

Abstract
Interferon (IFN) in combination with ribavirin has been the standard of care (SOC) for chronic hepatitis C for the past few decades. Although the current SOC lacks the desired efficacy, and 4 new direct-acting antiviral agents have been recently approved, interferons are still likely to remain the cornerstone of therapy for some time. Moreover, as an important cytokine system of innate immunity, host interferon signaling provides a powerful antiviral response. Nevertheless, the mechanisms by which HCV infection controls interferon production, and how interferons, in turn, trigger anti-HCV activities as well as control the outcome of HCV infection remain to be clarified. In this report, we review current progress in understanding the mechanisms of IFN against HCV, and also summarize the knowledge of induction of interferon signaling by HCV infection.
Key words: Interferon, Hepatitis C virus, Antiviral agent, Molecular mechanism
Abbreviations: CHC, chronic hepatitis C; DCs, dendritic cells; DNAM1, DNAX accessory molecule-1; dsRNA, double-stranded RNA; E2, envelop 2; GAS, IFN-γ-activated site; GWAS, genome-wide association studies; IFN, interferon; IFN-α, interferon-α; IFNAR1, interferon-alpha receptor 1; IFNAR2, interferon-alpha receptor 2; IFNGR1, interferon gamma receptor 1; IFNGR2, interferon gamma receptor 2; IFNL4, IFN-lambda 4; IL-10R2, interleukin-10 receptor 2; IL-29, interleukin-29; IRF-3, interferon regulatory factor 3; IRGs, IFN regulatory genes; ISG15, interferon-stimulated gene 15; ISGs, IFN-stimulated genes; ISGF3, IFN-stimulated gene factor 3; ISREs, IFN-stimulated response elements; JAKs, Janus activated kinases; MAVS, mitochondrial antiviral signaling protein; MDA-5, melanoma differentiation-associated gene-5; MHC, major histocompatibility complex; NKCs, natural killer cells; NKTCs, natural killer T cells; OAS, 2′-5′-oligoadenylate synthetase; PAMPs, pathogen-associated molecular patterns; PBMCs, peripheral blood mononuclear cells; pDC, plasmacytoid dendritic cell; PKR, protein kinase R; PRRs, pattern recognition receptors; RdRp, RNA dependent RNA polymerase; RIG-I, retinoic acid-inducible gene-I; RLRs, RIG-I-like receptors; SNPs, single-nucleotide polymorphisms; SOC, standard of care; STAT1, signal transducer and activator of transcription 1; STAT2, signal transducer and activator of transcription 2; SVR, sustained virological response; TH1, T-helper-1; TH2, T-helper-2; TLRs, Toll-like receptors; TYK2, tyrosine kinase 2; USP18, ubiquitin specific peptidase 18
Graphical abstract
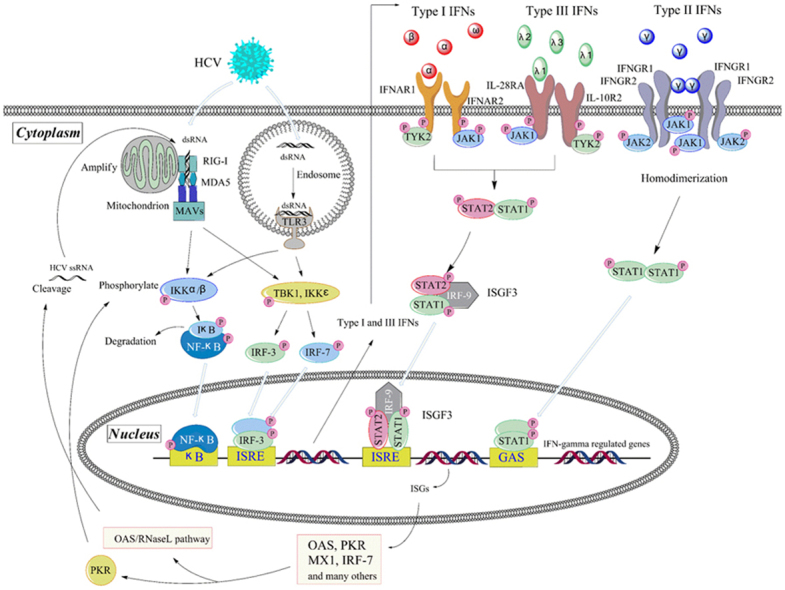
This review summarized pathways of interferon induction and regulation by HCV infection, and how interferon in turn triggers anti-HCV activities as well as control the outcome of HCV infection.
1. Introduction
Interferons (IFNs) are a family of cytokines secreted by host cells in response to various pathogens like viruses, bacteria, fungi, or parasites which trigger the protective defenses of the immune system1. To defend against invading viral pathogens, the interferon signatures activate the transcription of numerous IFN-stimulated genes (ISGs) and thus promote a broad-spectrum of antiviral responses2.
In 1986, interferon-α (IFN-α) was first approved as a treatment for patients with non-A non-B (now renamed type C) chronic hepatitis. After this landmark study, further improvements in research technologies led to increased sustained virological response (SVR) rates. These included addition of the guanosine analog ribavirin to IFN treatment3, followed by pegylation of the IFN molecule. Pegylated IFN-α, which was developed to extend the interdose interval to at least one week, offered improved convenience, decreased side effects, and superior clinical efficacy when compared with IFN-α. Presently, the standard treatment of chronic hepatitis C remains the combination of pegylated IFN-α and RBV. Recently, three NS3-4A protease inhibitors, telaprevir (Vertex/Janssen)4, boceprevir (Merck) and simeprevir (Janssen)5, 6, and one NS5B RNA dependent RNA polymerase (RdRp) inhibitor sofosbuvir (Gilead)6, have been approved in combination with pegylated IFN-α and ribavirin for the treatment of chronic HCV infection. These agents will improve the rate of SVR, but their toxicities combined with Pegylated IFN-α and RBV may limit their overall efficacy. For this reason, interferons are still likely to remain the cornerstone of therapy for some time.
Since the first use of the standard therapeutic paradigm against chronic hepatitis C more than 20 years ago, fundamental biomedical research has greatly improved our understanding of the underlying molecular mechanisms of IFN production, regulation and antiviral action. This has been accompanied by substantial progress in understanding the HCV life cycle.
Presently, we review the current information on the mechanisms of IFN actions against HCV, and summarize the latest knowledge of induction of interferon signal by HCV infection (Fig. 1).
Figure 1.

Pathways of interferon induction and effect in HCV infection. HCV RNA leads to the transcription of interferon genes through TLR3 in endosome and/or RIG-I in cytoplasm. Type I IFNs and type III IFNs bind to respective receptors, activate the same JAK-STAT pathway and then induce the overlapping ISGs. IFN-γ activates STAT1 and induces a partially overlapping but distinct ISGs. As the members of ISGs, PKR and/or OAS/RNaseL pathways play important roles in the anti-HCV action.
2. Induction of interferon
It is well known that the innate immune response is the first line of defense against viral infections, and IFNs are the central cytokines responsible for the induction, activation and regulation of the antiviral state in innate immune cells like natural killer cells (NKCs)7. Based on their structural features, receptor usage and biological activities, three distinct types of IFNs are now recognized. Type I IFNs (IFN-α, IFN-β and IFN-ω) and type III IFNs (IFN-λ1, IFN-λ2 and IFN-λ3; also named as IL29, IL28A, and IL28B respectively) are produced by most types of cells which are infected with virus and also by key sentinel cells of the innate immune system: macrophages and dendritic cells (DCs). However, the type II IFNs (IFN-γ) are only produced by certain cells of the immune system, including NKCs and natural killer T cells (NKTCs), which belong to a part of the innate immune response, as well as antigen-specific T cells (both CD4+ T helper 1 and CD8+ cytotoxic T cells).
It is now clear that there are several families of pattern recognition receptors (PRRs) surveying the cellular micro-environment for viral infection. The two important characterized families of PRRs that detect viral genomes and then induce type I and type III IFNs are the Toll-like receptors (TLRs) and the retinoic acid-inducible gene-I (RIG-I)8. After recognizing viral infection, these receptors initiate intracellular signaling cascades, which are responsible for the production of type I and type III IFNs and proinflammatory cytokines, and mediate the antiviral immune response finally. TLRs are a family of transmembrane pattern recognition receptors that recognize microbial pathogen-associated molecular patterns (PAMPs) and activate the expression of genes involved in inflammatory and immune responses9. There are at least 10 human TLRs, and 5 of them are involved in the recognition of viral infections. TLR3, TLR7 and TLR9 represent a TLR subfamily that recognize viral nucleic acids and induce an endogenous interferon response, while viral glycoproteins have been shown to interact with TLR2 and TLR4. TLRs are expressed on several types of immune cells including macrophages, DCs, B cells, as well as fibroblasts and epithelial cells. When the HCV invades, the TLR2 will recruit TLR1 and TLR6 as coreceptors for HCV core- and NS3-mediated activation of macrophages and innate immunity in humans10. Meanwhile, HCV infection directly induces TLR4 expression and thereby activates B cells, which may contribute to the host׳s innate immune responses11. Along with TLR3, TLR7 and TLR9 are localized in intracellular compartments such as endosomes. The latest reports show that TLR3 and TLR7 play highly significant roles in the recognition of HCV RNA12, 13, 14, 15; TLR3 senses double-stranded RNA (dsRNA), which is an essential intermediate in the HCV replication cycle, and then initiates signaling that activates NF-κB and interferon regulatory factor 3 (IRF-3), thereby inducing the synthesis of type I IFN16. TLR7, expressed on the DCs and Kupffer cells, shows a very good ability to sense the HCV RNA, subsequently leading to the synthesis of type I IFNs12, 17.
Importantly, macrophages and DCs do not have to directly infect themselves by HCV in order to produce IFNs. Instead, they constantly screen materials from the outside, including intact viral particles and cellular remnants containing viral fractions. After the degradation in the endosomes, viral nucleic acids are exposed and recognized by TLRs. In addition, viruses escaping the endosomal entry pathway can be detected by the cytosolic pathways and then induce types I and III IFNs. In general, viral RNA genomes or viral replication intermediates can be sensed in cytoplasm or endosomes by the related cytoplasmic RNA helicases RIG-I and melanoma differentiation-associated gene-5 (MDA-5). Accumulating evidence suggests that RIG-I-like receptors (RLRs), particularly RIG-I and MDA5, are responsible for the recognition of HCV RNA18. It has been reported that RIG-I senses HCV RNA in the early stage of infection and activates downstream pathways of the innate immune response19. After sensing the viral RNA, these sensors utilize the mitochondrial antiviral signaling protein (MAVS, also known as IPS-1, VISA, or Cardif)20, which initiates a series of signaling cascades. MAVS then propagates the signal to the closely related kinases TBK1 and IKKε, or the IKKα and IKKβ kinases. These kinases phosphorylate and thus activate the critical transcription factors IRF-3 and IRF-7 respectively, which bind to the response elements, eventually promoting expression of types I and III IFNs׳ genes21.
All types of IFNs induce an antiviral micro-environment in host cells by the transcriptional activation of hundreds of genes. The specific set of genes differs among IFNs and target cell types. In general, type I and type III IFNs induce a largely overlapping set of genes in cells22, whereas the gene set induced by type II IFNs is more distinct23. Meanwhile, the number of genes regulated by IFNs also differs among cell types. For instance, pegylated IFN-α induces 200–300 genes in liver cells, but nearly 2000 genes in peripheral blood mononuclear cells (PBMCs)24.
3. Anti-HCV mechanism of interferons
3.1. Anti-HCV mechanisms of type I interferon
Type I interferons belong to a family of major innate immune cytokines produced by host cells in response to viral infections. Since their discovery, their antiviral actions have been studied extensively, leading to the development of the first cytokine-based therapy. This treatment is licensed worldwide for several viral diseases, malignancies, and even immune disorders25, 26. Type I IFNs bind a unique ubiquitous heterodimeric receptor consisting of interferon-alpha receptor 1/2 (IFNAR1/IFNAR2), leading to the activation of signaling pathways and the subsequent induction of a large number of ISGs. ISG-encoded proteins mediate the antiviral and other effects of interferons27. The downstream signaling pathways for these receptors have been extensively described: IFNAR1 and IFNAR2 are associated with the Janus activated kinases (JAKs) tyrosine kinase 2 (TYK2) and JAK1, respectively. Binding of type I IFNs to their heterodimeric receptors leads to activation of JAKs, which results in tyrosine phosphorylation of signal transducer and activator of transcription 2 (STAT2) and STAT1; STAT1/STAT2 migrates into the nucleus and associates with the IFN regulatory factor 9 (IRF9), to form the STAT1–STAT2–IRF9 complex (also known as IFN-stimulated gene factor 3 [ISGF3]). This complex then binds IFN-stimulated response elements (ISREs) inside DNA to initiate the transcription of hundreds of different ISGs, which are known to mediate the diverse cellular effects in the infected cell28. Collectively, these genes facilitate both clearance of virus from infected cells and protection of neighboring uninfected cells from incoming viral progeny. The past 50 years of IFN research resulted in identification of a number of key IFN regulatory genes (IRGs). Most notably, the antiviral-associated proteins 2′-5′-oligoadenylate synthetase (OAS) and protein kinase R (PKR) have been intensely studied to understand their roles in the antiviral effects regulated through IFN signaling. Among the genes induced by type I IFNs, perhaps the most intensely studied is PKR. This kinase plays an important role in the antiviral response through phosphorylation of elF2α and inhibition of host cellular mRNA translation and protein synthesis. Recently, Zhang et al.29 showed that the host cell can employ PKR activation to restrict HCV 1a replication through regulating the NF-κB pathway. Moreover, the IFN-inducible OAS/RNase L pathway blocks multiple viral infections through cleaving the viral and cellular single-stranded RNA30. Type I IFNs induced by HCV infection initiate signaling via the OAS genes; both OAS and RNaseL are implicated in the antiviral response against hepatitis C virus. Some scientists have suggested that, among the members of the OAS family, OAS1 p46 and OAS3 p100 mediate the RNaseL-dependent antiviral activity against HCV31. It is very interesting that RNase L and PKR are effectors of the interferon antiviral response that share homology in their pseudokinase and protein kinase domains. Very recent research showed that the anti-cancer drug sunitinib, a potent inhibitor of both RNase L and PKR in cell cultures as well as in mice, eventually prevented antiviral innate immune responses mediated by RNase L and PKR32. Sunitinib is thought to modulate IRE1, an endoplasmic reticulum protein involved in the unfolded protein response that is closely related to RNase L. In addition to its direct antiviral actions, IFN has important interactions with the adaptive and innate immune responses. For example, type I IFNs can promote memory T-cell proliferation, prevent T-cell apoptosis and stimulate NKC activation and DC maturation as well. They can also up-regulate the production of major histocompatibility complex (MHC) class-I and class-II peptides, and might promote a T-helper-1 (TH1) over a T-helper-2 (TH2) phenotype. In addition to its direct immune-stimulating mechanisms (by decreasing HCV RNA replication), IFN might prevent immune exhaustion and enhance the adaptive HCV-specific immune response33.
In HCV infection, the type I IFN signaling pathway is controlled by a number of negative signatures; the interferon-stimulated gene 15/ubiquitin specific peptidase 18 (ISG15/USP18) pathway belongs to one of them. The role of ISG15/USP18 pathway in the host innate immune response is complex. Many studies have suggested that this pathway provides an important protective role against several viruses34, 35. However, HCV seems to utilize the pathway to enhance its replication inversely. By utilizing microarray-expression profiling, Chen et al.36 identified three genes, ISG15, USP18 and HERC5, which represent members of the ubiquitously IFN-regulated ISG15 group, out of a core set of 18 genes thought to be predictive of this response. Initially, these authors hypothesized that ISG15/ISGylation was antiviral, and supposed that silencing the USP18 would lead to increasing of ISGylation and potentiating the anti-viral effect of exogenous IFN-α37. However, increasing ISGylation independent of USP18 actually promoted efficient viral replication, such that increasing the level USP18 blunted the anti-viral effects of IFN-α, promoting HCV production in a protease-independent fashion38. Based on the above findings, the authors concluded that ISGylation promotes HCV production, decreases the anti-HCV effects of IFN-α and functions in the downstream steps of HCV RNA replication. ISG15, however, is generally considered to be antiviral. Furthermore, the authors pointed out that ISG15 is a novel mechanism for virus persistence, and ISGylation is a possible target for therapy of HCV infection38.
3.2. The anti-HCV mechanisms of type II interferon
Unlike the types I and III interferon, type II interferon (IFN-γ) is secreted predominantly by T cells, NKCs and other cells such as macrophage, DCs, and B cells. In the host, IFN-γ always stimulates cell-mediated immune responses that are critical for the development of host protection against pathogenic intracellular microorganisms. The IFN-γ receptor, widely expressed in host cells, is composed of two subunits, interferon gamma receptors 1 and 2 (IFNGR1 and IFNGR2, respectively). Each receptor subunit interacts with a member of the JAK family. Unlike the type I IFN receptor, IFNGR1 associates with JAK1, and IFNGR2 is associated with JAK2. After IFN-γ binds to the type II IFN receptor, JAK1 and JAK2 are activated and subsequently phosphorylate only STAT1. Phosphorylated STAT1 self associates to form homodimers that translocate to the nucleus and where they bind the IFN-γ-activated site (GAS) elements. The GAS elements are present in the promoter regions of IFN-γ-regulated genes21, 39. IFN-γ is structurally unrelated to type I and type III IFNs, which binds to a different receptor encoded by separate chromosomal locus. However, there are remarkable overlaps in the many hundreds of genes regulated by both of them40.
There is no convincing evidence that HCV infection can induce IFN-γ release from NKCs directly. However, some studies show that type I IFN can increase NKC cytotoxicity directed toward the HCV infected cells41, 42. Although previous reports suggested that NKCs do not directly respond to HCV virions secreted from HCV-infected cells43, a recent study reported that NKCs produce IFN-γ in response to HCV infected cells in a plasmacytoid dendritic cell (pDC)-and type I IFN-dependent manner. Monocyte-derived IL-15 can augment this process44. Another report demonstrates that IFN-α activates NKCs in a DNAX accessory molecule-1(DNAM1)-dependent manner; this leads to efficient recognition of HCV-infected hepatoma cells and secretion of IFN-γ by NKCs45. Furthermore, Jia et al.46 reported that IFN-γ has synergistic antiviral effects with IFN-α.
Importantly, IFN-γ has been demonstrated to be a key cytokine for clearance of acute and chronic HCV infection47, 48. Using gene transfer technology to produce sustained IFN-γ in the human hepatocyte chimeric mice infected with genotype 1b HCV, Takahashi et al. showed that injection of the human IFN-γ-expressing plasmid vector pCpG-huIFNγ could produce long-term elimination of HCV RNA in chimeric mice49. As compared with the cytotoxicity of NKCs, the toxicity of IFN-γ secreted from NKCs is considered to be a more efficient mechanism against HCV. The former requires a 1:1 interaction between NKCs and infected cells, whereas IFN-γ molecules secreted by an individual NKC may reach hundreds of hepatocytes50.
As mentioned above, the USP18 may participate in the inhibition of IFN-α signatures, but it does not seem to impact IFN-γ or IFN-λ signaling 51. One previous study has shown that the HCV envelop 2 (E2) glycoprotein may impair IFN-γ production by NKCs for its binding to the tetraspanin CD81 on the cell surface52. However, there are some controversial results. One of study suggests that it is HCV E1 but not E2 which is responsible for modulation of the antiviral function of NKCs43. Although activated NKCs can recognize and kill hepatoma cells containing HCV replicon53, there is another suggestion that cellular contact with hepatoma cells may impair the killing capabilities and IFN-γ response of NKCs54. The underlying mechanisms leading to such differences are not entirely clear, and considerable additional research is needed.
3.3. Anti-HCV mechanisms of type III interferon
Two research teams independently claimed the discovery and initial description of type III interferon (IFN-λ) in 2003, opening a brand new area of IFN research55, 56. Presently 3 different IFN-λ genes are known to encode 3 distinct but closely related proteins. These are designated as IFN-λ1, -λ2 and -λ3 or interleukin-29 (IL-29), IL-28A, and IL-28B. Although IFN-λ is functionally an interferon, it is clearly structurally related to members of the IL-10 family57. Meanwhile, the IFN-λ genes phylogenetically reside somewhere between the type I IFN and IL-10 gene families. Amino acid sequence comparisons show that the type III IFNs exhibit about 5%–18% homology with either type I IFNs or the IL-10-related cytokines. Therefore, it is not surprising that the antiviral activity of type III IFN resembles that of type I IFN.
Similar to type I IFN, the three IFNλs can be triggered by viral infection, then induce anti-viral and anti-tumor activities through both innate and adaptive immune system pathways58, 59. And yet, the IFN-λ proteins bind to and signal through a distinct receptor complex composed of the unique IFN-λR1 chain (also known as IL-28RA) and interleukin-10 receptor 2 (IL-10R2) receptor chain (also a part of the receptor complexes for IL-10, IL-22, and IL-26)60. Moreover, IL-10R2 is only expressed on a few restricted cell types, including hepatocytes, epithelial cells and pDCs61, 62. Although the receptor complex of IFN-λ is different from that of type I IFN, signaling through either IFN-λ or IFN-α receptor complexes results in the activation of the same JAK-STAT signal transduction cascades63, and result in activation of overlapping sets of ISGs64. Consequently, the biological activities induced by type III IFNs are very similar to those induced by type I, including induction of antiviral activity and up-regulation of MHC class I antigen expression on many cell types.
As described above, the IFN-λ signal transduction cascades are very similar to those induced by type I IFNs, so the mechanisms of HCV inhibition are likely to be the same between the types I and III IFNs. However, some differences between them are known. For example, the relative level of gene expression induced by IFN-λ is always weaker than that induced by IFN-α in many cell types64. Meanwhile, another study showed that IFN-α triggers an early peak in ISG expression, which is much greater than that produced by IFN-λ, followed by a rapid decline22. Furthermore, it has been suggested that the lower but sustained anti-HCV activity of IFN-λ could be preferable, since IFN-based therapies are administered over a relatively long period62.
Convincing evidence supports the suggestion that IFN-λs control HCV replication through the innate immune pathway22. Moreover, combinations of IFN-λ1 and IFN-α had a greater inhibitory effect on HCV replication as compared with the individual agents65. Interestingly, based on the important differences in time-dependent antiviral responses and ISGs׳ up-regulation between type I and type III IFN, the latest research found that a combination of IFN-α and IFN-λ3 produced synergistic anti-HCV activity66.
Three genome-wide association studies (GWAS) in 2009 identified three variant genes which strongly associated with both spontaneous HCV clearance and SVR to pegIFN-α/RBV treatment for chronic hepatitis C67, 68, 69. These gene variants, identified as rs12979860, rs12980275 and rs8099917, are single-nucleotide polymorphisms (SNPs) on chromosome 19q13.13 near the IFNL3 gene (IL28B)67, 68, 69. Additionally, the patients with these variants were more likely to spontaneously resolve acute HCV infection without treatment70. Recently, one group has identified a new inducible human protein-coding gene, IFN-λ 4 (IFNL4). The polymorphism of IFNL4 was shown to be in high linkage disequilibrium with that of genes close to IL28B, and more strongly associated with spontaneous or treatment-induced HCV clearance than IL28B genotypes, especially in individuals of African ancestry71. These results could provide a new and rational dimension for HCV research, including more basic studies on IFN-λs. Furthermore, they suggest the potential for more personalized novel, effective therapies for HCV infections.
4. Conclusions
Modern studies of the induction and biological activity of interferon in HCV infections have not only promoted our understanding of the pathogenesis of the most important liver disease worldwide, but also made important contributions to uncover the mysteries of virus–host interactions. The interferon system provides a powerful antiviral response, which inhibits the replication of several viruses including HCV, stimulates apoptosis of infected cells, and modulates the immune system as well. Although much has been learned about the biological activities of interferon, our understanding is far from complete. For example, how does HCV escape from the host immune system and what are the relevant molecular mechanisms? How can we provide more personalized chronic hepatitis C (CHC) treatment based on interferon therapy in the future? Why does interferon treatment fail to control CHC in some patients, even if there is a very high level of endogenous interferon? Moreover, what are the underlying molecular mechanisms of synergistic anti-HCV activity involving combinations of different IFNs? Future research is needed to address these important questions. Gaining new insights into IFNs׳ anti-HCV activities, increases in understanding how HCV infection leads to the induction of interferon signaling are essential for progress in understanding and treating HCV. Further research is imperative to develop novel highly effective, personalized treatments for this devastating liver disease.
Acknowledgments
This work was supported by the National Natural Science Foundation for Excellent Young Scholars of China (81322050), the National S&T Major Special Project on Major New Drug Innovation (2012ZX09301-002-001), and the Program for New Century Excellent Talents in University of Ministry of Education of China (NCET-12-0072).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.

References
- 1.de Weerd N.A., Samarajiwa S.A., Hertzog P.J. Type I interferon receptors: biochemistry and biological functions. J Biol Chem. 2007;282:20053–20057. doi: 10.1074/jbc.R700006200. [DOI] [PubMed] [Google Scholar]
- 2.Fensterl V., Sen G.C. Interferons and viral infections. Biofactors. 2009;35:14–20. doi: 10.1002/biof.6. [DOI] [PubMed] [Google Scholar]
- 3.Poynard T., Marcellin P., Lee S.S., Niederau C., Minuk G.S., Ideo G. Randomised trial of interferon α2b plus ribavirin for 48 weeks or for 24 weeks versus interferon α2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. International Hepatitis Interventional Therapy Group (IHIT) Lancet. 1998;352:1426–1432. doi: 10.1016/s0140-6736(98)07124-4. [DOI] [PubMed] [Google Scholar]
- 4.McHutchison J.G., Manns M.P., Muir A.J., Terrault N.A., Jacobson I.M., Afdhal N.H. Telaprevir for previously treated chronic HCV infection. New Engl J Med. 2010;362:1292–1303. doi: 10.1056/NEJMoa0908014. [DOI] [PubMed] [Google Scholar]
- 5.Bacon B.R., Gordon S.C., Lawitz E., Marcellin P., Vierling J.M., Zeuzem S. Boceprevir for previously treated chronic HCV genotype 1 infection. New Engl J Med. 2011;364:1207–1217. doi: 10.1056/NEJMoa1009482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chao D., Botwin G.J., Morgan T.R. Update on recently approved treatments for hepatitis C. Curr Treat Options Gastroenterol. 2014;12:211–228. doi: 10.1007/s11938-014-0013-z. [DOI] [PubMed] [Google Scholar]
- 7.Stetson D.B., Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 8.Wilkins C., Gale M., Jr. Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol. 2010;22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawai T., Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21:317–337. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang S., Dolganiuc A., Szabo G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J Leukoc Biol. 2007;82:479–487. doi: 10.1189/jlb.0207128. [DOI] [PubMed] [Google Scholar]
- 11.Machida K., Cheng K.T., Sung V.M., Levine A.M., Foung S., Lai M.M. Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of β interferon and interleukin-6. J Virol. 2006;80:866–874. doi: 10.1128/JVI.80.2.866-874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dreux M., Garaigorta U., Boyd B., Decembre E., Chung J., Whitten-Bauer C. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe. 2012;12:558–570. doi: 10.1016/j.chom.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Negash A.A., Ramos H.J., Crochet N., Lau D.T., Doehle B., Papic N. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013;9:e1003330. doi: 10.1371/journal.ppat.1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li K., Li N.L., Wei D., Pfeffer S.R., Fan M., Pfeffer L.M. Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on Toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology. 2012;55:666–675. doi: 10.1002/hep.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seki E., Brenner D.A. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–335. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 16.Wang N., Liang Y., Devaraj S., Wang J., Lemon S.M., Li K. Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J Virol. 2009;83:9824–9834. doi: 10.1128/JVI.01125-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi K., Asabe S., Wieland S., Garaigorta U., Gastaminza P., Isogawa M. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci USA. 2010;107:7431–7436. doi: 10.1073/pnas.1002301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loo Y.M., Gale M., Jr. Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnaud N., Dabo S., Akazawa D., Fukasawa M., Shinkai-Ouchi F., Hugon J. Hepatitis C virus reveals a novel early control in acute immune response. PLoS Pathog. 2011;7:e1002289. doi: 10.1371/journal.ppat.1002289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koshiba T., Yasukawa K., Yanagi Y., Kawabata S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci Signal. 2011;4:ra7. doi: 10.1126/scisignal.2001147. [DOI] [PubMed] [Google Scholar]
- 21.Heim M.H. Innate immunity and HCV. J Hepatol. 2013;58:564–574. doi: 10.1016/j.jhep.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Marcello T., Grakoui A., Barba-Spaeth G., Machlin E.S., Kotenko S.V., MacDonald M.R. Interferons α and λ inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131:1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 23.Dill M.T., Makowska Z., Duong F.H., Merkofer F., Filipowicz M., Baumert T.F. Interferon-γ-stimulated genes, but not USP18, are expressed in livers of patients with acute hepatitis C. Gastroenterology. 2012;143:777–786. doi: 10.1053/j.gastro.2012.05.044. [DOI] [PubMed] [Google Scholar]
- 24.Sarasin-Filipowicz M., Oakeley E.J., Duong F.H., Christen V., Terracciano L., Filipowicz W. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci USA. 2008;105:7034–7039. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borden E.C., Sen G.C., Uze G., Silverman R.H., Ransohoff R.M., Foster G.R. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trinchieri G. Type I interferon: friend or foe? J Exp Med. 2010;207:2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mogensen K.E., Lewerenz M., Reboul J., Lutfalla G., Uze G. The type I interferon receptor: structure, function, and evolution of a family business. J Interferon Cytokine Res. 1999;19:1069–1098. doi: 10.1089/107999099313019. [DOI] [PubMed] [Google Scholar]
- 28.Zhao W., Lee C., Piganis R., Plumlee C., de Weerd N., Hertzog P.J. A conserved IFN-α receptor tyrosine motif directs the biological response to type I IFNs. J Immunol. 2008;180:5483–5489. doi: 10.4049/jimmunol.180.8.5483. [DOI] [PubMed] [Google Scholar]
- 29.Zhang L., Alter H.J., Wang H., Jia S., Wang E., Marincola F.M. The modulation of hepatitis C virus 1a replication by PKR is dependent on NF-κB mediated interferon β response in Huh7.5.1 cells. Virology. 2013;438:28–36. doi: 10.1016/j.virol.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakrabarti A., Jha B.K., Silverman R.H. New insights into the role of RNase L in innate immunity. J Interferon Cytokine Res. 2011;31:49–57. doi: 10.1089/jir.2010.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwon Y.C., Kang J.I., Hwang S.B., Ahn B.Y. The ribonuclease L-dependent antiviral roles of human 2′,5′-oligoadenylate synthetase family members against hepatitis C virus. FEBS Lett. 2013;587:156–164. doi: 10.1016/j.febslet.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 32.Jha B.K., Polyakova I., Kessler P., Dong B., Dickerman B., Sen G.C. Inhibition of RNase L and RNA-dependent protein kinase (PKR) by sunitinib impairs antiviral innate immunity. J Biol Chem. 2011;286:26319–26326. doi: 10.1074/jbc.M111.253443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feld J.J., Hoofnagle J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature. 2005;436:967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- 34.Lenschow D.J., Lai C., Frias-Staheli N., Giannakopoulos N.V., Lutz A., Wolff T. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc Natl Acad Sci USA. 2007;104:1371–1376. doi: 10.1073/pnas.0607038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai C., Struckhoff J.J., Schneider J., Martinez-Sobrido L., Wolff T., Garcia-Sastre A. Mice lacking the ISG15 E1 enzyme UbE1L demonstrate increased susceptibility to both mouse-adapted and non-mouse-adapted influenza B virus infection. J Virol. 2009;83:1147–1151. doi: 10.1128/JVI.00105-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen L., Borozan I., Feld J., Sun J., Tannis L.L., Coltescu C. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology. 2005;128:1437–1444. doi: 10.1053/j.gastro.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 37.Randall G., Chen L., Panis M., Fischer A.K., Lindenbach B.D., Sun J. Silencing of USP18 potentiates the antiviral activity of interferon against hepatitis C virus infection. Gastroenterology. 2006;131:1584–1591. doi: 10.1053/j.gastro.2006.08.043. [DOI] [PubMed] [Google Scholar]
- 38.Chen L., Sun J., Meng L., Heathcote J., Edwards A.M., McGilvray I.D. ISG15, a ubiquitin-like interferon-stimulated gene, promotes hepatitis C virus production in vitro: implications for chronic infection and response to treatment. J Gen Virol. 2010;91:382–388. doi: 10.1099/vir.0.015388-0. [DOI] [PubMed] [Google Scholar]
- 39.Platanias L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 40.Hertzog P., Forster S., Samarajiwa S. Systems biology of interferon responses. J Interferon Cytokine Res. 2011;31:5–11. doi: 10.1089/jir.2010.0126. [DOI] [PubMed] [Google Scholar]
- 41.Ahlenstiel G., Titerence R.H., Koh C., Edlich B., Feld J.J., Rotman Y. Natural killer cells are polarized toward cytotoxicity in chronic hepatitis C in an interferon-α-dependent manner. Gastroenterology. 2010;138:325–335. doi: 10.1053/j.gastro.2009.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Golden-Mason L., Cox A.L., Randall J.A., Cheng L., Rosen H.R. Increased natural killer cell cytotoxicity and NKp30 expression protects against hepatitis C virus infection in high-risk individuals and inhibits replication in vitro. Hepatology. 2010;52:1581–1589. doi: 10.1002/hep.23896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoon J.C., Shiina M., Ahlenstiel G., Rehermann B. Natural killer cell function is intact after direct exposure to infectious hepatitis C virions. Hepatology. 2009;49:12–21. doi: 10.1002/hep.22624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang S., Saha B., Kodys K., Szabo G. IFN-γ production by human natural killer cells in response to HCV-infected hepatoma cells is dependent on accessory cells. J Hepatol. 2013;59:442–449. doi: 10.1016/j.jhep.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stegmann K.A., Bjorkstrom N.K., Ciesek S., Lunemann S., Jaroszewicz J., Wiegand J. Interferon α-stimulated natural killer cells from patients with acute hepatitis C virus (HCV) infection recognize HCV-infected and uninfected hepatoma cells via DNAX accessory molecule-1. J Infect Dis. 2012;205:1351–1362. doi: 10.1093/infdis/jis210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jia Y., Wei L., Jiang D., Wang J., Cong X., Fei R. Antiviral action of interferon-α against hepatitis C virus replicon and its modulation by interferon-γ and interleukin-8. J Gastroenterol Hepatol. 2007;22:1278–1285. doi: 10.1111/j.1440-1746.2007.04957.x. [DOI] [PubMed] [Google Scholar]
- 47.Thimme R., Oldach D., Chang K.M., Steiger C., Ray S.C., Chisari F.V. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194:1395–1406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flynn J.K., Dore G.J., Hellard M., Yeung B., Rawlinson W.D., White P.A. Early IL-10 predominant responses are associated with progression to chronic hepatitis C virus infection in injecting drug users. J Viral Hepat. 2011;18:549–561. doi: 10.1111/j.1365-2893.2010.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takahashi Y., Ando M., Nishikawa M., Hiraga N., Imamura M., Chayama K. Long-term elimination of hepatitis C virus from human hepatocyte chimeric mice following interferon-γ gene transfer. Hum Gene Ther Clin Dev. 2014;25:28–39. doi: 10.1089/humc.2013.066. [DOI] [PubMed] [Google Scholar]
- 50.Ahlenstiel G. The natural killer cell response to HCV infection. Immune Netw. 2013;13:168–176. doi: 10.4110/in.2013.13.5.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Makowska Z., Duong F.H., Trincucci G., Tough D.F., Heim M.H. Interferon-β and interferon-λ signaling is not affected by interferon-induced refractoriness to interferon-α in vivo. Hepatology. 2011;53:1154–1163. doi: 10.1002/hep.24189. [DOI] [PubMed] [Google Scholar]
- 52.Tseng C.T., Klimpel G.R. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J Exp Med. 2002;195:43–49. doi: 10.1084/jem.20011145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Larkin J., Bost A., Glass J.I., Tan S.L. Cytokine-activated natural killer cells exert direct killing of hepatoma cells harboring hepatitis C virus replicons. J Interferon Cytokine Res. 2006;26:854–865. doi: 10.1089/jir.2006.26.854. [DOI] [PubMed] [Google Scholar]
- 54.Yoon J.C., Lim J.B., Park J.H., Lee J.M. Cell-to-cell contact with hepatitis C virus-infected cells reduces functional capacity of natural killer cells. J Virol. 2011;85:12557–12569. doi: 10.1128/JVI.00838-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kotenko S.V., Gallagher G., Baurin V.V., Lewis-Antes A., Shen M., Shah N.K. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 56.Sheppard P., Kindsvogel W., Xu W., Henderson K., Schlutsmeyer S., Whitmore T.E. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 57.Gad H.H., Dellgren C., Hamming O.J., Vends S., Paludan S.R., Hartmann R. Interferon-λ is functionally an interferon but structurally related to the interleukin-10 family. J Biol Chem. 2009;284:20869–20875. doi: 10.1074/jbc.M109.002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoshimoto K., Kishida T., Nakano H., Matsui M., Shin-Ya M., Shimada T. Interleukin-28B acts synergistically with cisplatin to suppress the growth of head and neck squamous cell carcinoma. J Immunother. 2011;34:139–148. doi: 10.1097/CJI.0b013e318204ed70. [DOI] [PubMed] [Google Scholar]
- 59.Morrow M.P., Yan J., Pankhong P., Shedlock D.J., Lewis M.G., Talbott K. IL-28B/IFN-λ 3 drives granzyme B loading and significantly increases CTL killing activity in macaques. Mol Ther. 2010;18:1714–1723. doi: 10.1038/mt.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Donnelly R.P., Kotenko S.V. Interferon-λ: a new addition to an old family. J Interferon Cytokine Res. 2010;30:555–564. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Witte K., Gruetz G., Volk H.D., Looman A.C., Asadullah K., Sterry W. Despite IFN-λ receptor expression, blood immune cells, but not keratinocytes or melanocytes, have an impaired response to type III interferons: implications for therapeutic applications of these cytokines. Genes Immun. 2009;10:702–714. doi: 10.1038/gene.2009.72. [DOI] [PubMed] [Google Scholar]
- 62.Pagliaccetti N.E., Robek M.D. Interferon-λ in the immune response to hepatitis B virus and hepatitis C virus. J Interferon Cytokine Res. 2010;30:585–590. doi: 10.1089/jir.2010.0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang L., Jilg N., Shao R.X., Lin W., Fusco D.N., Zhao H. IL28B inhibits hepatitis C virus replication through the JAK-STAT pathway. J Hepatol. 2011;55:289–298. doi: 10.1016/j.jhep.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou Z., Hamming O.J., Ank N., Paludan S.R., Nielsen A.L., Hartmann R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J Virol. 2007;81:7749–7758. doi: 10.1128/JVI.02438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pagliaccetti N.E., Eduardo R., Kleinstein S.H., Mu X.J., Bandi P., Robek M.D. Interleukin-29 functions cooperatively with interferon to induce antiviral gene expression and inhibit hepatitis C virus replication. J Biol Chem. 2008;283:30079–30089. doi: 10.1074/jbc.M804296200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shindo H., Maekawa S., Komase K., Miura M., Kadokura M., Sueki R. IL-28B (IFN-λ3) and IFN-α synergistically inhibit HCV replication. J Viral Hepat. 2013;20:281–289. doi: 10.1111/j.1365-2893.2012.01649.x. [DOI] [PubMed] [Google Scholar]
- 67.Ge D., Fellay J., Thompson A.J., Simon J.S., Shianna K.V., Urban T.J. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 68.Tanaka Y., Nishida N., Sugiyama M., Kurosaki M., Matsuura K., Sakamoto N. Genome-wide association of IL28B with response to pegylated interferon-α and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 69.Suppiah V., Moldovan M., Ahlenstiel G., Berg T., Weltman M., Abate M.L. IL28B is associated with response to chronic hepatitis C interferon-α and ribavirin therapy. Nat Genet. 2009;41:1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 70.Thomas D.L., Thio C.L., Martin M.P., Qi Y., Ge D., O׳Huigin C. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prokunina-Olsson L., Muchmore B., Tang W., Pfeiffer R.M., Park H., Dickensheets H. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013;45:164–171. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]