

Abstract
Inflammatory mediators released in acute lung injury (ALI) trigger the disruption of interendothelial junctions, leading to loss of vascular barrier function, protein-rich pulmonary edema, and severe hypoxemia. Genetic signatures that predict patient recovery or disease progression are poorly defined, but recent genetic screening of ALI patients has identified an association between lung inflammatory disease and a single nucleotide polymorphism (SNP) in the gene for the actin-binding and barrier-regulatory protein cortactin. This study investigated the impact of this disease-linked cortactin variant on wound healing processes that may contribute to endothelial barrier restoration. A microfabricated platform was used to quantify wound healing in terms of gap closure speed, lamellipodia dynamics, and cell velocity. Overexpression of wild-type cortactin in endothelial cells (ECs) improved directional cell motility and enhanced lamellipodial protrusion length, resulting in enhanced gap closure rates. By contrast, the cortactin SNP impaired wound closure and cell locomotion, consistent with the observed reduction in lamellipodial protrusion length and persistence. Overexpression of the cortactin SNP in lung ECs mitigated the barrier-enhancing activity of sphingosine 1-phosphate. These findings suggest that this common cortactin variant may functionally contribute to ALI predisposition by impeding endothelial wound healing.
Keywords: acute lung injury, endothelial wound healing, wound healing assay, cortactin
acute lung injury (ALI), manifested clinically as the acute respiratory distress syndrome (ARDS), is a devastating syndrome that results in respiratory failure with substantial morbidity and mortality (24). There are approximately 200,000 new ARDS patients every year in the US. The mortality rate is often 40% or higher, and there is no effective therapeutic treatment except for supportive mechanical ventilation (24). Causes of ALI/ARDS include sepsis, aspiration, multiple trauma, and/or shock. Although mechanical ventilation can be life saving, it also poses a risk for ventilator-induced lung injury (VILI), which results from a complex interplay between various mechanical forces acting on lung structures during lung hyperinflation, followed by the activation of severe protective biological responses and inflammation (9). Inflammatory mediators trigger the disruption of interendothelial junctions and compromise endothelial barrier integrity. The pulmonary microvasculature is particularly susceptible to these perturbations (5, 29, 41, 43), and resulting disruption of endothelial barrier function causes protein-rich pulmonary edema and activates additional inflammatory responses and severe endothelial degradation, to produce refractory hypoxemia and respiratory failure (11). Conversely, gap formation triggers vascular endothelial cell locomotion, which may contribute to endothelial resealing by promoting wound healing responses that rapidly restore endothelial barrier function (7, 11). Although the genetic and other patient-specific factors responsible for determining the ∼60% of ALI/ARDS patients who survive are unclear, recovery may be due in part to biochemical processes that restore barrier function.
Multiple genetic screens in recent years have identified disease-linked single nucleotide polymorphisms (SNPs) in various genes from subsets of patients predisposed to ALI (10, 13, 14, 17, 26–28, 33). Several of these ALI-associated SNPs involve the genes for cortactin and myosin light chain kinase (MLCK), which are critical cytoskeletal proteins that regulate endothelial cell mechanics, cell locomotion, and the mechanical integrity of cell-cell junctions (10, 13, 14). One identified, common human genetic variant (rs56162978) corresponds to a coding substitution in the cortactin sequence that changes serine to asparagine at site 484 (Ser484Asn), which is located very near a functionally important phosphorylation site, Tyr486 (10, 13). The effects of this disease-linked cortactin SNP on endothelial barrier function at the molecular and cellular levels remain unclear and are the focus of the present study. Possibilities include rendering interendothelial junctions more susceptible to mechanical damage or impairing actin-dependent processes necessary for vascular barrier recovery.
Cortactin contributes to wound healing by regulating the dynamic actin network underlying lamellipodia extensions and cell spreading (19, 27), which in turn enable cells to rapidly fill wounds. Cortactin activates the actin-related protein 2/3 (Arp2/3) complex (37), which nucleates the actin branching structures that underlie lamellipodia protrusions and cell locomotion (16). After tyrosine phosphorylation, cortactin coordinates actin-driven membrane protrusions in response to multiple signals at adhesive contacts (37). Cortactin plays a key role in regulating pulmonary endothelial barrier integrity by participating in cytoskeletal changes that enhance barrier-protective functions of agonists such as sphingosine 1-phosphate (S1P) and adenosine triphosphate (ATP), as well as barrier recovery after disruptive stimuli such as thrombin (2, 12, 23).
This study investigated the impact of wild-type (WT) human cortactin and the ALI-associated cortactin SNP (rs56162978) on endothelial gap closure. Quantitative analyses of endothelial wound healing dynamics were performed based on time-lapse images of gap closure using a microfabricated wound assay platform (1). Analyses focused on the impact of WT cortactin and the cortactin SNP on lamellipodia dynamics and cell migration during wound healing. We quantified lamellipodial protrusion and retraction as a first step toward defining how this cortactin variant might predispose patients to ALI (1, 32). Results obtained with these approaches together with single-cell migration and transendothelial electrical resistance (TER) reveal a possible physiological role of this cortactin SNP in inflammatory lung disease.
MATERIALS AND METHODS
Cell culture.
Bovine aortic endothelial cells (BAECs) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum (FBS) in a humidified 5% CO2 atmosphere at 37°C. Human pulmonary artery endothelial cells (HPAECs) and human lung microvascular endothelial cells (HLMVECs) were obtained from Lonza (Walkersville, MD) and cultured in EGM-2 and EGM-2MV (Lonza), respectively, supplemented with 10% (vol/vol) FBS in a humidified 5% CO2 atmosphere at 37°C.
Generation of cortactin constructs.
A green fluorescent protein (GFP)-tagged cortactin construct was generated by subcloning full-length human cortactin as EcoRI-KpnI fragments into the pEGFP-C1 plasmid (Life Technologies, Carlsbad, CA). The QuikChange Multi Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) was used according to the manufacturer's protocol to introduce into human cortactin a single guanine-to-adenine substitution resulting in a serine-to-asparagine amino acid change at residue number 484. This cortactin mutant simulates an SNP (rs56162978) identified in the human genome that is associated with clinical disease (10, 13). DsRed-tagged cortactin constructs used in this study were generated by subcloning WT cortactin and cortactin SNP rs56162978 into the pDsRed-N1 plasmid (Clontech, Mountain View, CA).
Cell transfection.
BAECs were harvested from tissue culture flasks with 0.1% trypsin, centrifuged at 250 g for 5 min, and resuspended with DMEM. Then 2 ml of a BAEC suspension (1.5 ×105 cells/ml) in supplemented DMEM was seeded in a tissue culture dish. After 1 day, these cells were transfected with DsRed-cortactin with Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. Transfected BAECs were incubated at 37°C in culture medium for an additional 1–2 days before use in experiments.
HPAECs and HLMVECs were transfected with DsRed-cortactin constructs by electroporation (Neon Transfection System; Life Technologies) according to the manufacturer's protocol. The parameters optimized for HPAEC transfection are as follows: pulse voltage of 1,350 V, pulse width of 30 ms, and pulse number of 1. For HLMVEC transfection, the pulse voltage was 1,150 V. Otherwise, all other parameters were the same. In the TER experiments, HPAECs were transiently transfected with GFP-cortactin constructs using FuGENE 6 (Promega, Madison, WI) per manufacturer's protocol as we described previously (12). HPAECs were then incubated in complete culture medium for an additional 1–2 days to allow protein expression. Because the transfection efficiency achieved with this method ranged from 15% to 20%, an LSR Fortessa flow cytometer (Becton-Dickinson, Franklin Lakes, NJ) was used 1–2 days after transfection to isolate GFP-expressing HPAECs for analysis in subsequent monolayer permeability measurements.
Western blot.
Western blots were used to confirm similar overexpression of the DsRed-tagged WT cortactin and cortactin SNP constructs. BAECs were transfected with the cortactin constructs as described above, and the WT cortactin- and cortactin SNP-transfected BAECs were lysed after 30 h. A Bradford assay (Coomassie Plus Bradford Assay Kit; Thermo Scientific Pierce) determined the total protein concentrations in cell lysates, and appropriate volumes of the cell lysates were loaded onto 4–20% gradient precast polyacrylamide gels (Bio-Rad, Hercules, CA). Gels were run on a Mini-PROTEAN electrophoresis cell (Bio-Rad). After transfer to a PVDF membrane (Invitrolon; Invitrogen, Carlsbad, CA) with a semidry transfer cell (Trans-Blot SD; Bio-Rad), the membrane was cut at different molecular weight ranges, blocked, and stained overnight for cortactin and actin with primary mouse anti-cortactin antibody (Clone 4F11; EMD Millipore, Billerica, MA) and primary mouse anti-actin antibody Ab-5 (Clone C4/actin; BD Biosciences, San Jose, CA), respectively. The secondary antibody anti-mouse IgG HRP conjugate (Promega) was incubated with the membranes for 1 h, and antibody binding was visualized with ECL Western blotting substrate (Thermo Scientific).
Polydimethylsiloxane micropillars.
A silicon wafer was cleaned for 1 h in piranha solution [H2SO4-H2O2 mixture, 3:1 (vol/vol)] at 120°C (Caution: Piranha is highly corrosive and should be used with proper protection), then rinsed with deionized water, and dried under a stream of N2. To achieve an ∼100-μm pillar height, the photoresist (SU8-2050; MicroChem, Westborough, MA) was dispensed on the cleaned wafer surface and air bubbles remaining in the photoresist were removed. The spin speed was next ramped up to 500 rpm for 10 s, held at 1,700 rpm for 30 s, and then ramped down to stop, followed by a 5-min bake at 65°C and a 20-min bake at 95°C. Next, a chrome mask with micropillar patterns was placed on the coated wafer. To transfer the micropillar patterns onto the wafer, the coated wafer was exposed to 230 mJ/cm2 of i-line (365 nm) UV light with a mask aligner in contact mode. The wafer was baked for 2 min at 65°C and then for 8 min at 95°C. Finally, the wafer was immersed in SU-8 developer (1-methoxy-2-propyl acetate; MicroChem) for 15 min at room temperature with gentle shaking. An elastomer stencil was fabricated by the casting method as described previously (36, 46). Briefly, a polydimethylsiloxane (PDMS) base and a curing agent (Sylgard 184; Dow Corning, Midland, MI) [8:1 (wt/wt)] were mixed thoroughly, degassed, poured on the silicon template, degassed under vacuum, and then cured at 140°C for 24 h in a vacuum oven. The PDMS stamp was then peeled from the silicon template in ethanol.
Gap creation on a cell monolayer with micropillars.
The procedure used to generate circular gaps in a confluent endothelial monolayer, but without cell damage, is illustrated in Fig. 1A. A 35-mm glass-bottomed dish was coated with collagen I (Sigma) at 20 μg/ml for 40 min at room temperature. Then BAECs (5 × 104 cells/cm2) were first incubated in the dish for 12 h before the stencil was placed with a small iron plate attached to the top onto the glass-bottomed dish with cells. Before use, the stencil was submerged in a 1% (wt/vol) Pluronic solution (Pluronic F108 Prill; BASF) for 1 h at room temperature and rinsed with phosphate-buffered saline (PBS), in order to prevent cells from adhering to the micropillars. After 1- to 2-day incubation, a magnetic bar was used to lift off the stencil, in order to minimize monolayer damage during stencil removal, resulting in undamaged gaps. Damaged gaps were also generated by placing the stencil directly onto a confluent monolayer, crushing the cells underneath.
Fig. 1.
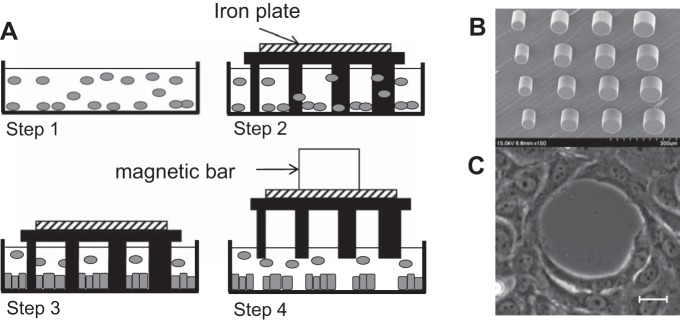
Gap creation with polydimethylsiloxane (PDMS) micropillars. A: experimental procedure for undamaged gap creation. Cells are seeded onto a collagen-coated glass surface (step 1). Then a PDMS stencil coated with a Pluronic block copolymer is placed on the dish (step 2) and incubated for 1 day to enable cells to attach to the glass surrounding the pillars (step 3). Finally, stencil detachment with a magnetic bar creates undamaged gaps of different sizes (step 4). B: scanning electron microscopy of a stencil of micropillars. C: gap pattern on a bovine aortic endothelial cell (BAEC) monolayer. Scale bar, 10 μm.
Time-lapse imaging of closure of stencil-generated gaps.
Before gap closure, cell migration, or lamellipodia dynamics was observed, the culture medium was replaced with CO2-independent medium (Life Technologies) with 10% FBS. Live-cell imaging for all experiments was performed with a Zeiss Axiovert 200M microscope (Carl Zeiss, Thornwood, NY) equipped with a temperature-controlled stage. Phase-contrast images of gaps in an endothelial monolayer were taken with a ×20 objective every 1.5–3 min for 3–4 h. The time evolution of the gap area was quantified from time-lapse images using AxioVision (Carl Zeiss) and ImageJ (version 1.48e; NIH). The following parameters were calculated from time-lapse images, in order to quantify gap closure dynamics as a function of initial gap diameter: gap closure time, rate of area decrease, radial velocity, and the initial radial velocity (calculated from the slope of the gap diameter vs. time based on the data obtained within the first 30 min).
Random cell migration.
Random cell migration measurements were carried out with both BAECs and HPAECs. Before cells were seeded, a glass-bottomed petri dish was incubated with 20 μg/ml collagen I for 40 min at room temperature. The cells were harvested with 0.1% trypsin, pelleted by centrifugation in a bench top centrifuge, and finally resuspended in culture medium. Then 2 ml of a cell suspension (2 × 104 cells/ml) in culture medium supplemented with 10% FBS was added to each collagen I-coated petri dish, which was then placed in a humidified 5% CO2 atmosphere for 3–5 h at 37°C. Next, multiple positions of interest (n > 20) were manually defined for sequential imaging. With the live-cell imaging setup described above, phase-contrast images of adhered cells were taken with a ×10 objective every 10 or 15 min for 12–16 h. Movements of individual cells that were expressing fluorescently tagged WT cortactin and cortactin SNP were tracked with the ImageJ plug-in “Manual Tracking” (6).
Cell velocity was defined in the following ways: 1) average cell velocity, which is the total cell migration distance divided by the migration period, 2) overall cell velocity, which is the net cell displacement during the migration period divided by the migration period, and 3) nonlinear least-squares fits of cell paths to a random walk model (8, 42). Also, the cell directionality (35) was calculated by the net displacement during the migration period divided by the total cell path length. The following criteria for selecting cells to analyze are based on published protocols (8). Nonmotile cells were excluded (8, 21). Only isolated individual cells that did not pause, contact other cells, or divide during the observation period were selected for quantitative analyses. Then cell paths were fitted to a persistent random walk model, 〈d2〉 = 2S2P[t − P(1 − e−t/P)], where 〈d2〉 is the average squared displacement over time, S is the cell speed, and P is the cell persistence time, as previously described (8). Parameters S and P were estimated from nonlinear least-squares fits of the data to this equation. For each case, the mean and SE were calculated. Values of the fitted parameters were compared by Student's t-test with unequal variance (34).
Kymograph analysis.
Kymographs were generated and analyzed for BAECs and HLMVECs. Transfected cells were seeded on collagen I glass-bottomed dishes and serum starved (0.5% FBS) for 5–10 h before the measurements. With the live-cell imaging setup described above, phase-contrast images of individual cells were taken with a ×20 objective and Optovar (×1.6) every 6 s for 20 min. To investigate the effect of EC barrier-enhancing or -disrupting agents on lamellipodia dynamics, BAECs were treated with S1P (1 μM; Sigma Aldrich) or thrombin (50 nM; Sigma Aldrich) and observed 5 min later. From time-lapse images of individual cells, lamellipodia that showed consistent and dynamic protrusion/retraction at the leading edge were selected for analysis. To generate kymographs, a line with a width of 1 pixel, orthogonal to the leading edge, was defined in the image stack. Then these lines from all images in the time lapse were stacked beside each other, to form a graph displaying lamellipodia position vs. time. Each lamellipodia protrusion and retraction cycle appears as a “peak.” The four parameters calculated from the kymographs were protrusion rate, retraction rate, persistence time, and protrusion length. The “peak height” defines the protrusion length, and the “peak width” before descending is the lamellipodia persistence. The ascending and descending slopes of the peak are the protrusion and retraction rates, respectively. These four parameters were quantified for each peak and averaged over all peaks analyzed in the kymographs.
Immunofluorescence imaging.
To visualize subcellular distributions of actin during gap closure after stencil removal, BAEC monolayers with gaps were fixed at different time points with 4% (vol/vol) paraformaldehyde (Sigma) for 30 min at room temperature, permeabilized with 0.1% (wt/vol) Triton X-100 (Sigma) in PBS for 5 min, and then blocked in 1% (wt/vol) BSA (Sigma) in PBS for 20 min. Actin was visualized with Alexa Fluor 488-conjugated phalloidin (Invitrogen) at 1:100 (3 U/ml) in the blocking solution. Images were captured with a Zeiss Axiovert 200M microscope (Carl Zeiss). To quantify the colocalization of WT cortactin and the cortactin SNP with actin at the leading edge of single migrating cells, transfected BAECs were seeded on glass at single-cell densities, allowed to attach and spread over 12 h, and then fixed and stained for actin as above. Correlation coefficients for the spatial distributions of cortactin and actin fluorescence intensities were calculated with the ImageJ plug-in “Colocalization Analysis” (Wright Cell Imaging Facility, University Network Research).
Transendothelial monolayer electrical resistance.
HPAECs transfected with GFP-cortactin constructs were grown to confluence in polycarbonate wells containing evaporated gold microelectrodes, and TER measurements were performed with an electrical cell-substrate impedance sensing system (ECIS) (Applied Biophysics, Troy, NY) as previously described (12, 15). HPAECs transfected with GFP vector, WT GFP-cortactin, or SNP GFP-cortactin were plated onto gold microelectrodes for measurements of monolayer barrier function with TER. After a stable baseline resistance was established, S1P (1 μM) was added. These ECIS experiments were performed with a frequency of 4,000 Hz, which is the standard used by us (15) and recommended by others (40). TER values from each microelectrode were expressed as normalized resistance, pooled as discrete time points, and then plotted as means ± SE vs. time. Analysis of variance (ANOVA) statistical comparisons were performed for the ECIS data for the maximal resistance levels obtained after S1P for each condition.
RESULTS
ALI-related cortactin SNP retards endothelial gap closure.
Circular gaps with 10- to 90-μm diameters were reproducibly created on cell monolayers with a PDMS micropillar stencil (Fig. 1B). The size and shape of the gaps were well defined. Coating pillars with a Pluronic block copolymer prevented cell adhesion on the sides of the pillars. There was no visible cell damage after stencil removal from the monolayer, resulting in clearly defined wound boundaries (Fig. 1C). On the other hand, “crushed gaps” with diameters >40 μm could also be created by placing the stencil directly on confluent cell monolayers. Smaller crushed gaps could not be reliably created by crushing the monolayer because the thinner pillars either buckled or left the gap filled with excess cell debris. The closure of 40- to 90-μm-diameter crushed gaps by nontransfected BAECs was ∼30% slower than the closure of undamaged gaps (Fig. 2). Otherwise, the overall trends were qualitatively similar. All subsequent experiments were done with undamaged gaps.
Fig. 2.
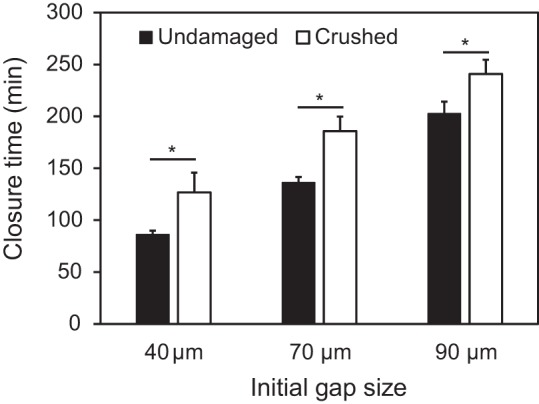
Comparison of closure time of undamaged and crushed gaps in a BAEC monolayer. The closure time of crushed gaps with initial diameter of 40 μm, 70 μm, and 90 μm is 46%, 36%, and 19% longer than that of undamaged gaps, respectively. Error bars indicate SE. *P < 0.05.
In transfected BAECs, the levels of overexpression of DsRed WT cortactin and cortactin SNP were comparable, as confirmed by a Western blot of whole cell lysates (Fig. 3A). The expression of DsRed cortactin was confirmed from fluorescence images of transfected BAECs, and the transfection efficiency was 70–80% for both DsRed WT cortactin and cortactin SNP. This ensured that the majority of cells surrounding analyzed monolayer “wounds” overexpressed DsRed WT cortactin or cortactin SNP.
Fig. 3.

Analysis of gap closure in a BAEC monolayer. A: Western blot of BAECs overexpressing DsRed-cortactin; densitometry performed with actin as loading control revealed equal overexpression of DsRed cortactin [single nucleotide polymorphism (SNP): 1.03, wild type (WT): 1] and equal expression of endogenous cortactin (BAECs: 1.09, SNP: 0.97, WT: 1). B: gap closure time as a function of the initial size (diameter, μm) of circular gaps. C: gap closure time in BAECs overexpressing the cortactin SNP relative to WT cortactin (expressed in %). D: radial velocity of gap closure vs. initial gap size. E: lamellipodia initiation time after starting observation of 20- and 90-μm gaps. F: initial radial closure velocity in 20- and 90-μm gaps. For each size of gaps, n = 10–60. *P < 0.05, **P < 0.01.
The cortactin SNP slowed endothelial closure of undamaged gaps after stencil removal. Relative to nontransfected BAECs, WT cortactin overexpression reduced the gap closure time (P < 0.01 for all gap sizes) (Fig. 3B). In contrast, the closure of 70- to 90-μm-diameter gaps by BAECs overexpressing the cortactin SNP was slower than cells overexpressing WT cortactin. Closure times were, respectively, 120 ± 4.2 min vs. 107 ± 4.0 min (P < 0.05) for 70-μm gaps, 158 ± 5.4 min vs. 123 ± 3.8 min (P < 0.01) for 80-μm gaps, and 205 ± 11.4 min vs. 163 ± 5.1 min (P < 0.01) for 90-μm gaps (Fig. 3, B and C).
The average rate of the decrease in gap area increased with the initial gap size for both transfected and nontransfected BAECs (data not shown). On the other hand, the radial velocity at which WT cortactin- or cortactin SNP-transfected cells reduced the gaps decreased with the initial gap size, whereas the radial velocity at which nontransfected cells closed the gap was independent of the initial gap size (Fig. 3D).
The overexpression of either WT cortactin or cortactin SNP promoted earlier lamellipodia formation in BAECs, compared with nontransfected cells (Fig. 3E). For a 20-μm gap, the onset of lamellipodia formation was at t = 5 min for transfected cells surrounding the gap. In contrast, lamellipodia of nontransfected BAECs formed at t = 17 min at the gap boundary. The formation time for lamellipodia in BAECs overexpressing WT cortactin was statistically similar for 90-μm and 20-μm gaps within the same monolayer (P = 0.29). Although lamellipodia appeared to form later in BAECs overexpressing the cortactin SNP (9 ± 2 min, 90-μm gap) relative to cells overexpressing WT cortactin (5 ± 2 min, 90-μm gap), these values were not statistically different at the 95% confidence level (P = 0.2). The earlier lamellipodia formation (Fig. 3E) correlated with faster initial radial velocities (Fig. 3F).
As shown by immunofluorescence images, immediately after gap formation F-actin fibers accumulated in a purse string-like structure at the boundary of gaps in WT cortactin- and cortactin SNP-overexpressing BAEC monolayers (Fig. 4A). During the early stage of gap closure, time-lapse imaging also revealed a reduction in gap area prior to lamellipodia formation (Supplemental Videos S1–S6).1 During gap closure, the cells changed their shape and actively rearranged relative to one another. After 25 min, F-actin at the gap boundary became less apparent, as the actin belt remodeled after lamellipodia extension into the gaps. F-actin accumulation at the gap boundary followed by its remodeling could be observed in both the WT cortactin- and cortactin SNP-overexpressing cells, around small (Fig. 4B) as well as large (Fig. 4C) gaps.
Fig. 4.
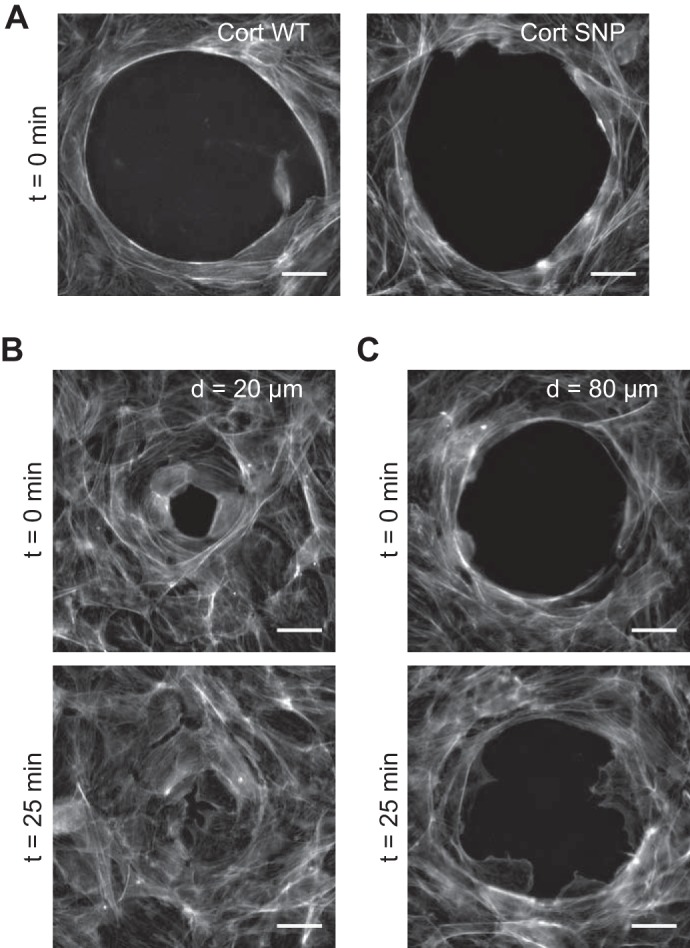
Formation and disappearance of actin belts around gaps. A: gaps on WT cortactin- and cortactin SNP-overexpressing BAEC monolayers, showing actin stained with Alexa Fluor 488-phalloidin just after large gap formation (t = 0 min). This actin belt morphology was found around both small gaps as well. B and C: actin belt morphology around small gaps (B) and large gaps (C) in a BAEC monolayer overexpressing cortactin SNP. Samples fixed at t = 0 min and 25 min. After the actin belt disappears, small gaps close by cell rearrangement and large gaps close by lamellipodial extension and cell migration (Supplemental Videos S1–S6). This trend was also seen with WT cortactin. Scale bars, 20 μm.
Cortactin SNP decreases both cell migration speed and directionality.
The velocity and directionality of randomly migrating BAECs and HPAECs were calculated based on the trace of cell movements for 12–16 h. Wind rose plots (8, 18) of BAEC migration show that cells moved isotropically (Fig. 5A). As defined in materials and methods, average velocity, overall velocity, and directionality of BAECs were calculated and compared (Fig. 5, B–D). The overexpression of WT cortactin in BAECs increased the overall velocity (P = 0.013) and the directionality (P = 0.014) relative to nontransfected cells. The overexpression of the cortactin SNP retarded the average velocity (P = 0.0007) and overall velocity (P = 0.01) relative to cells overexpressing WT cortactin. On the other hand, although the cortactin SNP appeared to decrease the directionality of BAECs (0.34 ± 0.03) compared with WT cortactin (0.40 ± 0.04), the difference was not statistically significant at the 95% confidence level (P = 0.16).
Fig. 5.

Quantified mobility of BAECs on collagen I-coated glass. A: wind rose plots for cell migration trajectories. Scale bars, 100 μm. B–D: average velocity (B), overall velocity (C), and directionality (D) were obtained as described in materials and methods. E and F: cell speed (E) and persistence (F) were obtained by fitting experimental cell migration data to a persistent random walk model. Error bars indicate SE. *P < 0.05, **P < 0.01. n = 41 nontransfected BAECs, n = 35 BAECs overexpressing WT cortactin, n = 41 BAECs overexpressing cortactin SNP.
Additionally, characteristic parameters of single-cell migration were also determined by fitting individual cell trajectories to the persistent random walk model (Fig. 5, E and F). For both transfected and nontransfected cells 35–50% of the migration trajectories met the selection criteria for analysis (see materials and methods), and their trajectories were analyzed further to determine the cell speed and persistence (8). The speed and persistence of cells overexpressing WT cortactin were statistically similar to nontransfected cells (P = 0.66 for cell speed and P = 0.16 for persistence). The persistence of cells overexpressing the cortactin SNP was similar to cells overexpressing WT cortactin (P = 0.9), but the cell speed was lower (P = 0.045) (Fig. 5, E and F).
The HPAEC migration behavior was qualitatively similar to that of the BAECs (Fig. 6). HPAECs overexpressing WT cortactin migrated faster (overall velocity 0.087 ± 0.008 μm/min) with greater directionality (0.34 ± 0.03) than nontransfected cells (overall velocity 0.056 ± 0.005 μm/min, directionality 0.26 ± 0.02). Comparison of WT cortactin-transfected vs. nontransfected cells shows that the parameters are statistically different (P = 0.001 for overall velocity and P = 0.015 for directionality). Compared with cells overexpressing WT cortactin, the cortactin SNP slowed the overall HPAEC velocity to 0.034 ± 0.004 μm/h and reduced the directionality to 0.248 ± 0.025 (P < 10−6 for overall velocity, P = 0.02 for directionality). Based on fits to the random walk model, SNP overexpression reduced the cell speed to 0.21 ± 0.02 μm/min compared with 0.36 ± 0.04 μm/min for cells overexpressing WT cortactin (P = 0.0027; Fig. 6, E and F).
Fig. 6.

Quantified mobility of human pulmonary artery endothelial cells (HPAECs) on collagen I-coated glass. A: wind rose plots for cell migration trajectories. Scale bars, 100 μm. B–D: average velocity (B), overall velocity (C), and directionality (D) were obtained as described in materials and methods. E and F: cell speed (E) and persistence (F) were obtained by fitting experimental cell migration data to a persistent random walk model. Error bars indicate SE. *P < 0.05, **P < 0.01. n = 44 nontransfected HPAECs, n = 44 HPAECs overexpressing WT cortactin, n = 43 HPAECs overexpressing cortactin SNP.
Cortactin SNP alters lamellipodia dynamics.
We also quantified the lamellipodia dynamics of individual BAECs and HLMVECs. In both cases, a time interval of 6 s was sufficient to capture lamellipodia dynamics in serum-starved cells—that is, the lamellipodia protrusion-retraction cycle was longer than the 6-s time interval.
In BAECs, WT cortactin overexpression increased the lamellipodia persistence to 1.48 ± 0.09 min relative to 0.96 ± 0.05 min in nontransfected cells (P < 0.0001; Fig. 7A). It also increased the lamellipodia protrusion length to 2.4 ± 0.1 μm compared with 1.7 ± 0.1 μm in nontransfected cells (P < 0.001; Fig. 7B). However, WT cortactin had no significant effect on either the protrusion or retraction rates of BAECs (Fig. 7, C and D). On the other hand, the cortactin SNP reduced the persistence to 1.06 ± 0.06 min relative to WT cortactin (1.48 ± 0.09 min, P = 0.002; Fig. 7A). Similarly, cortactin SNP overexpression reduced the protrusion length of lamellipodia in BAECs from 2.4 ± 0.1 μm to 1.46 ± 0.09 μm (P < 10−5; Fig. 7B), but it did not change the protrusion or retraction rates compared with WT cortactin (Fig. 7, C and D).
Fig. 7.

Kymograph analysis of lamellipodia of WT cortactin-overexpressing, cortactin SNP-overexpressing, and nontransfected BAECs. For each condition, 10–14 cells were observed every 6 s for 20–40 min. A: persistence. B: protrusion length. C: protrusion rate. D: retraction rate. Error bars indicate SE. *P < 0.05, **P < 0.01. S1P, sphingosine 1-phosphate.
To determine whether the barrier-enhancing agent S1P (15) mitigates the observed effects of the cortactin SNP on BAEC lamellipodia, cells were stimulated with S1P and observed after 5 min. S1P is an endogenous phospholipid postulated to enhance barrier function by activating Rac and Src kinase, which both regulate actin and lamellipodia dynamics (12, 15, 31). Pretreatment with S1P increased lamellipodia activity in transfected cells. Both the lamellipodial persistence and protrusion length in BAECs overexpressing either WT cortactin or the cortactin SNP increased upon response to S1P (Fig. 7, A and B). However, after S1P treatment, both the lamellipodial persistence and protrusion length still remained lower in BAECs overexpressing the cortactin SNP relative to WT cortactin (P < 0.01 for both; Fig. 7, A and B). Also, S1P decreased the protrusion rate of WT cortactin-overexpressing cells slightly, whereas it increased that of nontransfected cells (Fig. 7C).
In addition, nontransfected and transfected BAECs were exposed to the barrier-disrupting agonist thrombin. Thrombin treatment of BAECs overexpressing WT cortactin reduced both lamellipodial persistence and protrusion length to levels statistically similar to those in nontransfected cells (Fig. 7, A and B). On the other hand, thrombin treatment had a milder effect on the lamellipodia dynamics of nontransfected BAECs and of BAECs overexpressing the cortactin SNP. However, the effects of the cortactin SNP on lamellipodial persistence and protrusion length were still noticeable. Upon thrombin treatment, BAECs overexpressing the cortactin SNP showed lower lamellipodial persistence (0.91 ± 0.07 min) compared with WT cortactin (1.12 ± 0.06 min) (P = 0.028; Fig. 7A) and they also exhibited lower protrusion lengths (1.68 ± 0.10 μm) compared with BAECs overexpressing WT cortactin (2.02 ± 0.10 μm) (P = 0.039; Fig. 7B).
Compared with BAECs and HPAECs, HLMVECs are a better model of endothelial cells lining the capillaries around alveoli. The pulmonary microvasculature is susceptible to inflammatory mediators that can cause gap formation in ALI (5, 29, 39). Because cortactin-driven lamellipodia dynamics contribute to gap closure in endothelial monolayers, kymography was the preferred tool for investigating the effects of this SNP on HLMVEC lamellipodia.
Kymographs of nontransfected BAECs and HLMVECs are shown in Fig. 8, and lamellipodia dynamics in Supplemental Video S7 reveal notable differences between BAEC and HLMVEC lamellipodia. The slopes for the HLMVEC kymographs are significantly higher than those for the BAEC kymographs, and this difference is reflected in the increased protrusion and retraction rates. The protrusion and retraction cycles for HLMVECs are also narrower than those for BAECs, resulting in reduced lamellipodial persistence.
Fig. 8.
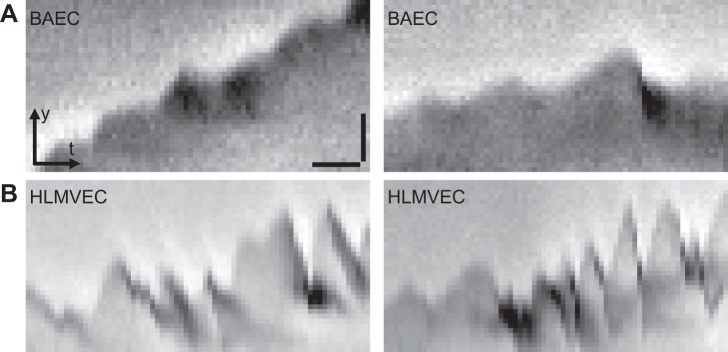
Kymographs of nontransfected BAECs (A) and nontransfected human lung microvascular endothelial cells (HLMVECs; B) after 5–10 h of serum starvation (0.5% FBS). Each kymograph represents the temporal evolution of the leading edge of a cell along a line drawn perpendicular to it, each pixel on the x-axis representing 6 s. A protrusion and retraction cycle of a lamellipodium thus appears as a peak. The HLMVEC kymographs appear jagged relative to BAECs, with some peaks being only 1–2 pixels wide and <4 pixels tall. x-Scale bar (10 pixels), 60 s; y-scale bar (10 pixels), 2 μm.
Kymography results in Fig. 9 show that the overexpression of WT cortactin or the cortactin SNP in HLMVECs did not affect the lamellipodial persistence, unlike in BAECs. However, as was the case with BAECs, WT cortactin overexpression in HLMVECs significantly increased the lamellipodial protrusion length to 1.85 ± 0.10 μm compared with 1.60 ± 0.08 μm for nontransfected HLMVECs (P = 0.024). Cortactin SNP overexpression hardly induced increased protrusion length (1.58 ± 0.08 μm), compared with WT cortactin overexpression (P = 0.018). Also, WT cortactin-overexpressing HLMVECs had a higher protrusion rate (6.15 ± 0.31 μm/min) compared with cortactin SNP (5.30 ± 0.29 μm/min, P = 0.027). Interestingly, the retraction rate of lamellipodia in WT cortactin-overexpressing HLMVECs (5.99 ± 0.30 μm/min) was lower than that in nontransfected cells (6.82 ± 0.37 μm/min, P = 0.046), but it was similar to that in cortactin SNP-overexpressing cells (5.82 ± 0.42 μm/min). Although cortactin regulates cortical actin and thus lamellipodia dynamics, it is currently not completely understood how cortactin influences the parameters used to quantify these dynamics.
Fig. 9.
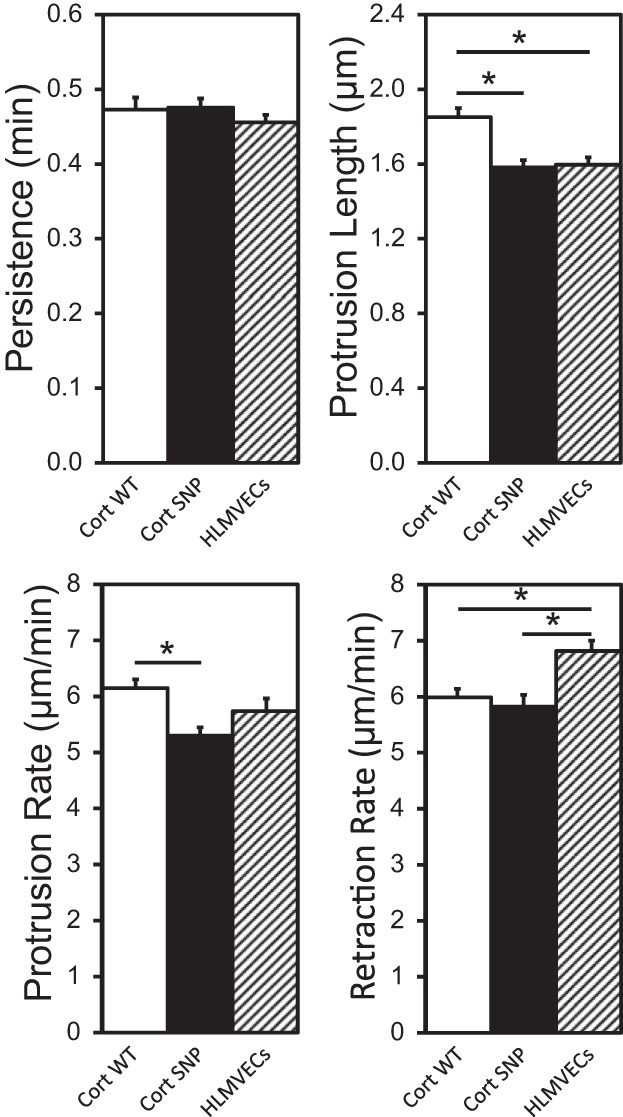
Kymograph analysis of lamellipodia of WT cortactin-overexpressing, cortactin SNP-overexpressing, and nontransfected HLMVECs. n = 23 cells analyzed for each condition, every 6 s for 20 min. Error bars indicate SE. *P < 0.05.
Cortactin SNP colocalizes with actin at the leading edge.
Cortactin is known to localize at the leading edge of migrating cells. We found that both DsRed WT and SNP cortactin accumulate at the leading edge of motile BAECs (Fig. 10A, Supplemental Videos S8 and S9) but distribute throughout the cytosol in nonmigrating cells. In migrating BAECs, the average cortactin fluorescence intensity at the leading edge (Fig. 10B) was about three times greater than 5 μm from the edge for both WT cortactin and cortactin SNP. Additionally, we found that the exogenous cortactin constructs, both WT and SNP, colocalized equally with endogenous cortactin in immunostained single BAECs, with Pearson correlation coefficients ρ ∼ 0.95 (data not shown).
Fig. 10.

Colocalization of cortactin with actin in BAECs. A: localization of actin (green) and DsRed cortactin (red). Scale bars, 10 μm. B: fluorescence intensity measurements along the line scan perpendicular to the moving direction of lamellipodia. n = 9 BAECs overexpressing WT cortactin or cortactin SNP. C: correlation coefficient between fluorescence intensities of actin and DsRed cortactin. The subregion of a cell for the correlation analysis was defined to include the cell membrane boundary region.
The colocalization of cortactin and actin was determined by correlating Alexa 488-phalloidin-labeled actin fluorescence in BAECs with either DsRed WT cortactin or cortactin SNP. There was no difference in F-actin morphology between transfected BAEC monolayers overexpressing WT cortactin or cortactin SNP, with actin bundles found at the cell cortex as well as the cell body in both cases (data not shown). Merged images of actin and cortactin showed a significant overlap along the cell boundary in cells overexpressing either WT cortactin or cortactin SNP (Fig. 10A). The correlation coefficients between actin and cortactin intensities were 0.83 ± 0.01 and 0.84 ± 0.01 for WT cortactin and cortactin SNP, respectively. These values were statistically indistinct (P = 0.38) and demonstrate that the cortactin SNP does not affect its colocalization with actin, relative to WT cortactin (Fig. 10C).
Cortactin SNP impairs endothelial barrier enhancement by S1P.
Given the inhibitory effects of the cortactin SNP on endothelial gap closure, migration speed, and lamellipodia dynamics, we next determined whether these differences in behaviors related to wound healing translated into functional changes in endothelial permeability. HPAECs were transfected with GFP-labeled WT cortactin or cortactin SNP (or GFP vector control) and then analyzed with TER, as described in materials and methods (12, 15). No significant differences in the baseline TER were observed among these conditions (Fig. 11). However, S1P treatment induced a rapid and sustained increase in TER values of WT cortactin-overexpressing cells indicative of enhanced HPAEC barrier function. No statistically significant differences were observed between HPAECs transfected with GFP vector and WT cortactin. However, cells overexpressing the cortactin SNP exhibited significantly lower maximal TER than those transfected with WT cortactin (n = 5–8; P < 0.04) (Fig. 11). These results demonstrate that this ALI-associated cortactin SNP impairs lung endothelial barrier function and suggest that it may contribute to increased vascular leak in patients.
Fig. 11.

Cortactin SNP impairs endothelial barrier enhancement by S1P. Here S1P was added at t = 0, and the traces show the endothelial resistance as a function of time after S1P addition. HPAECs were transfected with green fluorescent protein (GFP) vector (orange line), WT GFP-cortactin (blue line), or SNP GFP-cortactin (red line) as described in materials and methods. Black line represents control HPAECs treated with vehicle only. Transendothelial electrical resistance (TER) traces represent pooled results ± SE from n = 5–8 independent experiments per condition. The maximal resistance level of WT GFP-cortactin was higher than that of SNP GFP-cortactin (P < 0.04). GFP vector-transfected HPAEC values (orange) are shown without SE bars as a reference. Mean baseline TER values for each conditions are as follows: vehicle control (no transfection) = 1,126 Ω, GFP vector control = 990 Ω, WT GFP-cortactin = 1,019 Ω, SNP GFP-cortactin = 915 Ω.
DISCUSSION
These findings reveal the influence of an ALI-associated SNP on cellular processes that contribute to endothelial wound healing and barrier function, namely, endothelial gap closure rates, cell locomotion, and lamellipodia dynamics. The well-controlled shape and size of gaps created with micropillar stencils enabled quantitative comparisons of gap closure and lamellipodia dynamics in different cell monolayers subject to different perturbations.
Cells close gaps either by a purse string mechanism or by cell migration into a gap after lamellipodia extension and cell crawling (22). In addition to lamellipodia extensions followed by cell crawling, we found that ECs also closed gaps by contracting the circumferential actin at the apical boundaries surrounding the wound (Fig. 4 and Supplemental Videos S1–S6). Actin belt formation (Fig. 4) is an initial signature preceding cell contraction and subsequent gap closure (3). The observed accumulation of circumferential actin suggests that the purse string mechanism contributed to early stages of EC gap closure. At later stages, the gap closure mechanism depends on the gap size. For small gaps (10–20 μm), the dominant mechanism involves rearrangement of neighboring cells with relatively low lamellipodial activity (Supplemental Videos S1–S3). The dominant mechanism closing large gaps involved lamellipodia protrusions and subsequent cell crawling into the wound (Supplemental Videos S4–S6). Cell crawling, rather than purse string closure, similarly dominated the closure of undamaged epithelial gaps (1).
Results obtained with the cortactin SNP demonstrated that this genetic variant impedes lamellipodia dynamics and cell locomotion in different endothelial cell types, both factors contributing to gap closure. Cortactin regulates actin polymerization by associating with the Arp2/3 complex, and thus contributes to cell motility (20, 25). The increased gap closure rate of BAECs overexpressing WT cortactin (Fig. 3) correlates with both enhanced directional cell motility (Fig. 5) and lamellipodial persistence (Fig. 7). The latter processes appear to be linked causally. Cortactin-deficient fibrosarcoma cells were defective in their ability to assemble integrin adhesions in cellular protrusions, which would in turn reduce lamellipodia persistence and cell motility (4). Although lamellipodial persistence in HLMVECs overexpressing WT cortactin was not enhanced relative to cells overexpressing the cortactin SNP, they did show significantly higher protrusion lengths and protrusion rates (Fig. 9). This behavior could also aid rapid gap closure in cell monolayers, suggesting that the cortactin SNP may adversely affect monolayer recovery in injured pulmonary microvasculature in ALI.
Random cell migration measurements were also performed with HLMVECs to determine the effect of the cortactin SNP on cell motility, but the basal cell motility of these cells on collagen I- and fibronectin-coated glass was too slow to detect statistically significant differences between WT cortactin and cortactin SNP overexpression. However, gap diameters in the microvasculature associated with ALI are of the order of ∼1 μm. Thus gap closure would be independent of random cell migration but dependent on lamellipodial dynamics, which our results showed are impaired by the cortactin SNP (30).
In this study, although the cortactin SNP adversely affected lamellipodia dynamics of different cell types relative to WT cortactin, the cortactin SNP and WT cortactin similarly colocalized with actin at the leading edge. This is consistent with a previous finding that the COOH-terminal domain of cortactin, where the SNP is located, was not necessary for Rac-induced translocation to the cell cortex and F-actin binding (45).
Cortactin depletion in human umbilical vein endothelial cells (HUVECs) significantly increased endothelial barrier permeability and attenuated S1P-induced endothelial barrier enhancement, whereas WT cortactin overexpression augmented barrier resistance (12). Our findings similarly suggest that S1P does not mitigate efficiently against the effects of the cortactin SNP, such that therapeutic approaches utilizing this pathway may not be effective for ALI patients harboring this SNP. However, the cAMP analog 8-pCPT-2Me-cAMP (also known as 007) activated the Epac/Rap1 pathway and restored barrier function in monolayers of cortactin-knockdown HUVECs to levels similar to WT HUVECs (38). The latter result suggests an alternative to S1P for treating ALI patients with the cortactin SNP.
The detailed biochemical basis for the impaired cortactin function is currently unknown. However, the substitution (Ser484Asn) is near the critical p60Src-targeted Tyr486. Cortactin tyrosine-phosphorylation increases collective cell migration (20) and was necessary for maximal S1P-induced barrier enhancement in TER measurements (12). Preliminary studies suggest that the cortactin SNP decreases Tyr486 phosphorylation 40–60% below baseline and S1P-stimulated conditions, and this reduction correlated with a 25% reduction in actin polymerization compared with WT cortactin (13). Cortactin phosphorylation is required for Arp2/3-dependent actin branching, and cortactin also prevents the disassembly of branched actin (44). The role of potentially impaired Tyr486 phosphorylation in ALI remains to be determined. Nevertheless, our findings further quantified the impact of this disease-linked SNP on dynamic cellular processes that affect endothelial barrier function, and identified potential mechanisms linking this SNP to ALI pathogenesis.
GRANTS
This work was supported by National Institutes of Health Grants 5RO1 GM-097443 (D. E. Leckband), R01 HL-88144 (S. M. Dudek), and P01 HL-58064 (J. G. N. Garcia).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.C., A.D., J.G.G., S.M.D., and D.E.L. conception and design of research; S.C., S.M.C., and A.D. performed experiments; S.C., S.M.C., A.D., and D.E.L. analyzed data; S.C., S.M.C., A.D., S.M.D., and D.E.L. interpreted results of experiments; S.C., S.M.C., and A.D. prepared figures; S.C., A.D., S.M.D., and D.E.L. drafted manuscript; S.C., A.D., S.M.D., and D.E.L. edited and revised manuscript; D.E.L. approved final version of manuscript.
Supplementary Material
Footnotes
Supplemental Material for this article is available online at the Journal website.
REFERENCES
- 1.Anon E, Serra-Picamal X, Hersen P, Gauthier NC, Sheetz MP, Trepat X, Ladoux B. Cell crawling mediates collective cell migration to close undamaged epithelial gaps. Proc Natl Acad Sci USA 109: 10891–10896, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arce FT, Whitlock JL, Birukova AA, Birukov KG, Arnsdorf MF, Lal R, Garcia JG, Dudek SM. Regulation of the micromechanical properties of pulmonary endothelium by S1P and thrombin: role of cortactin. Biophys J 95: 886–894, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bement WM, Forscher P, Mooseker MS. A novel cytoskeletal structure involved in purse string wound closure and cell polarity maintenance. J Cell Biol 121: 565–578, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bryce NS, Clark ES, Leysath JM, Currie JD, Webb DJ, Weaver AM. Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr Biol 15: 1276–1285, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Chiang E, Wang T, Garcia JN. Acute lung injury: the injured lung endothelium, therapeutic strategies for barrier protection, and vascular biomarkers. In: Textbook of Pulmonary Vascular Disease, edited by Yuan JX, Garcia JG, West JB, Hales CA, Rich S, Archer SL. New York: Springer US, 2011, p. 197–222. [Google Scholar]
- 6.Cordelières FP. Manual Tracking. National Institutes of Health. http://rsbweb.nih.gov/ij/plugins/track/Manual%20Tracking%20plugin.pdf [9 Jul. 2015]. [Google Scholar]
- 7.Csortos C, Kolosova I, Verin AD. Regulation of vascular endothelial cell barrier function and cytoskeleton structure by protein phosphatases of the PPP family. Am J Physiol Lung Cell Mol Physiol 293: L843–L854, 2007. [DOI] [PubMed] [Google Scholar]
- 8.DiMilla PA, Quinn JA, Albelda SM, Lauffenburger DA. Measurement of individual cell migration parameters for human tissue cells. AIChE J 38: 1092–1104, 1992. [Google Scholar]
- 9.Dos Santos CC, Slutsky AS. Mechanisms of ventilator-induced lung injury: a perspective. J Appl Physiol 89: 1645–1655, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Dudek SM, Camp SM, Desai A, Ma SF, Garcia JG. An acute lung injury-associated polymorphism in the human cortactin gene alters protein tyrosine phosphorylation and function (Abstract). Am J Respir Crit Care Med 177: A844, 2008. [Google Scholar]
- 11.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 91: 1487–1500, 2001. [DOI] [PubMed] [Google Scholar]
- 12.Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X, Garcia JG. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem 279: 24692–24700, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Dudek SM, Ma SF, Wade MS, Camp SM, Desai A, Gordeuk VR, Garcia JG. A single nucleotide polymorphism in the human cortactin gene associates with sickle cell disease and alters protein function (Abstract). 181: A1040, 2010. [Google Scholar]
- 14.Gao L, Grant A, Halder I, Brower R, Sevransky J, Maloney JP, Moss M, Shanholtz C, Yates CR, Meduri GU. Novel polymorphisms in the myosin light chain kinase gene confer risk for acute lung injury. Am J Respir Cell Mol Biol 34: 487–495, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 108: 689–701, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goley ED, Welch MD. The Arp2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol 7: 713–726, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Gong MN, Wei Z, Xu LL, Miller DP, Thompson BT, Christiani DC. Polymorphism in the surfactant protein-B gene, gender, and the risk of direct pulmonary injury and ARDS. Chest 125: 203–211, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Goodman SL, Risse G, von der Mark K. The E8 subfragment of laminin promotes locomotion of myoblasts over extracellular matrix. J Cell Biol 109: 799–809, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gross SR. Actin binding proteins: their ups and downs in metastatic life. Cell Adh Migr 7: 199–213, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang C, Liu J, Haudenschild CC, Zhan X. The role of tyrosine phosphorylation of cortactin in the locomotion of endothelial cells. J Biol Chem 273: 25770–25776, 1998. [DOI] [PubMed] [Google Scholar]
- 21.Isenberg BC, DiMilla PA, Walker M, Kim S, Wong JY. Vascular smooth muscle cell durotaxis depends on substrate stiffness gradient strength. Biophys J 97: 1313–1322, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacinto A, Martinez-Arias A, Martin P. Mechanisms of epithelial fusion and repair. Nat Cell Biol 3: E117–E123, 2001. [DOI] [PubMed] [Google Scholar]
- 23.Jacobson JR, Dudek SM, Singleton PA, Kolosova IA, Verin AD, Garcia JG. Endothelial cell barrier enhancement by ATP is mediated by the small GTPase Rac and cortactin. Am J Physiol Lung Cell Mol Physiol 291: L289–L295, 2006. [DOI] [PubMed] [Google Scholar]
- 24.Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv 23: 243–252, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kowalski JR, Egile C, Gil S, Snapper SB, Li R, Thomas SM. Cortactin regulates cell migration through activation of N-WASP. J Cell Sci 118: 79–87, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Lin Z, Pearson C, Chinchilli V, Pietschmann SM, Luo J, Pison U, Floros J. Polymorphisms of human SP-A, SP-B, and SP-D genes: association of SP-B Thr131Ile with ARDS. Clin Genet 58: 181–191, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Lodish H, Baltimore D, Berk A, Zipursky SL, Matsudaira P, Darnell J. Molecular Cell Biology (3rd ed). New York: Scientific American Press, 1995. [Google Scholar]
- 28.Marshall RP, Webb S, Bellingan GJ, Montgomery HE, Chaudhari B, McAnulty RJ, Humphries SE, Hill MR, Laurent GJ. Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am J Respir Crit Care Med 166: 646–650, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 295: L379–L399, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDonald DM, Thurston G, Baluk P. Endothelial gaps as sites for plasma leakage in inflammation. Microcirculation 6: 7–22, 1999. [PubMed] [Google Scholar]
- 31.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 86: 279–367, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Nobes CD, Hall A. Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81: 53–62, 1995. [DOI] [PubMed] [Google Scholar]
- 33.O'Keefe GE, Hybki DL, Munford RS. The G → A single nucleotide polymorphism at the −308 position in the tumor necrosis factor-alpha promoter increases the risk for severe sepsis after trauma. J Trauma 52: 817–825, 2002. [DOI] [PubMed] [Google Scholar]
- 34.Pagano M, Gauvreau K. Principles of Biostatistics. Pacific Grove, CA: Duxbury, 2000. [Google Scholar]
- 35.Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat Rev Mol Cell Biol 10: 538–549, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin D, Xia Y, Whitesides GM. Soft lithography for micro- and nanoscale patterning. Nat Protoc 5: 491–502, 2010. [DOI] [PubMed] [Google Scholar]
- 37.Ren G, Crampton MS, Yap AS. Cortactin: coordinating adhesion and the actin cytoskeleton at cellular protrusions. Cell Motil Cytoskeleton 66: 865–873, 2009. [DOI] [PubMed] [Google Scholar]
- 38.Schnoor M, Lai FP, Zarbock A, Kläver R, Polaschegg C, Schulte D, Weich HA, Oelkers JM, Rottner K, Vestweber D. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J Exp Med 208: 1721–1735, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strieter RM, Kunkel SL. Acute lung injury: the role of cytokines in the elicitation of neutrophils. J Investig Med 42: 640–651, 1994. [PubMed] [Google Scholar]
- 40.Szulcek R, Bogaard HJ, van Nieuw Amerongen GP. Electric cell-substrate impedance sensing for the quantification of endothelial proliferation, barrier function, and motility. J Vis Exp 85: 51300, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomashefski JF, Davies P, Boggis C, Greene R, Zapol WM, Reid LM. The pulmonary vascular lesions of the adult respiratory distress syndrome. Am J Pathol 112: 112–126, 1983. [PMC free article] [PubMed] [Google Scholar]
- 42.Tranquillo RT, Lauffenburger DA, Zigmond SH. A stochastic model for leukocyte random motility and chemotaxis based on receptor binding fluctuations. J Cell Biol 106: 303–309, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turnage RH, Guice KS, Oldham KT. Pulmonary microvascular injury following intestinal reperfusion. New Horiz 2: 463–475, 1994. [PubMed] [Google Scholar]
- 44.Weaver AM, Karginov AV, Kinley AW, Weed SA, Li Y, Parsons JT, Cooper JA. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol 11: 370–374, 2001. [DOI] [PubMed] [Google Scholar]
- 45.Weed SA, Karginov AV, Schafer DA, Weaver AM, Kinley AW, Cooper JA, Parsons JT. Cortactin localization to sites of actin assembly in lamellipodia requires interactions with F-actin and the Arp2/3 complex. J Cell Biol 151: 29–40, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitesides GM, Ostuni E, Takayama S, Jiang X, Ingber DE. Soft lithography in biology and biochemistry. Annu Rev Biomed Eng 3: 335–373, 2001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.