

Abstract
Inhibitors of sodium-glucose cotransporter (SGLT)2 are a new class of oral drugs for type 2 diabetic patients that reduce plasma glucose levels by inhibiting renal glucose reabsorption. There is increasing evidence showing the beneficial effect of SGLT2 inhibitors on glucose control; however, less information is available regarding the impact of SGLT2 inhibitors on cardiovascular outcomes. The present study was designed to determine whether SGLT inhibitors regulate vascular relaxation in mouse pulmonary and coronary arteries. Phlorizin (a nonspecific SGLT inhibitor) and canagliflozin (a SGLT2-specific inhibitor) relaxed pulmonary arteries in a dose-dependent manner, but they had little or no effect on coronary arteries. Pretreatment with phlorizin or canagliflozin significantly inhibited sodium nitroprusside (SNP; a nitric oxide donor)-induced vascular relaxation in pulmonary arteries but not in coronary arteries. Phlorizin had no effect on cGMP-dependent relaxation in pulmonary arteries. SNP induced membrane hyperpolarization in human pulmonary artery smooth muscle cells, and pretreatment of cells with phlorizin and canagliflozin attenuated SNP-induced membrane hyperpolarization by decreasing K+ activities induced by SNP. Contrary to the result observed in ex vivo experiments with SGLT inhibitors, SNP-dependent relaxation in pulmonary arteries was not altered by chronic administration of canagliflozin. On the other hand, canagliflozin administration significantly enhanced SNP-dependent relaxation in coronary arteries in diabetic mice. These data suggest that SGLT inhibitors differentially regulate vascular relaxation depending on the type of arteries, duration of the treatment, and health condition, such as diabetes.
Keywords: diabetic vascular complications, membrane potential, vasodilatation, sodium-glucose cotransporters, nitric oxide
sodium-glucose cotransporters (SGLTs) are a family of glucose transporters, and six isoforms have been identified (SGLT1–SGLT6) (5). Of these isoforms, SGLT1 and SGLT2 play a critical role in renal glucose reabsorption; SGLT2 is responsible for the majority of renal glucose reabsorption (nearly 90%), whereas SGLT1 accounts for ∼10% of the reabsorption (4). Inhibition of SGLTs leads to a significant decrease in blood glucose levels by preventing glucose reabsorption in the kidneys. Therefore, SGLT inhibitors have been proposed as a new treatment for type 2 diabetic (T2D) patients.
Phlorizin, a nonselective SGLT inhibitor, was the first SGLT inhibitor tested for blood glucose control. Chronic administration of phlorizin decreases the plasma glucose level in diabetic animal models (40, 41); however, this drug was not developed further clinically because of several disadvantages (e.g., nonspecific reaction to SGLT1, low bioavailability, and the potential inhibitory effect on glucose transporter 1) (10). On the other hand, the SGLT2-selective inhibitors dapagliflozin, canagliflozin, and empagliflozin went through clinical trials and were approved for T2D patients in Europe first. Canagliflozin (Invokana) was approved by the United States Federal Drug Administration in March 2013 followed by the approval of dapagliflozin (Farxiga) in January 2014 and empagliflozin (Jardiance) in August 2014. These drugs exhibit a significant effect by reducing blood glucose levels in both diabetic animal models and patients with T2D (17, 21, 30). However, to the best of our knowledge, there are no reports that have investigated the effect of SGLT inhibitors on vascular reactivity in normal conditions and diabetes.
Micro- and macrovascular complications contribute to mortality and morbidity in patients with diabetes (23, 24). Coronary artery disease is the seventh leading cause of mortality in diabetes, and it is mainly caused by atherosclerotic plaque formation in the coronary artery. Other potential causes for coronary artery disease include sustained coronary arterial contraction in diabetic patients (26, 35). Decreased nitric oxide (NO) bioavailability and impaired NO-dependent vascular relaxation are implicated in the development and progression of many cardiovascular diseases. Attenuated coronary arterial relaxation leads to a decrease in the blood supply to the heart and subsequently induces myocardial ischemia. NO donors, such as nitroglycerin, are used clinically for the treatment of angina pectoris as a result of myocardial ischemia or intermittent vasospasm of coronary vessels (27, 39). Pulmonary arterial dysfunction in T2D patients is not common, but there are several clinical reports that have indicated a link between diabetes and pulmonary hypertension (2, 28, 37). Attenuated pulmonary arterial relaxation increases pulmonary vascular resistance and induces pulmonary hypertension. Aerosol inhalation of a NO donor or NO gas is an effective treatment for pulmonary hypertension and respiratory distress in both adult and pediatric patients (7, 13, 20). Some diabetic patients who use a NO donor as a treatment for their heart or lung problems might consider taking SGLT2 inhibitors for glycemic control. Therefore, there is a matter of great urgency to test the vascular response to this new drug. In the present study, we examined the effect of the SGLT inhibitors phlorizin and canagliflozin on the regulation of vascular tone as well as NO-induced vascular relaxation in mouse coronary and pulmonary arteries. The observations from this study will help us understand the mechanisms by which SGLT inhibitors affect the cardiovascular system and provide important information about the potential beneficial and/or adverse effects of SGLT inhibitors on diabetic patients.
MATERIALS AND METHODS
Biological materials and reagents.
Canagliflozin was purchased from Selleck Chemicals (Houston, TX). Human pulmonary artery endothelial cells (PAECs), human pulmonary artery smooth muscle cells (PASMCs), human coronary artery endothelial cells (CAECs), and human coronary artery smooth muscle cells (CASMCs) were obtained from Cell Applications (San Diego, CA). Kidney total RNA was purchased from Life Technologies (Grand Island, NY), and small intestine total RNA was from ZYAGEN (San Diego, CA). All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
Animal preparation.
All investigations conformed with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The present study was approved by and conducted in accordance with guidelines established by the Institutional Animal Care and Use Committee of the University of Illinois at Chicago and the University of Arizona. Male C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). T2D mice were generated by a single injection of streptozotocin (STZ; 75 mg/kg ip, dissolved in citrate buffer) at 6 wk of age and given a high-fat diet (60% kcal) from the day of STZ injection (6). All surgery was performed under anesthesia with a mixture of ketamine (100 mg/kg ip) and xylazine (5 mg/kg ip), and all efforts were made to minimize suffering.
Assay of SGLT mRNA levels.
mRNA levels of SGLT subtypes in the small intestine, kidney, PAECs, PASMCs, CAECs, and CASMCs from humans and mice were detected by real-time RT-PCR. Mouse PAECs and CAECs were isolated using CD31-coated magnetic beads, as previously described (6, 11), and mouse CASMCs were isolated by NG2-coated magnetic beads (25). Mouse PASMCs were obtained as follows: mouse pulmonary arteries were isolated from the lungs, and the endothelial layer was removed by physical rubbing of the inner layer of the artery with a 40-μm wire. mRNA from the cells or tissues was isolated using the miRNeasy Mini Kit (QIAGEN, Chatsworth, CA). The intestine sample was used as a positive control for SGLT1 and SGLT3, and the kidney sample was used for SGLT2, SGLT4, SGLT5, and SGLT6. cDNA was made by reverse transcription of DNase-free RNA templates using SuperScript III First strand Synthesis SuperMix (Life Technologies). Primer sequences are shown in Table 1. 18S rRNA was used as an endogenous reference gene. Measurements were made in triplicate with a Bio-Rad Real Time PCR System. The efficiency correlated cycle threshold (ΔCT) method was used to determine the level (in arbitrary units) of SGLTs relative to 18S rRNA. Values are shown as ratios to each positive control correspondingly.
Table 1.
Primer information
| Forward | Reverse | |
|---|---|---|
| Mouse SGLT1 | 5′-GACATCTCAGTCATCGTCATC-3′ | 5′-TGTGATTGTATAAAGGGCAGTG-3′ |
| Mouse SGLT2 | 5′-TGGTGTTGGCTTGTGGTCTATG-3′ | 5′-GGCACAAAAAGCCATCCGAG-3′ |
| Mouse SGLT3 | 5′-TGCTGAAGACGAACCGAAGC-3′ | 5′-GCAAACGAATGTGATTCAAACGC-3′ |
| Mouse SGLT4 | 5′-CGGAGCTTGTGGCAATGGA-3′ | 5′-TGCACGGATGGATGACCAAAT-3′ |
| Mouse SGLT5 | 5′-GGCATATGGTCGGCTTGTCG-3′ | 5′-TGGGTACAAACACCCATGCC-3′ |
| Mouse SGLT6 | 5′-GCATTTCTGTCGCTGCCTATG-3′ | 5′-ATTCTGGCATCGTGGTTACCT-3′ |
| Human SGLT1 | 5′-GGACTGTGGGCTATGTTTTCCA-3′ | 5′-AACCACCAAAACCAGGGCAT-3′ |
| Human SGLT2 | 5′-TCCTGCTGACATCCTAGTCATT-3′ | 5′-GAAGAGCGCATTCCACTCG-3′ |
| Human SGLT3 | 5′-TTTCTGGTGGTGATGGCTGTT-3′ | 5′-ATGGTCATCACCCCCGACTTG-3′ |
| Human SGLT4 | 5′-CTTCGAGTGGAACGCAACCT-3′ | 5′-TCCCTGTGGAGAGGTACAGG-3′ |
| Human SGLT5 | 5′-GGGGCTGTCATCCTGACAAT-3′ | 5′-ATAGACATGAGCACCTGGGAAC-3′ |
| Human SGLT6 | 5′-CATATTTTCCGAGATCCGCTGA-3′ | 5′-CGCTGGACAATCACCTGATCC-3′ |
| 18S rRNA | 5′-GTAACCCGTTGAACCCCATT-3′ | 5′-CCATCCAATCGGTAGTAGCG-3′ |
SGLT, sodium-glucose cotransporter.
Isometric tension measurements in coronary and pulmonary arteries.
Isometric tension was measured to evaluate vascular function as previously described (6, 11). Third-order small coronary arteries or pulmonary arteries were isolated in Krebs-Henseleit solution [containing (in mmol/l) 118.0 NaCl, 25.0 NaHCO3, 1.2 NaH2PO4, 1.2 MgSO4, 1.8 CaCl2, 4.7 KCl, and 11.1 glucose and equilibrated with carbogen (95% O2-5% CO2)]. Arteries were then cleaned of any adherent connective tissues and cut into 1- to 1.2-mm segments (1 arterial ring/mouse was used). Arterial rings were mounted in a dual-chambered myograph (DMT-USA) with 20-μm wires and set at a resting tension of 0.1 g. All segments were equilibrated for 45 min with intermittent washes every 15 min. After equilibration, each arterial ring was contracted by treatment with PGF2α, and SGLT inhibitors were administrated in a dose-dependent manner. For the pretreatment with SGLT inhibitors, the inhibitors were added 20 min before the contraction induced by PGF2α. The degree of relaxation is shown as a percentage of PGF2α-induced contraction. In Figs. 2 and 3, the degree of vasodilatation was normalized by vehicle-induced relaxation.
Fig. 2.
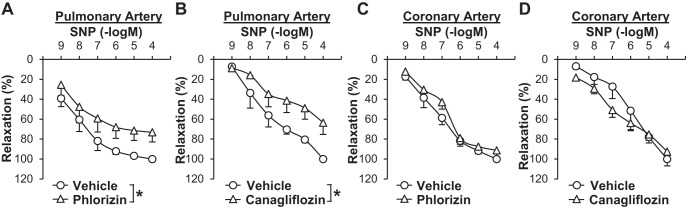
Pretreatment of SGLT inhibitors significantly attenuates sodium nitroprusside (SNP)-induced vascular relaxation in pulmonary arteries. A: dose-response curves of SNP-induced relaxation in the presence or absence of phlorizin in pulmonary arteries (vehicle: 0.05% methanol, phlorizin: 100 µmol/l). Data are means ± SE; n = 4 mice/group. *P < 0.05 vs. vehicle. B: dose-response curves of SNP-induced relaxation in the presence or absence of canagliflozin in pulmonary arteries (vehicle: 0.1% DMSO, canagliflozin: 10 µmol/l). Data are means ± SE; n = 3 mice/group. *P < 0.05 vs. vehicle. C: dose-response curves of SNP-induced relaxation in the presence or absence of phlorizin in coronary arteries. Data are means ± SE; n = 5 mice/group. D: dose-response curves of SNP-induced relaxation in the presence or absence of canagliflozin in coronary arteries. Data are means ± SE; n = 3 mice/group.
Fig. 3.
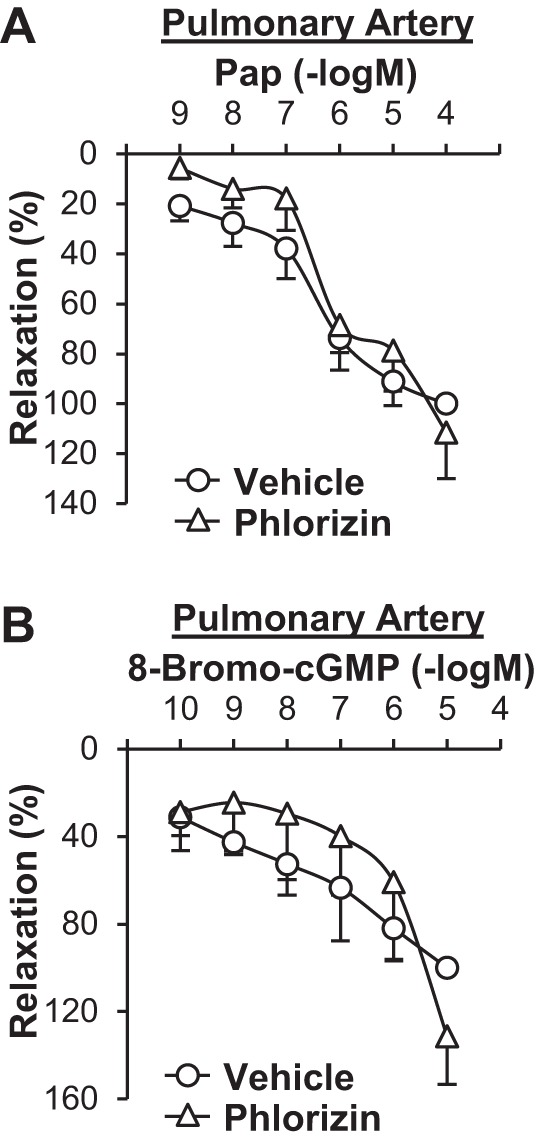
Pretreatment with phlorizin does not affect papaverine (Pap)- or 8-bromo-cGMP-induced relaxation in pulmonary arteries. A: dose-response curves of Pap-induced relaxation in the presence or absence of phlorizin in pulmonary arteries. n = 3 mice/group. B: dose-response curves of 8-bromo-cGMP-induced relaxation in the presence or absence of phlorizin in pulmonary arteries. n = 3 mice/group.
Electrophysiological measurements.
Human PASMCs were used for electrophysiological experiments. Whole cell K+ currents were recorded using conventional patch-clamp techniques. Cells were perfused at a rate of 2 ml/min with Ca2+-free physiological extracellular solution containing (in mmol/l) 141 NaCl, 4.7 KCl, 3 MgCl2, 10 HEPES, 1 EGTA, and 10 glucose (pH 7.4). The intracellular (pipette) solution was composed of (in mmol/l) 135 KCl, 4 MgCl2, 10 HEPES, 10 EGTA, and 5 Na2ATP (pH 7.2). All experiments were performed at room temperature (22–24°C). Patch pipettes (2–3 MΩ) were used to make high-resistance seals with the cell membrane for whole cell and current-clamp recordings. Series resistance compensation was performed in most of the whole cell experiments. Leak and capacitative currents were subtracted using the P/4 protocol in pCLAMP software. K+ currents were elicited by 500-ms step depolarization at potentials ranging between −60 and +60 mV from a holding potential of −70 mV. The resting membrane potential was recorded under the whole cell configuration in the current-clamp mode (current = 0).
Chronic canagliflozin administration.
Canagliflozin was formulated in 0.5% hydroxypropyl methylcellulose (HPMC) (21). Twelve weeks after the diabetic induction, canagliflozin (30 mg·10 ml−1·kg body wt−1) or vehicle (0.5% HPMC) was administered to diabetic or control mice, respectively, via oral gavage every day for 4 wk.
Metabolic characterization.
At the end of the 4-wk treatment with canagliflozin, total cholesterol, HDL, and triglyceride in plasma were measured with kits from Wako Chemicals USA (Richmond, VA). The plasma insulin level was assessed by a kit from ALPCO Diagnostics (Salem, NH). An oral glucose tolerance test was performed as previously reported (6).
Statistical analysis.
Values are expressed as means ± SE. Statistical comparisons within or between dose-response curves were made by one-way or two-way ANOVA with a Bonferroni correction, respectively. Student's t-test for unpaired samples was carried out to identify significant differences. Differences were considered to be statistically significant at P < 0.05.
RESULTS
Expression levels of SGLT in mouse and human PASMCs, PAECs, CASMCs, and CAECs.
Six SGLT isoforms (SGLT1–SGLT6) have been identified in mammals, and SGLT1 and SGLT2 are the major isoforms responsible for renal glucose reabsorption. We used real-time PCR to examine mRNA expression levels of SGLT subtypes in mouse and human PASMCs, PAECs, CASMCs, and CAECs. Samples from the small intestine and kidney were used as positive controls. SGLT2 and SGLT5 were not detected in PASMCs, PAECs, CASMCs, and CAECs with real-time PCR, whereas SGLT1, SGLT3, SGLT4, and SGLT6 were detected but had much lower expression levels compared with the intestine and kidney (Table 2). SGLT1 and SGLT4 were expressed more in PASMCs than in CASMCs, whereas SGLT3 expression levels were less in PASMCs than in CASMCs in mice. In human samples, SGLT1 was detected in PASMCs but not in CASMCs.
Table 2.
mRNA expression levels of SGLTs
| SGLT1 | SGLT2 | SGLT3 | SGLT4 | SGLT5 | SGLT6 | |
|---|---|---|---|---|---|---|
| Mouse Kidney | 0.14 | 1.0 | 25.6E-04 | 1.0 | 1.0 | 1.0 |
| Mouse SI | 1.1 | ND | 1.1 | 0.3 | ND | 0.8 |
| Mouse PASMC | 42.5E-05 | ND | 0.7E-04 | 0.4E-03 | ND | 50.8E-05 |
| Mouse PAEC | 1.8E-05 | ND | 0.1E-04 | ND | ND | 0.4E-05 |
| Mouse CASMC | 1.0E-05 | ND | 5.5E-04 | ND | ND | 50.9E-05 |
| Mouse CAEC | 112.3E-05 | ND | 45.9E-04 | ND | ND | 191.9E-05 |
| Human Kidney | 28.4E-04 | 1.0 | 8.0E-04 | 1.0 | 1.0 | 1.0 |
| Human SI | 1.0 | ND | 1.1 | 12.3 | 0.3 | 9.13 |
| Human PASMC | 2.8E-05 | ND | 10.2E-04 | ND | ND | 2.8E-02 |
| Human PAEC | 2.2E-05 | ND | 27.6E-04 | ND | ND | 12.7E-02 |
| Human CASMC | ND | ND | 6.3E-04 | ND | ND | 2.1E-02 |
| Human CAEC | ND | ND | ND | ND | ND | 18.1E-02 |
Averaged data are shown. SI, small intestine; PASMC, pulmonary artery smooth muscle cell; PAEC, pulmonary artery endothelial cell; CASMC, coronary artery smooth muscle cell; CAEC, coronary artery endothelial cell. mRNA expression was confirmed by semiquantitative RT-PCR at least two to three times per individual SGLT subtype. mRNA expression levels were normalized by the level in kidney samples except SGLT1 and SGLT3. SGLT1 and SGLT3 expression levels were normalized using the level of SI samples. ND, not determined.
Effect of SGLT inhibitors on vascular relaxation in pulmonary and coronary arteries.
We first tested whether SGLT inhibitors caused vasodilatation in mouse pulmonary and coronary arteries. As shown in Fig. 1, A and B, both phlorizin and canagliflozin significantly decreased vascular tone in pulmonary arteries in a dose-dependent manner. In coronary arteries, however, SGLT inhibitors had a small vasodilatory effect, but it was not significant (Fig. 1, C and D).
Fig. 1.
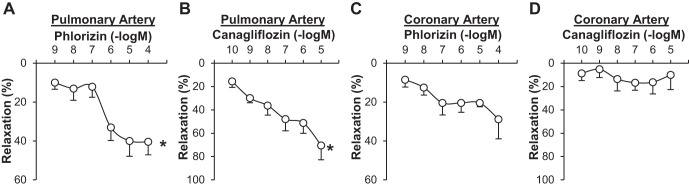
Sodium-glucose cotransporter (SGLT) inhibitors lead to vascular relaxation in pulmonary arteries. A: dose-response curve of phlorizin-induced relaxation in pulmonary arteries. Data are means ± SE; n = 4 mice. *P < 0.05 vs. 1 nmol/l. B: dose-response curve of canagliflozin-induced relaxation in pulmonary arteries. Data are means ± SE; n = 3 mice. *P < 0.05 vs. 100 pmol/l. C: dose-response curve of phlorizin-induced relaxation in coronary arteries. Data are means ± SE; n = 3 mice. D: dose-response curve of canagliflozin-induced relaxation in coronary arteries. Data are means ± SE; n = 4 mice.
Next, we examined whether pretreatment with SGLT inhibitors affected vascular relaxation induced by sodium nitroprusside (SNP; a NO donor). Pretreatment of isolated arterial rings with phlorizin (100 μmol/l, 20 min) or canagliflozin (10 μmol/l, 20 min) significantly inhibited SNP-induced vascular relaxation in pulmonary arteries compared with control arterial rings that were pretreated with vehicle (0.05% methanol for phlorizin and 0.1% DMSO for canagliflozin; Fig. 2, A and B). In coronary arteries, however, pretreatment with SGLT inhibitors had no effect on SNP-induced vascular relaxation (Fig. 2, C and D). NO relaxes vascular smooth muscle cells through the cGMP-PKG cascade and/or by inducing membrane hyperpolarization in smooth muscle cells by activating K+ channels (29, 48, 53). To define the molecular mechanisms by which phlorizin attenuates SNP-induced vascular relaxation in pulmonary arteries, we examined the effect of phlorizin on vascular relaxation induced by the cGMP-PKG cascade, which is a major cascade of NO-induced vascular relaxation. 8-Bromo-cGMP, a membrane-permeable cGMP analog, and papaverine (Pap), an inhibitor of a nonspecific phosphodiesterase that hydrolyzes cGMP, both cause vasodilatation by increasing cGMP concentration in SMCs. 8-Bromo-cGMP and Pap relaxed pulmonary arteries in a dose-dependent manner, whereas pretreatment with phlorizin had no effect on either 8-bromo-cGMP- or Pap-induced vascular relaxation in pulmonary arteries (Fig. 3). These data imply that the inhibitory effect of phlorizin on SNP-induced relaxation is independent of the cGMP-PKG cascade.
Effects of SGLT inhibitors on resting membrane potential and SNP-induced membrane hyperpolarization.
Since phlorizin did not affect 8-bromo-cGMP- or Pap-induced vascular relaxation (Fig. 3), we examined another potential mechanism for NO-induced vascular relaxation: smooth muscle cell membrane hyperpolarization. Indeed, phlorizin (100 μmol/l) induced a marked membrane hyperpolarization in human PASMCs (Fig. 4A), and pretreatment of cells with phlorizin significantly attenuated SNP-dependent membrane hyperpolarization (Fig. 4C). Canagliflozin treatment (10 μmol/l) also led to membrane hyperpolarization in human PASMCs (Fig. 4B), and pretreatment of cells with canagliflozin attenuated SNP-induced membrane hyperpolarization (Fig. 4D). These data suggest that SGLT inhibitors attenuate SNP-induced relaxation in the pulmonary artery due, at least in part, to inhibition of SNP-induced membrane hyperpolarization in pulmonary smooth muscle cells.
Fig. 4.

Effects of SGLT inhibitors on SNP-induced hyperpolarization and K+ channel activation in human pulmonary artery smooth muscle cells. A: averaged data of phlorizin (100 µmol/l)-induced hyperpolarization. Data are means ± SE; n = 5 cells. B: averaged data of canagliflozin (10 µmol/l)-induced hyperpolarization in human pulmonary artery smooth muscle cells. Data are means ± SE; n = 7 cells. C: summarized data of SNP-induced hyperpolarization in the presence or absence of phlorizin (vehicle: 0.05% methanol, n = 6 cells; phlorizin: 100 µmol/l, n = 6 cells). Data are means ± SE. *P < 0.05 vs. vehicle. D: summarized data of SNP-induced hyperpolarization in the presence or absence of canagliflozin (vehicle: 0.1% DMSO, n = 5 cells; and canagliflozin: 10 µmol/l, n = 5 cells). Data are means ± SE. E: representative K+ currents in the presence or absence of SNP and phlorizin. F: summarized current-voltage (I–V) relationship curves. Data are means ± SE; n = 6 cells/group. *P < 0.05 vs. vehicle; #P < 0.05 vs. Vehicle + SNP. G: representative K+ currents in the presence or absence of SNP and canagliflozin. H: summarized I–V relationship curves. Data are means ± SE; n = 4 cells/group. *P < 0.05 vs. vehicle; #P < 0.05 vs. Vehicle + SNP.
Effects of SGLT inhibitors on SNP-induced K+ channel activation.
NO-induced hyperpolarization is mainly due to the activation of K+ channels (29, 48, 53). To examine how SGLT inhibitors attenuated SNP-induced hyperpolarization, we tested the effect of SGLT inhibitors on SNP-induced K+ channel activation. Figure 4, E–H, showed that SNP activates K+ channels and that both phlorizin and canagliflozin significantly attenuated SNP-induced K+ channel activation.
Effect of chronic treatment of canagliflozin on SNP-induced relaxation in pulmonary and coronary arteries.
Our T2D mice induced by a high-fat diet and a low dose of STZ injection exhibit impaired glucose tolerance and hyperlipidemia (Fig. 5 and Table 3). This model is a well-established T2D mouse model and is very close to human T2D induced by a Western diet (6). We measured and compared SGLT mRNA levels in PASMCs and CASMCs isolated from control and T2D mice (Table 4). In PASMCs, there was no difference in mRNAs of SGLT 1, SGLT4, and SGLT6 between control and T2D samples. However, in CASMCs, SGLT6 mRNA expression was significantly decreased in T2D compared with control samples.
Fig. 5.

Effect of chronic administration of canagliflozin on the oral glucose tolerance test, plasma insulin level, and SNP-induced vascular relaxation in pulmonary and coronary arteries. A: for the oral glucose tolerance test, mice were fasted for 6 h before the experiment [control + 0.5% hydroxypropyl methylcellulose (HPMC): n = 5 mice; type 2 diabetes (T2D) + HPMC: n = 7 mice, and T2D + canagliflozin: n = 7 mice]. Data are means ± SE. *P < 0.05 vs. control + HPMC; #P < 0.05 vs. T2D + HPMC. B: plasma samples were collected for the measurement of plasma insulin concentration without fasting the mice. Data are means ± SE; n = 7 mice/group. *P < 0.05 vs. control + HPMC. C: dose-response curves of SNP-induced relaxation in pulmonary arteries dissected from control or diabetic mice treated with HPMC or canagliflozin (control + HPMC: n = 6 mice, T2D + HPMC: n = 6 mice, and T2D + canagliflozin: n = 5 mice). Data are means ± SE. *P < 0.05 vs. control + HPMC; #P < 0.05 vs. T2D + HPMC. D: dose-response curves of SNP-induced relaxation in coronary arteries dissected from control or diabetic mice treated with HPMC or canagliflozin (control + HPMC: n = 3 mice, T2D + HPMC: n = 3 mice, and T2D + canagliflozin: n = 5 mice). Data are means ± SE. *P < 0.05 vs. control + HPMC; #P < 0.05 vs. T2D + HPMC. E: dose-response curves of Pap-induced relaxation in pulmonary arteries dissected from control or diabetic mice treated with HPMC or canagliflozin (control + HPMC: n = 6 mice, and T2D + HPMC: n = 6 mice, and T2D + canagliflozin: n = 5 mice). Data are means ± SE. F: dose-response curves of Pap-induced relaxation in coronary arteries dissected from control or diabetic mice treated with HPMC or canagliflozin (control + HPMC: n = 3 mice, T2D + HPMC: n = 3 mice, and T2D + canagliflozin: n = 5 mice). Data are means ± SE.
Table 3.
Metabolic characterization
| Control + HPMC | T2D + HPMC | T2D + Canagliflozin | |
|---|---|---|---|
| Body weight, g | 28.2 ± 0.8 | 31.8 ± 0.9 | 33.7 ± 1.7* |
| Total cholesterol, mg/dl | 64.6 ± 7.5 | 186.3 ± 10.7* | 124.6 ± 14.3*† |
| Triglyceride, mg/dl | 21.6 ± 4.0 | 40.8 ± 3.5 | 47.7 ± 5.7 |
| HDL, mg/dl | 36.7 ± 3.1 | 85.0 ± 4.8* | 63.1 ± 8.4* |
| LDL, mg/dl | 32.2 ± 6.2 | 93.1 ± 9.5* | 53.0 ± 9.5† |
Data are means ± SE; n ≥ 7 mice/group. HPMC, hydroxypropyl methylcellulose; T2D, type 2 diabetes.
P < 0.05 vs. control + HPMC;
P < 0.05 vs. T2D + HPMC.
Table 4.
mRNA expression levels of SGLTs in pulmonary arterial and coronary arterial smooth muscle cells isolated from control and T2D mice
| Control | T2D | |
|---|---|---|
| Mouse pulmonary arterial smooth muscle cells | ||
| SGLT1 | 1.19 ± 0.13 | 1.06 ± 0.23 |
| SGLT4 | 1.04 ± 0.01 | 1.41 ± 0.47 |
| SGLT6 | 1.04 ± 0.03 | 1.13 ± 0.05 |
| Mouse coronary arterial smooth muscle cells | ||
| SGLT1 | 1.05 ± 0.03 | 1.29 ± 0.37 |
| SGLT6 | 1.03 ± 0.01 | 0.52 ± 0.18* |
Data are means ± SE; n = 3 mice/group.
P < 0.05 vs. control.
Canagliflozin and HPMC (vehicle) administration in mice was started at 12 wk after diabetic induction and continued for 4 wk. Chronic canagliflozin treatment significantly improved glucose tolerance in T2D mice (Fig. 5A). Our T2D mice exhibited hyperinsulinemia (6), and chronic canagliflozin treatment further increased the plasma insulin level in these mice (Fig. 5B). Chronic canagliflozin treatment also showed a beneficial effect on lipid concentrations; total cholesterol and LDL were significantly decreased, whereas plasma triglyceride was slightly increased by canagliflozin treatment in T2D mice (Table 3).
Pulmonary arteries from diabetic mice exhibited a slight but statistically significant decrease in SNP-induced relaxation compared with pulmonary arteries from control mice. Chronic canagliflozin treatment, however, did not improve SNP-induced vasodilatation in diabetic pulmonary arteries (Fig. 5C). In coronary arteries, there was no difference in SNP-induced vasodilation between control and diabetic mice (Fig. 5D), and this result was consistent with our previous data (6, 11). Contrary to the ex vivo data in the coronary artery (Fig. 2), chronic canagliflozin treatment significantly augmented SNP-induced coronary vasodilation in diabetic mice (Fig. 5D). There was no difference in Pap-induced vascular relaxation among control, T2D, and canagliflozin-treated T2D mice in coronary and pulmonary arteries (Fig. 5, E and F).
DISCUSSION
T2D is a chronic disease characterized by a resistance to the actions of insulin and impaired insulin secretion, which results in hyperglycemia. The treatment for T2D patients is primarily focused on enhancing insulin secretion and/or improving insulin sensitivity. Despite the many treatments available for diabetic patients on the market, the mortality of diabetic patients is still increasing every year. Therefore, it is necessary to develop new therapeutic approaches for diabetic patients. SGLT2 inhibitors are one of the targets as a new class of oral drugs for T2D patients that have shown a significant effect on reducing blood glucose levels in animal models and patients with T2D (17, 21, 30). Cardiovascular complications are commonly seen in diabetic patients and are the risk factor associated with their mortality. However, there are limited studies that have examined the beneficial or adverse effects of SGLT inhibitors on the cardiovascular system. Several clinical reports have indicated that canagliflozin treatment decreases blood pressure in diabetic patients (3, 31, 42), but it is still not clear whether the reduction of blood pressure is because of secondary effects of glycemic control by canagliflozin or a direct effect of canagliflozin on the vascular system. Recently, Kashiwagi et al. (18) demonstrated that preconditioning with phlorizin aggravated ischemia-reperfusion injury in mouse hearts. This adverse effect of phlorizin was due to, at least in part, decreased glucose uptake and the subsequent reduction of ATP synthesis in cardiac myocytes rather than affecting the coronary circulatory system (i.e., the coronary flow rate was not altered by phlorizin infusion). The present study is the first to investigate the effect of SGLT inhibitors on vascular relaxation in mouse pulmonary and coronary arteries.
SGLT1 is a low-capacity, high-affinity transporter that is found essentially on the apical membranes of small intestinal absorptive cells and renal proximal straight tubules. The principle role of SGLT1 is dietary glucose absorption in the small intestine (50). SGLT2 is a high-capacity, low-affinity transporter that is mainly expressed in the early convoluted segment of renal proximal tubules and functions as a major renal glucose transporter (4, 5). Several investigations have demonstrated that SGLT1 and/or STLT2 are also expressed in other organs and tissues, including the heart (5, 49, 54), lungs (36, 54), and pulmonary and coronary arterial endothelial cells (45). Taubert et al. (45) showed that the glucose-induced increase in cytosolic Ca2+ concentration is enhanced, whereas NO production in endothelial cells is significantly inhibited, by pretreatment of cells with phlorizin (100 µmol/l), implying that SGLT1/SGLT2 may play a potential role in regulating the vascular tone. Figures 1 and 2 from the present study show that phlorizin and canagliflozin exerted a vasodilatory effect on the pulmonary artery (Fig. 1, A and B) and had a significant inhibitory effect on vascular relaxation induced by SNP (Fig. 2, A and B). Since phlorizin pretreatment had little effect on either 8-bromo-cGMP-induced or Pap-induced pulmonary vasodilation (Fig. 3), phlorizin might not directly regulate the cGMP-PKG cascade in the pulmonary artery. Indeed, our pilot studies showed that the cGMP concentration after SNP treatment was not altered by phlorizin pretreatment in human PASMCs (vehicle: 0.73 ± 0.03 pmol/l and phlorizin: 0.75 ± 0.03 pmol/l, n = 9 in each group).
Two reports have shown that phlorizin exhibited a nonspecific effect on sarco(endo)plasmic reticulum Ca2+-ATPase at a concentration above 50 μmol/l (32, 33). To elucidate the effect of SGLT inhibitors on SNP-induced relaxation at a concentration lower than 50 μmol/l, we tested concentrations of 1 and 10 μmol/l for phlorizin and 100 nmol/l for canagliflozin (data not shown). Even at a concentration that is 100 times lower than that used in Fig. 2, SNP-induced vascular relaxation was significantly inhibited by these inhibitors in pulmonary arteries, suggesting that the inhibitory effect of SGLT is independent of sarco(endo)plasmic reticulum Ca2+-ATPase activation (which required higher dosage).
It is well established that high glucose treatment leads to membrane depolarization in different cell types and that SGLT inhibitors attenuate glucose-induced membrane depolarization (9, 14). Nevertheless, the direct effect of SGLT inhibitors on membrane potential has not been explored in arterial smooth muscle cells. Figure 4 shows that both phlorizin and canagliflozin induced membrane hyperpolarization in human PASMCs, implying that SGLT inhibitors might relax pulmonary arteries by inducing smooth muscle cell membrane hyperpolarization. The resting membrane potential in smooth muscle cells is mainly determined by the activity of Na+-K+-ATPase and K+ channels in the plasma membrane. Activation of K+ channels (by NO or other endothelium-derived hyperpolarizing factors) induces membrane hyperpolarization by driving the resting membrane potential close to the K+ equilibrium potential (approximately −85 mV given that extracellular and intracellular K+ concentrations are about 5 and 140 mmol/l, respectively). Accordingly, when a cell is hyperpolarized (by NO or SNP) due to activation of K+ channels or an increase in K+ currents, the extracellular application of another hyperpolarizing factor would likely be unable to cause further membrane hyperpolarization by opening K+ channels. That is, when cells are treated with two hyperpolarizing factors that cause membrane hyperpolarization by opening K+ channels, the hyperpolarizing effect of one factor would be inhibited by the other. In other words, in the presence of one of the hyperpolarizing factors, the hyperpolarizing effect of the other hyperpolarizing factor should be significantly inhibited or decreased. The results from this study show that 1) SNP (by donating NO) and SGLT inhibitors both cause membrane hyperpolarization in human PASMCs and 2) SNP-mediated membrane hyperpolarization is significantly inhibited in human PASMCs pretreated with SGLT inhibitors (Fig. 4, A–D). Furthermore, we demonstrated that SNP-induced K+ activation was significantly attenuated by pretreatment with SGLT inhibitors (Fig. 4, E–H). Given the fact that SNP or NO causes membrane hyperpolarization by, at least partially, activating K+ channels (29, 48, 53), our data imply that the SGLT inhibitor-mediated membrane hyperpolarization in human PASMCs is likely due to activation of K+ channels, a similar mechanism by which SNP/NO causes membrane hyperpolarization.
In addition to functional experiments, we also examined the expression levels of SGLT subtypes in PASMCs, PAECs, CASMCs, and CAECs. We initially anticipated that PASMCs may express more SGLT2 than CASMCs based on functional experiments showing that pretreatment with SGLT inhibitors attenuated pulmonary vasodilation but not coronary vasodilation. Contrary to our expectations, the real-time PCR data shown in Table 2 indicate that SGLT2 was not expressed in PASMCs and CASMCs. These results suggest that 1) SGLT inhibitors affect smooth muscle cell functions (e.g., causing membrane hyperpolarization by activating K+ channels and vasodilation) through mechanisms independent of the classical transporter-mediated cascade (i.e., Na+-dependent glucose transportation) or have a nonspecific effect and/or 2) phlorizin and canagliflozin cause membrane hyperpolarization in PASMCs and vasodilation through the classical transporter-mediated cascade via other isoforms of the SGLT family (e.g., SGLT3–SGLT6).
The localization of SGLT3 varies depending on the species and tissue/cell types; SGLT3 is expressed in skeletal muscle in human, the small intestine in human and pig, and the kidney in pig and mouse (9, 43). Interestingly, SGLT3 does not function as a glucose transporter but as a glucose sensor (9, 19). SGLT4 is predominantly expressed in the kidney and small intestine and functions to transport glucose and 1,5-anhydro-d-glucitol (46). SGLT4 mainly transports 1,5-anhydro-d-glucitol under healthy conditions, whereas in the diabetic state, it transports excess amounts of glucose that cannot be reabsorbed by SGLT2 and SGLT1 (46, 51, 52). Human kidney also express SGLT5, which is characterized as a mannose and fructose transporter (15). SGLT6 has been cloned, but its structural and functional characterizations have not been fully explored. SGLT3 and SGLT6 have been found to be expressed in the kidney and small intestine, but very low expression levels were detected in vascular cells in mouse and human samples (Table 2). SGLT5 was predominantly expressed in the kidney but not in other tissues. Interestingly, SGLT4 was expressed in PASMCs but not in CASMCs in mouse samples, whereas SGLT4 was not detected in either PASMCs or CASMCs in human samples. These data make it difficult for us to reach a conclusion, and, at this stage, we do not know why acute treatment of SGLT inhibitors exhibited a significant effect in PASMCs but not in CASMCs. More importantly, the specificity of SGLT inhibitors to any of SGLT3–SGLT6 is not fully understood; we need to conduct further experiments to define the role of these isoforms in vascular function.
Chronic treatment with SGLT2 inhibitors significantly decreases fasting blood glucose and HbA1c levels and improves glucose tolerance in experimental diabetic animal models: high-fat diet-induced obese mice [canagliflozin (30 mg/kg), 4 wk] (21), ob/ob mice [dapagliflozin (1 mg/day), 4 wk] (44), and Zucker diabetic fatty rats (16, 21, 22). In the present study, we decided to use a high dose (30 mg/kg) of canagliflozin for our in vivo experiments in mice; this was the highest concentration of canagliflozin we could find in the literature (including the dosage used in acute treatment). The data shown in Fig. 5A demonstrate that canagliflozin treatment significantly improved glucose tolerance in diabetic mice. The renal SGLT2 expression level has been demonstrated to be increased in Akita mice (genetically modified type 1 diabetic mice) and db/db (genetically modified T2D) mice (47) and alloxan-induced type 1 diabetic rats (12). SGLT2 protein or mRNA expression levels are usually increased in proximal tubular cells isolated from diabetic patients compared with cells isolated from control subjects (8, 38). In ex vivo experiments, high glucose treatment increased SGLT2 protein expression in kidney cells (1). Based on these data, we hypothesized that SGLT2 inhibitors work more effectively under diabetic conditions in the kidneys because of higher expression levels of SGLT2 in diabetes than under control conditions. In addition, other SGLT isoforms in control mice might be balancing out the reabsorption of glucose in the kidney.
Our study showed that chronic treatment with canagliflozin significantly increased plasma insulin levels in diabetic mice (Fig. 5B), which is consistent with other investigators' data (21), although there is a report (22) showing that chronic dapagliflozin treatment decreased the plasma insulin level. It has been reported that canagliflozin improves insulin sensitivity by decreasing the demand of insulin secretion (34), implying that the SGLT2 inhibitor could decrease plasma insulin levels. The increase in the plasma insulin levels seen in our diabetic mice may require additional experiments to define the cause or underlying mechanisms. One of the demonstrated adverse effects of canagliflozin treatment is the increase in LDL concentration in diabetic patients (17). However, in our diabetic mice, we observed a decrease in LDL levels after canagliflozin treatment. It is possible that metabolic status differences between mice and humans are the reason for the canagliflozin-mediated increase in LDL only in diabetic patients (but not in diabetic mice).
One of the important questions was whether chronic canagliflozin treatment would 1) inhibit SNP-induced vascular relaxation in pulmonary arteries (as observed in our ex vivo experiments by pretreatment with SGLT inhibitors) or 2) improve SNP-induced relaxation in diabetic mice (that was attenuated 16 wk after diabetic induction) by decreasing plasma glucose levels. Our data indicate that chronic canagliflozin treatment did not inhibit SNP-induced relaxation in diabetic pulmonary arteries (Fig. 5C) but significantly enhanced the vascular sensitivity to SNP in diabetic coronary arteries (Fig. 5D). On the other hand, Pap-induced vascular relaxation was not affected by chronic canagliflozin treatment in T2D mice in either coronary or pulmonary arteries (Fig. 5, E and F), implying that the enhancement of SNP-induced relaxation by canagliflozin seen in coronary arteries is not mediated by an alteration of the cGMP-PKG signaling cascade. Our mRNA measurement data indicate that CASMCs in T2D mice significantly decreased SGLT6 mRNA expression compared with the control, but other subtypes were not changed (Table 4). Since the answer to the fundamental question of “what is the role of SGLT6” is not known, it would be necessary to investigate the signaling cascade and physiological role of SGLT6 in CASMCs together with the pathophysiological role of SGLT6 in T2D mice.
There might be several possibilities that lead to the different results of SNP-induced relaxation between ex vivo canagliflozin pretreatment in isolated arterial rings and in vivo chronic canagliflozin treatment in intact animals: 1) the concentration of SGLT inhibitors we used in the ex vivo experiment was higher than the actual concentration of the drug in the plasma after 4-wk chronic administration, 2) the adverse effect of SGLT inhibitors on vascular function was gradually desensitized or minimized during the 4 wk of drug administration, and 3) the inhibitory effect on NO-dependent relaxation by SGLT inhibitors in pulmonary arteries was masked by other beneficial effects of SGLT inhibitors after a 4-wk treatment in vivo (e.g., improvement of hemodynamic abnormalities in T2D mice).
In summary, the results from this study indicate that acute treatment with SGLT inhibitors exerts an adverse effect on pulmonary arterial function ex vivo (i.e., inhibits pulmonary vasodilation), whereas chronic treatment with SGLT inhibitors in vivo has a beneficial effect on coronary arteries (i.e., enhances coronary vasodilation) in T2D mice. When the SGLT2 inhibitor is used together with NO or a NO donor (e.g., SNP) in diabetic patients, it is important to time the individual treatment appropriately and use the right concentration to avoid undesired interactions of the two drugs.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant HL-115578 (to A. Makino).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Y.H. and A.M. conception and design of research; Y.H., Y.-E.C., R.A., R.G., K.D.Y., M.P., A.D., and A.M. performed experiments; Y.H., Y.-E.C., R.A., K.D.Y., and A.M. analyzed data; Y.H., Y.-E.C., and A.M. interpreted results of experiments; Y.H., Y.-E.C., and A.M. prepared figures; Y.H. and Y.-E.C. drafted manuscript; Y.H., Y.-E.C., R.A., R.G., K.D.Y., M.P., A.D., J.X.-J.Y., and A.M. approved final version of manuscript; K.D.Y., J.X.-J.Y., and A.M. edited and revised manuscript.
REFERENCES
- 1.Beloto-Silva O, Machado UF, Oliveira-Souza M. Glucose-induced regulation of NHEs activity and SGLTs expression involves the PKA signaling pathway. J Membr Biol 239: 157–165, 2011. [DOI] [PubMed] [Google Scholar]
- 2.Benson L, Brittain EL, Pugh ME, Austin ED, Fox K, Wheeler L, Robbins IM, Hemnes AR. Impact of diabetes on survival and right ventricular compensation in pulmonary arterial hypertension. Pulm Circ 4: 311–318, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cefalu WT, Stenlof K, Leiter LA, Wilding JP, Blonde L, Polidori D, Xie J, Sullivan D, Usiskin K, Canovatchel W, Meininger G. Effects of canagliflozin on body weight and relationship to HbA1c and blood pressure changes in patients with type 2 diabetes. Diabetologia 58: 1183–1187, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chao EC, Henry RR. SGLT2 inhibition–a novel strategy for diabetes treatment. Nat Rev Drug Discov 9: 551–559, 2010. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Williams S, Ho S, Loraine H, Hagan D, Whaley JM, Feder JN. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther 1: 57–92, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho YE, Basu A, Dai A, Heldak M, Makino A. Coronary endothelial dysfunction and mitochondrial reactive oxygen species in type 2 diabetic mice. Am J Physiol Cell Physiol 305: C1033–C1040, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Creagh-Brown BC, Griffiths MJ, Evans TW. Bench-to-bedside review: inhaled nitric oxide therapy in adults. Crit Care 13: 221, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeFronzo RA, Davidson JA, Del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab 14: 5–14, 2011. [DOI] [PubMed] [Google Scholar]
- 9.Diez-Sampedro A, Hirayama BA, Osswald C, Gorboulev V, Baumgarten K, Volk C, Wright EM, Koepsell H. A glucose sensor hiding in a family of transporters. Proc Natl Acad Sci USA 100: 11753–11758, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ehrenkranz JR, Lewis NG, Kahn CR, Roth J. Phlorizin: a review. Diabetes Metab Res Rev 21: 31–38, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Estrada IA, Donthamsetty R, Debski P, Zhou MH, Zhang SL, Yuan JX, Han W, Makino A. STIM1 restores coronary endothelial function in type 1 diabetic mice. Circ Res 111: 1166–1175, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freitas HS, Anhe GF, Melo KF, Okamoto MM, Oliveira-Souza M, Bordin S, Machado UF. Na+-glucose transporter-2 messenger ribonucleic acid expression in kidney of diabetic rats correlates with glycemic levels: involvement of hepatocyte nuclear factor-1α expression and activity. Endocrinology 149: 717–724, 2008. [DOI] [PubMed] [Google Scholar]
- 13.Frostell C, Fratacci MD, Wain JC, Jones R, Zapol WM. Inhaled nitric oxide. A selective pulmonary vasodilator reversing hypoxic pulmonary vasoconstriction. Circulation 83: 2038–2047, 1991. [DOI] [PubMed] [Google Scholar]
- 14.Futakuchi S, Ishiguro H, Naruse S, Ko SB, Fujiki K, Yamamoto A, Nakakuki M, Song Y, Steward MC, Kondo T, Goto H. High glucose inhibits HCO3− and fluid secretion in rat pancreatic ducts. Pflügers Arch 459: 215–226, 2009. [DOI] [PubMed] [Google Scholar]
- 15.Grempler R, Augustin R, Froehner S, Hildebrandt T, Simon E, Mark M, Eickelmann P. Functional characterisation of human SGLT-5 as a novel kidney-specific sodium-dependent sugar transporter. FEBS Lett 586: 248–253, 2012. [DOI] [PubMed] [Google Scholar]
- 16.Han S, Hagan DL, Taylor JR, Xin L, Meng W, Biller SA, Wetterau JR, Washburn WN, Whaley JM. Dapagliflozin, a selective SGLT2 inhibitor, improves glucose homeostasis in normal and diabetic rats. Diabetes 57: 1723–1729, 2008. [DOI] [PubMed] [Google Scholar]
- 17.Hasan FM, Alsahli M, Gerich JE. SGLT2 inhibitors in the treatment of type 2 diabetes. Diabetes Res Clin Pract 104: 297–322, 2014. [DOI] [PubMed] [Google Scholar]
- 18.Kashiwagi Y, Nagoshi T, Yoshino T, Tanaka TD, Ito K, Harada T, Takahashi H, Ikegami M, Anzawa R, Yoshimura M. Expression of SGLT1 in human hearts and impairment of cardiac glucose uptake by phlorizin during ischemia-reperfusion injury in mice. PLos One 10: e0130605, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kothinti RK, Blodgett AB, North PE, Roman RJ, Tabatabai NM. A novel SGLT is expressed in the human kidney. Eur J Pharmacol 690: 77–83, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar P; Committee on Fetus and Newborn. Use of inhaled nitric oxide in preterm infants. Pediatrics 133: 164–170, 2014. [DOI] [PubMed] [Google Scholar]
- 21.Liang Y, Arakawa K, Ueta K, Matsushita Y, Kuriyama C, Martin T, Du F, Liu Y, Xu J, Conway B, Conway J, Polidori D, Ways K, Demarest K. Effect of canagliflozin on renal threshold for glucose, glycemia, and body weight in normal and diabetic animal models. PLos One 7: e30555, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macdonald FR, Peel JE, Jones HB, Mayers RM, Westgate L, Whaley JM, Poucher SM. The novel sodium glucose transporter 2 inhibitor dapagliflozin sustains pancreatic function and preserves islet morphology in obese, diabetic rats. Diabetes Obes Metab 12: 1004–1012, 2010. [DOI] [PubMed] [Google Scholar]
- 23.Madonna R, De Caterina R. Cellular and molecular mechanisms of vascular injury in diabetes–part I: pathways of vascular disease in diabetes. Vascul Pharmacol 54: 68–74, 2011. [DOI] [PubMed] [Google Scholar]
- 24.Madonna R, De Caterina R. Cellular and molecular mechanisms of vascular injury in diabetes–part II: cellular mechanisms and therapeutic targets. Vascul Pharmacol 54: 75–79, 2011. [DOI] [PubMed] [Google Scholar]
- 25.Makino A, Wang H, Scott BT, Yuan JX, Dillmann WH. Thyroid hormone receptor-alpha and vascular function. Am J Physiol Cell Physiol 302: C1346–C1352, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mather KJ. The vascular endothelium in diabetes–a therapeutic target? Rev Endocr Metab Disord 14: 87–99, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller MR, Megson IL. Recent developments in nitric oxide donor drugs. Br J Pharmacol 151: 305–321, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moral-Sanz J, Moreno L, Cogolludo A, Perez-Vizcaino F. Pulmonary vascular function in insulin resistance and diabetes. Curr Vasc Pharmacol 12: 473–482, 2014. [DOI] [PubMed] [Google Scholar]
- 29.Murphy ME, Brayden JE. Nitric oxide hyperpolarizes rabbit mesenteric arteries via ATP-sensitive potassium channels. J Physiol 486: 47–58, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nomura S. Renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors for new anti-diabetic agent. Curr Top Med Chem 10: 411–418, 2010. [DOI] [PubMed] [Google Scholar]
- 31.Oliva RV, Bakris GL. Blood pressure effects of sodium-glucose co-transport 2 (SGLT2) inhibitors. J Am Soc Hypertens 8: 330–339, 2014. [DOI] [PubMed] [Google Scholar]
- 32.Olson ML, Kargacin ME, Honeyman TW, Ward CA, Kargacin GJ. Effects of phytoestrogens on sarcoplasmic/endoplasmic reticulum calcium ATPase 2a and Ca2+ uptake into cardiac sarcoplasmic reticulum. J Pharmacol Exp Ther 316: 628–635, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Olson ML, Kargacin ME, Ward CA, Kargacin GJ. Effects of phloretin and phloridzin on Ca2+ handling, the action potential, and ion currents in rat ventricular myocytes. J Pharmacol Exp Ther 321: 921–929, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Polidori D, Mari A, Ferrannini E. Canagliflozin, a sodium glucose co-transporter 2 inhibitor, improves model-based indices of beta cell function in patients with type 2 diabetes. Diabetologia 57: 891–901, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porter KE, Riches K. The vascular smooth muscle cell: a therapeutic target in type 2 diabetes? Clin Sci (Lond) 125: 167–182, 2013. [DOI] [PubMed] [Google Scholar]
- 36.Powell DR, DaCosta CM, Gay J, Ding ZM, Smith M, Greer J, Doree D, Jeter-Jones S, Mseeh F, Rodriguez LA, Harris A, Buhring L, Platt KA, Vogel P, Brommage R, Shadoan MK, Sands AT, Zambrowicz B. Improved glycemic control in mice lacking Sglt1 and Sglt2. Am J Physiol Endocrinol Metab 304: E117–E130, 2013. [DOI] [PubMed] [Google Scholar]
- 37.Pugh ME, Robbins IM, Rice TW, West J, Newman JH, Hemnes AR. Unrecognized glucose intolerance is common in pulmonary arterial hypertension. J Heart Lung Transplant 30: 904–911, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 54: 3427–3434, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Roberts BW, Mitchell J, Kilgannon JH, Chansky ME, Trzeciak S. Nitric oxide donor agents for the treatment of ischemia/reperfusion injury in human subjects: a systematic review. Shock 39: 229–239, 2013. [DOI] [PubMed] [Google Scholar]
- 40.Rossetti L, Lauglin MR. Correction of chronic hyperglycemia with vanadate, but not with phlorizin, normalizes in vivo glycogen repletion and in vitro glycogen synthase activity in diabetic skeletal muscle. J Clin Invest 84: 892–899, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rossetti L, Smith D, Shulman GI, Papachristou D, DeFronzo RA. Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J Clin Invest 79: 1510–1515, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stenlof K, Cefalu WT, Kim KA, Alba M, Usiskin K, Tong C, Canovatchel W, Meininger G. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab 15: 372–382, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tabatabai NM, Blumenthal SS, Lewand DL, Petering DH. Mouse kidney expresses mRNA of four highly related sodium-glucose cotransporters: regulation by cadmium. Kidney Int 64: 1320–1330, 2003. [DOI] [PubMed] [Google Scholar]
- 44.Tatarkiewicz K, Polizzi C, Villescaz C, D'Souza LJ, Wang Y, Janssen S, Parkes DG. Combined antidiabetic benefits of exenatide and dapagliflozin in diabetic mice. Diabetes Obes Metab 16: 376–380, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taubert D, Rosenkranz A, Berkels R, Roesen R, Schomig E. Acute effects of glucose and insulin on vascular endothelium. Diabetologia 47: 2059–2071, 2004. [DOI] [PubMed] [Google Scholar]
- 46.Tazawa S, Yamato T, Fujikura H, Hiratochi M, Itoh F, Tomae M, Takemura Y, Maruyama H, Sugiyama T, Wakamatsu A, Isogai T, Isaji M. SLC5A9/SGLT4, a new Na+-dependent glucose transporter, is an essential transporter for mannose, 1,5-anhydro-d-glucitol, and fructose. Life Sci 76: 1039–1050, 2005. [DOI] [PubMed] [Google Scholar]
- 47.Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, Koepsell H, Cunard R, Sharma K, Thomson SC, Rieg T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol 304: F156–F167, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wanstall JC, Homer KL, Doggrell SA. Evidence for, and importance of, cGMP-independent mechanisms with NO and NO donors on blood vessels and platelets. Curr Vasc Pharmacol 3: 41–53, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Wright EM, Hirayama BA, Loo DF. Active sugar transport in health and disease. J Intern Med 261: 32–43, 2007. [DOI] [PubMed] [Google Scholar]
- 50.Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev 91: 733–794, 2011. [DOI] [PubMed] [Google Scholar]
- 51.Yamanouchi T, Ogata N, Tagaya T, Kawasaki T, Sekino N, Funato H, Akaoka L, Miyashita H. Clinical usefulness of serum 1,5-anhydroglucitol in monitoring glycaemic control. Lancet 347: 1514–1518, 1996. [DOI] [PubMed] [Google Scholar]
- 52.Yamanouchi T, Shinohara T, Ogata N, Tachibana Y, Akaoka I, Miyashita H. Common reabsorption system of 1,5-anhydro-d-glucitol, fructose, and mannose in rat renal tubule. Biochim Biophys Acta 1291: 89–95, 1996. [DOI] [PubMed] [Google Scholar]
- 53.Yuan XJ, Tod ML, Rubin LJ, Blaustein MP. NO hyperpolarizes pulmonary artery smooth muscle cells and decreases the intracellular Ca2+ concentration by activating voltage-gated K+ channels. Proc Natl Acad Sci USA 93: 10489–10494, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou L, Cryan EV, D'Andrea MR, Belkowski S, Conway BR, Demarest KT. Human cardiomyocytes express high level of Na+/glucose cotransporter 1 (SGLT1). J Cell Biochem 90: 339–346, 2003. [DOI] [PubMed] [Google Scholar]