessed by qRT-PCR. Results are representative of 2 independent experiments; n = 4–7 per group; *P < 0.05 by Mann-Whitney test.
Bacterial LPS dephosphorylation is impaired in vitamin D3-deficient mice at day 10 pi with C. rodentium.
Lastly, we explored why the intestinal inflammatory response was exaggerated in the vitamin D3-deficient mice. We previously showed that the inflammatory response during C. rodentium infection is largely LPS dependent, as it is significantly reduced in TLR4-deficient mice (32). We therefore examined whether vitamin D3-deficient mice might have altered responses to bacterial LPS. Many of the immune-activating abilities of LPS can be attributed to the lipid A unit, which contains two phosphate groups coupled to glucosamines. Removal of one of the phosphate groups generates a monophosphoryl lipid A that is a 100-fold less toxic than the unmodified lipid A (44). Using the malachite green assay to measure LPS dephosphorylation, we found LPS dephosphorylation was significantly increased in vitamin D3-sufficient mice over that seen in uninfected mice both in the duodenum and ileum (Fig. 9A), likely as a means to limit inflammatory responses against C. rodentium. In contrast, this increase in LPS activity did not occur in vitamin D3-deficient mice during challenge with C. rodentium (Fig. 9A), indicating that vitamin D3-deficient mice may possess reduced LPS dephosphorylation activity during infection.
Fig. 9.
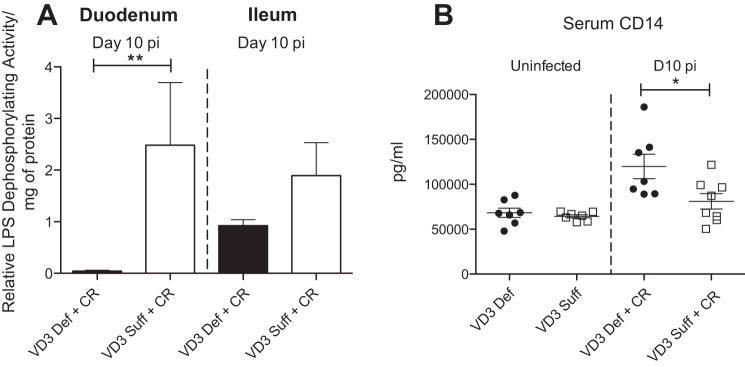
Vitamin D3-deficient mice have impaired detoxification of bacterial lipopolysaccharide and higher serum levels of CD14 at day 10 pi with C. rodentium. Weanling (3-wk-old) female C57Bl/6 mice were fed vitamin D3-deficient (0 IU) or vitamin D3-sufficient (1,000 IU) diets for 5 wk and then orally infected with C. rodentium. A: relative LPS dephosphorylating activity in duodenum and ileum at day 10 pi as determined by the malachite green assay, which measures free phosphate release. Results are representative of 2 independent experiments; n = 6 per group; **P < 0.0043 by Mann-Whitney test. B: serum CD14 during uninfected conditions and at day 10 pi. Results are representative of 3 independent experiments; n = 7–8 per group; *P < 0.05 by Mann-Whitney test.
Vitamin D3-deficient mice have higher levels of serum CD14 at day 10 pi with C. rodentium.
In keeping with the assessment of responses against bacterial LPS, we examined serum levels of CD14, a pattern recognition receptor responsible for the detection of several bacterial products including LPS (55). CD14 is found in two forms, membrane bound CD14 (mCD14) on the surface of monocytes, macrophages, and neutrophils (55), and soluble CD14 (sCD14), which is secreted into bodily fluids including tears, blood, and breast milk (52). In combination with TLR4, detection of LPS by CD14 results in a proinflammatory immune response, therefore, the more CD14, the greater the inflammation. While we noted no difference in serum CD14 levels between uninfected groups, at day 10 pi, however, vitamin D3-deficient mice carried significantly higher levels of serum CD14, compared with vitamin D3-sufficient mice (Fig. 9B), confirming that in the absence of vitamin D3, responses against bacterial LPS are exaggerated.
DISCUSSION
Vitamin D has been shown to modulate a wide variety of immune responses; however, its potential to impact host defense against enteric pathogens within the gastrointestinal (GI) tract is poorly understood. The most significant impact of vitamin D3 in our study was its effect on the host inflammatory response within the intestine. Even under uninfected conditions, vitamin D3-deficient mice showed a higher intestinal inflammatory tone, with elevated cecal expression of TNF-α, IL-1β, IL-6, and IL-17A gene transcripts. Similarly, Assa et al. (3) has shown that vitamin D3-deficient mice had elevated expression of IL-17A and IL-17F in the distal colon compared with vitamin D3-sufficient mice. Moreover, infection with C. rodentium was associated with an appropriate, but exaggerated, response with a further rise in cecal transcript levels of TNF-α, IL-1β, IL-6, IL-17A, and IL-17F with levels that were significantly elevated above vitamin D3-sufficient infected mice. It has previously been shown that TNF-α, IL-1β, and IL-6 play an important role in controlling C. rodentium burdens in the gut and preventing tissue injury during infection (1, 10, 21). However, excess TNF-α and IL-1β are also known to promote tissue injury during infection with C. rodentium (1, 21). Indeed, along with the heightened inflammatory response, vitamin D3-deficient mice also suffered worsened cecal histological damage at day 10 pi, compared with vitamin D3-sufficient mice. Taken together, our results demonstrate that vitamin D3 deficiency results in dysregulated inflammatory responses in the ceca during bacteria challenge.
The basis for this exaggerated inflammatory response was uncertain; however, the elevated levels of TNF-α, IL-1β, and IL-6, even under baseline conditions, suggested the involvement of innate signaling. Bacterial LPS is a major component of the cell wall of Gram-negative bacteria and is recognized by the host innate receptor TLR4, in combination with CD14, MD-2, and the LPS binding protein, found on the surface of monocytes, macrophages, and intestinal epithelial cells. Activation of TLR4 and its cofactors by bacterial LPS induce a signaling cascade that leads to the production of inflammatory mediators and localized recruitment of inflammatory cells. Considering that the active form of vitamin D, 1,25(OH)2D3, has previously been shown to inhibit LPS-induced inflammatory responses in the host through a number of mechanisms, including suppressing TLR4 expression, blocking NF-κB signaling, and targeting MAPK phosphatase-1 (42, 58), we decided to examine whether vitamin D3-deficient mice show exaggerated responses to LPS.
In the current study, we found that vitamin D3-deficient mice showed a defect in their ability to dephosphorylate bacterial LPS, compared with vitamin D3-sufficient mice, at day 10 pi, as determined by the malachite green assay. LPS dephosphorylation can occur by intestinal alkaline phosphatase (IAP), a small intestinal brush border enzyme that plays a critical role in host defense and in maintaining intestinal homeostasis. Indeed, IAP has been shown to reduce intestinal inflammation and limit bacterial translocation into systemic sites (36). Interestingly, although IAP is secreted mainly in the duodenum, it has been shown to retain its activity throughout the small intestine and colon (20) and increased IAP activity has been observed in the colon during infection with C. rodentium (17). A previous study showed that vitamin D-deficient rats exhibited lower total alkaline phosphatase activity in their duodenum, compared with vitamin D-sufficient rats (13). Furthermore, 1,25(OH)2D3 has been shown to stimulate the activity of IAP in the duodenum of chicks (38). Overall these findings indicate that vitamin D may play a role in detoxification of LPS and future studies should investigate IAP.
Along with impaired LPS dephosphorylation, we also noted an increase in serum levels of CD14, a pattern recognition receptor responsible for the detection of several bacterial products including LPS (55). Soluble CD14 (sCD14) is secreted into bodily fluids, and, in combination with TLR4, detection of LPS by CD14 can result in a proinflammatory immune response (52). However, soluble CD14 may also decrease immune responses to LPS by binding LPS and keeping it from mCD14-expressing cells and providing clearance of LPS through the liver (52). Interestingly, the active form of vitamin D, 1,25(OH)2D3, has been shown to increase the expression of CD14 in human monocytes, while suppressing the expression of TLR4 (11, 42). Furthermore, a vitamin D derivative, 1α,25-dihydroxy-22-oxavitamin D3 (Oxa-D3), has been shown to increase the release of soluble CD14 from intestinal HT-29 cells through ERK1/2 activation, in vitro (25). While we noted no difference in serum CD14 levels between uninfected groups, at day 10 pi vitamin D3-deficient mice carried significantly higher levels of serum CD14, compared with vitamin D3-sufficient mice, confirming that in the absence of vitamin D3, responses against bacterial LPS are exaggerated.
Despite their heightened baseline inflammatory tone, vitamin D3-deficient mice proved more susceptible to C. rodentium infection, carrying higher bacterial burdens both in the cecal lumen and at systemic sites. Interestingly, although several previous studies have found that vitamin D can help maintain the intestinal epithelial barrier (3, 16, 34, 59), we did not find any difference in barrier integrity (FITC/dextran assay) between dietary groups at either day 6 pi or day 10 pi, suggesting the translocation to systemic sites was not due to an overtly weaker gut barrier. Although an overt alteration in epithelial barrier integrity was not observed in the vitamin D3-deficient mice, bacteria can enter the systemic circulation through several routes including through intestinal-epithelial microfold cells (M cells), which are permeable to bacteria as well as macromolecules (24). More likely the increased systemic spread of C. rodentium reflects the impaired ability of vitamin D3-deficient mice to dephosphorylate LPS, permitting greater C. rodentium translocation out of the gut. With respect to their increased luminal burdens, previous studies showed that VDR−/− mice carried higher pathogen burdens in their ceca during infection with Salmonella typhimurium (56), although the basis for this was not defined. A potential explanation reflects the concept that pathogens such as C. rodentium actually benefit from intestinal inflammation, since it helps deplete competing commensal bacteria, creating a niche within the intestine where C. rodentium can colonize and proliferate. Furthermore, studies have shown that enteric bacterial pathogens can utilize nutrients and metabolites released within the inflamed intestine that are not used by commensal species (30, 54). We recently demonstrated that mice-deficient in SIGIRR, a negative regulator of innate signaling, not only developed greater inflammatory/antimicrobial responses during C. rodentium infection, but they also proved more susceptible to infection, carrying much higher C. rodentium burdens than wild-type mice (45). Notably, in the current study, we found no difference in the total number of commensal bacteria per gram between the dietary groups under uninfected conditions or at day 2 pi. However, by day 6 pi, vitamin D3-deficient mice showed a significant drop in commensal microbe populations, in keeping with the increased inflammatory response seen in these mice. Moreover, we found an exaggerated induction of the gene encoding the antimicrobial factor REG3γ in the vitamin D3-deficient mice. We have previously shown that 1,25(OH)2D3 treatment in vivo can increase susceptibility to C. rodentium infection by suppressing Th17-mediated immune responses, as well as REG3γ transcription, leading to increased tissue adherent pathogen burdens (41). Thus our studies show REG3γ is clearly dependent on vitamin D status and its upregulated expression in vitamin D3-deficient mice might explain the accelerated loss of commensal microbes in these mice, as well as selective increase in C. rodentium burdens in the cecal lumen, rather than at the mucosal surface where REG3γ is expressed.
While the increased inflammatory tone seen in the uninfected intestines of vitamin D3-deficient mice did not affect total commensal numbers, vitamin D3-deficient mice were found to carry more SFB in their gut luminal contents under both uninfected and infected conditions, compared with vitamin D3-sufficient mice. SFB can induce the differentiation of Th17 cells in the small intestine and can also potentially protect hosts against extracellular bacterial infections or promote inflammatory diseases (28). It is notable that by simply altering dietary vitamin D levels, we were able to alter SFB populations in the gut and change Th17 response. As for other commensals, we found no significant differences in Bacteroidetes or γ-Proteobacteria in the stool between dietary groups during uninfected conditions, and although there was a trend for vitamin D3-deficient mice to carry fewer Firmicutes, it did not reach statistical significance. At day 10 pi, vitamin D3-deficient mice had significantly higher levels of Actinobacteria as well as γ-Proteobacteria in the stool, in agreement with findings from Assa et al. (3). Interestingly, infected vitamin D3-deficient mice also carried significantly higher levels of Bifidobacteria, compared with vitamin D3-sufficient mice; however, we found no differences in the major bacteria phyla-Firmicutes or Bacteroides between dietary groups at day 10 pi, similar to findings by Assa et al. (3).
There is an established link between vitamin D deficiency and respiratory infections (18) and systemic infections (31). However, our understanding of the potential for vitamin D levels to impact host susceptibility to GI infections is limited. Our current study demonstrates that a short 5-wk dietary induced vitamin D3 deficiency increases bacterial overgrowth during infection with the enteric pathogen C. rodentium by altering host factors required for bacterial detoxification, indicating that vitamin D plays a protective role during enteric infection. However, these results should be interpreted with caution, since we have previously shown that treating C. rodentium-infected mice with active vitamin D, 1,25(OH)2D3, led to increased pathogen burdens and exaggerated tissue pathology. In association with their increased susceptibility, 1,25(OH)2D3-treated mice showed less expression of IL-6 and IL-17A and substantially reduced numbers of Th17 T cells within their infected colons. Th17 responses play a protective role during C. rodentium infection (27, 46, 47). Therefore, too much active vitamin D can suppress Th17-mediated inflammatory responses, which can impair host defense against C. rodentium. To summarize, during 1,25(OH)2D3 supplementation, there is suppression of Th17-mediated immune responses that are normally important for clearing C. rodentium infection. In contrast, during vitamin D deficiency, we showed there is a higher baseline intestinal inflammatory and antimicrobial tone, which leads to a faster loss of commensal microbes, reducing commensal microbial competition with C. rodentium,and allowing the pathogen to overgrow and cause increased intestinal damage. Overall these results suggest that to promote gut health there is likely an optimal range of vitamin D and that too little or too much of this vitamin may promote gut inflammation and/or increase susceptibility to enteric infections.
GRANTS
This work was supported by two Grants in Aid awarded by Crohn's and Colitis Canada (CCC; to B. A. Vallance and K. Jacobson) and a discovery grant from the National Science and Engineering Research Council (NSERC; to B. A. Vallance). N. R. Ryz and K. Bhullar were both supported by Vanier Canada Graduate Scholarship Doctoral Research Awards and by Four-Year Doctoral Fellowships from the University of British Columbia. B. A. Vallance is the Children with Intestinal and Liver Disorders (CHILD) Foundation Research Chair in Pediatric Gastroenterology. K. Jacobson is a Senior Clinician Scientist supported by the CHILD Foundation and the Child and Family Research Institute (CFRI) Clinician Scientists Award Program, University of British Columbia.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
N.R.R., K.J., and B.A.V. conception and design of research; N.R.R., A.L., K.B., C.M., T.H., G.B., E.B., and X.W. performed experiments; N.R.R. analyzed data; N.R.R. and B.A.V. interpreted results of experiments; N.R.R. prepared figures; N.R.R. drafted manuscript; N.R.R., S.M.I., K.J., and B.A.V. edited and revised manuscript; N.R.R., A.L., K.B., C.M., T.H., G.B., E.B., X.W., S.M.I., and B.A.V. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the staff at the Child and Family Research Institute animal care facility for help with animal care. We also thank Roger Dyer for help with HPLC analysis of 25(OH)D3.
REFERENCES
- 1.Alipour M, Lou Y, Zimmerman D, Bording-Jorgensen MW, Sergi C, Liu JJ, Wine E. A balanced IL-1beta activity is required for host response to Citrobacter rodentium infection. PLoS One 8: e80656, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ananthakrishnan AN, Cagan A, Gainer VS, Cheng SC, Cai T, Szolovits P, Shaw SY, Churchill S, Karlson EW, Murphy SN, Kohane I, Liao KP. Higher plasma vitamin D is associated with reduced risk of Clostridium difficile infection in patients with inflammatory bowel diseases. Aliment Pharmacol Ther 39: 1136–1142, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Assa A, Vong L, Pinnell LJ, Avitzur N, Johnson-Henry KC, Sherman PM. Vitamin D deficiency promotes epithelial barrier dysfunction and intestinal inflammation. J Infect Dis 210: 1296–1305, 2014. [DOI] [PubMed] [Google Scholar]
- 4.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, Huang T, Velcich A, Finlay BB, Chadee K, Vallance BA. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog 6: e1000902, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhinder G, Stahl M, Sham HP, Crowley SM, Morampudi V, Dalwadi U, Ma C, Jacobson K, Vallance BA. Intestinal epithelium-specific MyD88 signaling impacts host susceptibility to infectious colitis by promoting protective goblet cell and antimicrobial responses. Infect Immun 82: 3753–3763, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouillon R, Carmeliet G, Verlinden L, van Etten E, Verstuyf A, Luderer HF, Lieben L, Mathieu C, Demay M. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev 29: 726–776, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruce D, Yu S, Ooi JH, Cantorna MT. Converging pathways lead to overproduction of IL-17 in the absence of vitamin D signaling. Int Immunol 23: 519–528, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chalmers JD, McHugh BJ, Docherty C, Govan JR, Hill AT. Vitamin-D deficiency is associated with chronic bacterial colonisation and disease severity in bronchiectasis. Thorax 68: 39–47, 2013. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Waddell A, Lin YD, Cantorna MT. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal Immunol 8: 618–626, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dann SM, Spehlmann ME, Hammond DC, Iimura M, Hase K, Choi LJ, Hanson E, Eckmann L. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. J Immunol 180: 6816–6826, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dickie LJ, Church LD, Coulthard LR, Mathews RJ, Emery P, McDermott MF. Vitamin D3 down-regulates intracellular Toll-like receptor 9 expression and Toll-like receptor 9-induced IL-6 production in human monocytes. Rheumatology (Oxford) 49: 1466–1471, 2010. [DOI] [PubMed] [Google Scholar]
- 12.Ding S, Schoenmakers I, Jones K, Koulman A, Prentice A, Volmer DA. Quantitative determination of vitamin D metabolites in plasma using UHPLC-MS/MS. Anal Bioanal Chem 398: 779–789, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dupuis Y, Tardivel S, Ranivosoa A, Fournier P. Intestinal calcium transfer and alkaline phosphatase activity in relation with vitamin D and glucide diet. Arch Int Physiol Biochim Biophys 98: 141–148, 1990. [DOI] [PubMed] [Google Scholar]
- 14.Eckmann L. Animal models of inflammatory bowel disease: lessons from enteric infections. Ann NY Acad Sci 1072: 28–38, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Ferreira GB, van Etten E, Verstuyf A, Waer M, Overbergh L, Gysemans C, Mathieu C. 1,25-Dihydroxyvitamin D3 alters murine dendritic cell behaviour in vitro and in vivo. Diabetes Metab Res Rev 27: 933–941, 2011. [DOI] [PubMed] [Google Scholar]
- 16.Froicu M, Cantorna MT. Vitamin D and the vitamin D receptor are critical for control of the innate immune response to colonic injury. BMC Immunol 8: 5, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh S, DeCoffe D, Brown K, Rajendiran E, Estaki M, Dai C, Yip A, Gibson DL. Fish oil attenuates omega-6 polyunsaturated fatty acid-induced dysbiosis and infectious colitis but impairs LPS dephosphorylation activity causing sepsis. PLoS One 8: e55468, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibney KB, MacGregor L, Leder K, Torresi J, Marshall C, Ebeling PR, Biggs BA. Vitamin D deficiency is associated with tuberculosis and latent tuberculosis infection in immigrants from sub-Saharan Africa. Clin Infect Dis 46: 443–446, 2008. [DOI] [PubMed] [Google Scholar]
- 19.Gibson DL, Ma C, Rosenberger CM, Bergstrom KS, Valdez Y, Huang JT, Khan MA, Vallance BA. Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell Microbiol 10: 388–403, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Goldberg RF, Austen WG Jr, Zhang X, Munene G, Mostafa G, Biswas S, McCormack M, Eberlin KR, Nguyen JT, Tatlidede HS, Warren HS, Narisawa S, Millan JL, and Hodin RA. Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc Natl Acad Sci USA 105: 3551–3556, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goncalves NS, Ghaem-Maghami M, Monteleone G, Frankel G, Dougan G, Lewis DJ, Simmons CP, MacDonald TT. Critical role for tumor necrosis factor alpha in controlling the number of lumenal pathogenic bacteria and immunopathology in infectious colitis. Infect Immun 69: 6651–6659, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greene-Finestone LS, Berger C, de Groh M, Hanley DA, Hidiroglou N, Sarafin K, Poliquin S, Krieger J, Richards JB, Goltzman D; CaMos Research Group. 25-Hydroxyvitamin D in Canadian adults: biological, environmental, and behavioral correlates. Osteoporos Int 22: 1389–1399, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gropper Smith, Groff. The fat-soluble vitamins. In: Advanced Nutrition and Human Metabolism (5th ed). Belmont, CA: Wadsworth Cengage Learning, 2009, p. 373–416. [Google Scholar]
- 24.Hathaway LJ, Kraehenbuhl JP. The role of M cells in mucosal immunity. Cell Mol Life Sci 57: 323–332, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hidaka M, Wakabayashi I, Takeda Y, Fukuzawa K. Vitamin D(3) derivatives increase soluble CD14 release through ERK1/2 activation and decrease IL-8 production in intestinal epithelial cells. Eur J Pharmacol 721: 305–312, 2013. [DOI] [PubMed] [Google Scholar]
- 26.Ikeda U, Wakita D, Ohkuri T, Chamoto K, Kitamura H, Iwakura Y, Nishimura T. 1alpha,25-Dihydroxyvitamin D3 and all-trans retinoic acid synergistically inhibit the differentiation and expansion of Th17 cells. Immunol Lett 134: 7–16, 2010. [DOI] [PubMed] [Google Scholar]
- 27.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30: 108–119, 2009. [DOI] [PubMed] [Google Scholar]
- 28.Ivanov II, Littman DR. Segmented filamentous bacteria take the stage. Mucosal Immunol 3: 209–212, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kallay E, Bises G, Bajna E, Bieglmayer C, Gerdenitsch W, Steffan I, Kato S, Armbrecht HJ, Cross HS. Colon-specific regulation of vitamin D hydroxylases–a possible approach for tumor prevention. Carcinogenesis 26: 1581–1589, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol 13: 321–335, 2013. [DOI] [PubMed] [Google Scholar]
- 31.Kempker JA, Tangpricha V, Ziegler TR, Martin GS. Vitamin D in sepsis: from basic science to clinical impact. Crit Care 16: 316, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan MA, Ma C, Knodler LA, Valdez Y, Rosenberger CM, Deng W, Finlay BB, Vallance BA. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect Immun 74: 2522–2536, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khoo AL, Joosten I, Michels M, Woestenenk R, Preijers F, He XH, Netea MG, van der Ven AJ, Koenen HJ. 1,25-Dihydroxyvitamin D3 inhibits proliferation but not the suppressive function of regulatory T cells in the absence of antigen-presenting cells. Immunology 134: 459–468, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kong J, Zhang Z, Musch MW, Ning G, Sun J, Hart J, Bissonnette M, Li YC. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am J Physiol Gastrointest Liver Physiol 294: G208–G216, 2008. [DOI] [PubMed] [Google Scholar]
- 35.Lagishetty V, Misharin AV, Liu NQ, Lisse TS, Chun RF, Ouyang Y, McLachlan SM, Adams JS, Hewison M. Vitamin D deficiency in mice impairs colonic antibacterial activity and predisposes to colitis. Endocrinology 151: 2423–2432, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lalles JP. Intestinal alkaline phosphatase: novel functions and protective effects. Nutr Rev 72: 82–94, 2014. [DOI] [PubMed] [Google Scholar]
- 37.Lebeis SL, Sherman MA, Kalman D. Protective and destructive innate immune responses to enteropathogenic Escherichia coli and related A/E pathogens. Future Microbiol 3: 315–328, 2008. [DOI] [PubMed] [Google Scholar]
- 38.Moreno J, Cortes CS, Asteggiano CA, Pereira R, Tolosa N, Canas FM, Blanco A. Changes of intestinal alkaline phosphatase produced by cholecalciferol or 1,25-dihydroxyvitamin D3 in vitamin D-deficient chicks. Arch Biochem Biophys 240: 201–206, 1985. [DOI] [PubMed] [Google Scholar]
- 39.Ooi JH, Li Y, Rogers CJ, Cantorna MT. Vitamin D regulates the gut microbiome and protects mice from dextran sodium sulfate-induced colitis. J Nutr 143: 1679–1686, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peleg S, Sellin JH, Wang Y, Freeman MR, Umar S. Suppression of aberrant transient receptor potential cation channel, subfamily V, member 6 expression in hyperproliferative colonic crypts by dietary calcium. Am J Physiol Gastrointest Liver Physiol 299: G593–G601, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryz NR, Patterson SJ, Zhang Y, Ma C, Huang T, Bhinder G, Wu X, Chan J, Glesby A, Sham HP, Dutz JP, Levings MK, Jacobson K, Vallance BA. Active vitamin D (1,25-dihydroxyvitamin D3) increases host susceptibility to Citrobacter rodentium by suppressing mucosal Th17 responses. Am J Physiol Gastrointest Liver Physiol 303: G1299–G1311, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sadeghi K, Wessner B, Laggner U, Ploder M, Tamandl D, Friedl J, Zugel U, Steinmeyer A, Pollak A, Roth E, Boltz-Nitulescu G, Spittler A. Vitamin D3 down-regulates monocyte TLR expression and triggers hyporesponsiveness to pathogen-associated molecular patterns. Eur J Immunol 36: 361–370, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Samuel S, Sitrin MD. Vitamin D's role in cell proliferation and differentiation. Nutr Rev 66: S116–124, 2008. [DOI] [PubMed] [Google Scholar]
- 44.Schromm AB, Brandenburg K, Loppnow H, Zahringer U, Rietschel ET, Carroll SF, Koch MH, Kusumoto S, Seydel U. The charge of endotoxin molecules influences their conformation and IL-6-inducing capacity. J Immunol 161: 5464–5471, 1998. [PubMed] [Google Scholar]
- 45.Sham HP, Yu EY, Gulen MF, Bhinder G, Stahl M, Chan JM, Brewster L, Morampudi V, Gibson DL, Hughes MR, McNagny KM, Li X, Vallance BA. SIGIRR, a negative regulator of TLR/IL-1R signaling promotes Microbiota dependent resistance to colonization by enteric bacterial pathogens. PLoS Pathog 9: e1003539, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shiomi H, Masuda A, Nishiumi S, Nishida M, Takagawa T, Shiomi Y, Kutsumi H, Blumberg RS, Azuma T, Yoshida M. Gamma interferon produced by antigen-specific CD4+ T cells regulates the mucosal immune responses to Citrobacter rodentium infection. Infect Immun 78: 2653–2666, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simmons CP, Goncalves NS, Ghaem-Maghami M, Bajaj-Elliott M, Clare S, Neves B, Frankel G, Dougan G, MacDonald TT. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-gamma. J Immunol 168: 1804–1812, 2002. [DOI] [PubMed] [Google Scholar]
- 48.Simoneau T, Bazzaz O, Sawicki GS, Gordon C. Vitamin D status in children with cystic fibrosis. Associations with inflammation and bacterial colonization. Ann Am Thorac Soc 11: 205–210, 2014. [DOI] [PubMed] [Google Scholar]
- 49.Uaesoontrachoon K, Wasgewatte Wijesinghe DK, Mackie EJ, Pagel CN. Osteopontin deficiency delays inflammatory infiltration and the onset of muscle regeneration in a mouse model of muscle injury. Dis Model Mech 6: 197–205, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Umar S, Morris AP, Kourouma F, Sellin JH. Dietary pectin and calcium inhibit colonic proliferation in vivo by differing mechanisms. Cell Prolif 36: 361–375, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Wilden GM, Fagenholz PJ, Velmahos GC, Quraishi SA, Schipper IB, Camargo CA Jr. Vitamin D status and severity of clostridium difficile infections: a prospective cohort study in hospitalized adults. JPEN J Parenter Enteral Nutr 39: 465–470, 2015. [DOI] [PubMed] [Google Scholar]
- 52.Ward TL, Goto K, Altosaar I. Ingested soluble CD14 contributes to the functional pool of circulating sCD14 in mice. Immunobiology 219: 537–546, 2014. [DOI] [PubMed] [Google Scholar]
- 53.Williams B, Williams AJ, Anderson ST. Vitamin D deficiency and insufficiency in children with tuberculosis. Pediatr Infect Dis J 27: 941–942, 2008. [DOI] [PubMed] [Google Scholar]
- 54.Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, Roth JR, Baumler AJ. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467: 426–429, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249: 1431–1433, 1990. [DOI] [PubMed] [Google Scholar]
- 56.Wu S, Xia Y, Liu X, Sun J. Vitamin D receptor deletion leads to reduced level of IkappaBalpha protein through protein translation, protein-protein interaction, and post-translational modification. Int J Biochem Cell Biol 42: 329–336, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu XP, Bellido T, Manolagas SC. Down-regulation of NF-kappa B protein levels in activated human lymphocytes by 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci USA 92: 10990–10994, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Leung DY, Richers BN, Liu Y, Remigio LK, Riches DW, Goleva E. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J Immunol 188: 2127–2135, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao H, Zhang H, Wu H, Li H, Liu L, Guo J, Li C, Shih DQ, Zhang X. Protective role of 1,25(OH)2 vitamin D3 in the mucosal injury and epithelial barrier disruption in DSS-induced acute colitis in mice. BMC Gastroenterol 12: 57, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14: 282–289, 2008. [DOI] [PubMed] [Google Scholar]