

Abstract
In the present study, haloperidol (HP)-loaded solid lipid nanoparticles (SLNs) were prepared to enhance the uptake of HP to brain via intranasal (i.n.) delivery. SLNs were prepared by a modified emulsification–diffusion technique and evaluated for particle size, zeta potential, drug entrapment efficiency, in vitro drug release, and stability. All parameters were found to be in an acceptable range. In vitro drug release was found to be 94.16±4.78% after 24 h and was fitted to the Higuchi model with a very high correlation coefficient (R2=0.9941). Pharmacokinetics studies were performed on albino Wistar rats and the concentration of HP in brain and blood was measured by high performance liquid chromatography. The brain/blood ratio at 0.5 h for HP-SLNs i.n., HP sol. i.n. and HP sol. i.v. was 1.61, 0.17 and 0.031, respectively, indicating direct nose-to-brain transport, bypassing the blood–brain barrier. The maximum concentration (Cmax) in brain achieved from i.n. administration of HP-SLNs (329.17±20.89 ng/mL, Tmax 2 h) was significantly higher than that achieved after i.v. (76.95±7.62 ng/mL, Tmax 1 h), and i.n. (90.13±6.28 ng/mL, Tmax 2 h) administration of HP sol. The highest drug-targeting efficiency (2362.43%) and direct transport percentage (95.77%) was found with HP-SLNs as compared to the other formulations. Higher DTE (%) and DTP (%) suggest that HP-SLNs have better brain targeting efficiency as compared to other formulations.
KEY WORDS: Brain targeting, Haloperidol, Intranasal route, Pharmacokinetics, Solid lipid nanoparticles
Graphical abstract
The study indicates that SLNs represent a robust drug brain specific delivery system to overcome the blood–brain barrier via i.n. route. Ultimately, the current work may provide a successful concept for clinical application of HP via i.n. route in the treatment of psychiatric conditions like schizophrenia in the future.
1. Introduction
Nanotechnology has several applications in the ‘real world’1. It encompasses the production and application of physical, chemical and biological systems at submicron level as well as the integration of resulting nanostructures into larger systems2, 3.
Over the past two decades, there has been a marked improvement in our understanding of the underlying etiology and treatment of central nervous system (CNS) disorders4. However, many of the drugs used to treat these disorders lack an effective means for crossing the blood–brain barrier (BBB)5. The BBB presents a great obstacle to the transport of exogenous substances into the brain. Thus, various approaches like BBB disruption (osmotic and biochemical), drug manipulation (prodrug, lipophilic analogs, chemical drug delivery, carrier mediated drug delivery, and receptor/vector-mediated drug delivery) and alteration in the route of administration, including intracerebroventricular, intrathecal, and olfactory pathways (intranasal route) are used for the targeting of drugs to the brain6.
In the present scenario, the intranasal route to bypass the BBB is explored, as this route provides a novel, practical, simple and non-invasive approach to bypass the blood brain barrier and reduces the systemic exposure and thus systemic adverse effects7. Drug after intranasal administration reaches the olfactory epithelium region of the nasal mucosa that acts as a gateway for substances entering the CNS due to the neural connection between the nasal mucosa and the brain8.
Haloperidol (HP) is a dopamine inverse agonist of the typical antipsychotic class of medications that chemically belongs to butyrophenone group. It occurs as a white crystalline powder and is chemically known as 4-(4-chlorophenyl)-1-[4-(4-fluorophenyl)-4-oxobutyl]-4-piperidinol] with molecular weight of 375.86 g/mol and log P=3.36. Its mechanism of action is mediated by blockade of D2 dopamine receptors in brain9. It is used to treat certain psychiatric conditions including schizophrenia, manic states, medication-induced psychosis and neurological disorders with hyperkinesias10. It is also used to treat extreme behavior problems in children and to ease the symptoms of Tourette׳s syndrome11. After oral drug delivery, the drug undergoes first-pass metabolism followed by systemic distribution, resulting in only a small portion being able to reach the brain through the blood12, 13. The plasma–protein binding of HP is about 90%, thereby further affecting oral bioavailability12. Specific clinical complications associated with a high systemic concentration include respiratory disturbance (bronchospasm and increased depth of respiration), dermatological reactions (maculopapular and acneiform skin reactions), nausea, vomiting and musculoskeletal disorder14, 15, 16.
Therefore, a drug delivery system is required which not only provides rapid and targeted delivery to brain but also reduces systemic exposure. Attempts have been made to bypass the BBB, and it has been demonstrated in earlier studies that the intranasal route bypasses the BBB17.
Solid lipid nanoparticles (SLNs) were introduced in 1991 as submicron colloidal carriers (50–1000 nm). They are used for both hydrophilic and lipophilic drug(s) which are trapped in biocompatible lipid core made up of a lipid or combination of lipids like compritol 888 ATO, precirol ATO 5, glyceryl monostearate, palmitic acid, stearic acid, as well as others, and stabilized by surfactant present at the outer shell18. SLNs have the several advantages including targeted drug delivery and controlled release delivery, and increase bioavailability so as to reduce the dose and adverse effects19. SLNs with polymeric nanoparticles offer lower toxicity. They have good tolerability and biodegradability, lack of acute and chronic toxicity of the carrier, and scalability20, 21.
SLNs offer an improvement to traditional nose-to-brain drug delivery since they are able to protect the encapsulated drug from biological and/or chemical degradation and may also increase nasal retention time due to an occlusive effect, good application properties, and adhesion of the SLNs to mucous membranes17. The objectives of the current study were to evaluate HP-SLNs prepared by the modified solvent emulsification–diffusion technique, study the stability of the optimized formulation, and the pharmacokinetics of optimized HP-SLNs after intranasal administration.
2. Materials and methods
2.1. Materials
Haloperidol (HP) was received as a gift from Vamsi Labs Ltd. (Solapur, Maharashtra, India). Compritol ATO 888, glyceryl monostearate (GMS) and Precirol ATO 5 were obtained as a gift from Gattefosse (Witten, Germany). Stearic acid, palmitic acid, acetonitrile (ACN), triethylamine (TEA), o-phosphoric acid (o-PA) and Tween 80 along with all the other chemicals were purchased from Sigma-Aldrich (New Delhi, India). ACN, TEA, o-PA were high-performance liquid chromatography (HPLC)-grade while all other solvents and chemicals used were of analytical grade. Water was distilled and filtered before use through a 0.22 µm membrane filter.
2.2. Methods
2.2.1. Excipients selection
Solubility of drug in melted lipid is one of the most important factors that determine the encapsulation efficiency of a drug in lipid. However, equilibrium solubility studies cannot be carried out in this case. Hence, we used a modified method to identify the solid lipid having best solubilization potential for drug22. GMS, compritol ATO 888, precirol ATO 5, stearic acid and palmitic acid were screened for their potential to solubilize HP.
HP (20 mg) was placed in a screw-cap vial. The solid lipids were separately heated above their melting point. These lipid melts were gradually added in portions to the vial containing HP with continuous stirring using a vortex mixer and maintaining the same temperature (above the melting point of lipid). The end point was the formation of a clear, pale yellow solution of molten lipid. The amount of molten lipid required to solubilize the HP was noted visually. The experiment was performed in triplicate. The compatibility between the lipids and drug was identified by Fourier transform-infrared spectrophotometer (Alpha model Bruker ATR-FTIR spectrophotometer). IR spectra of drug, lipid and a physical mixture of drug and lipid (1:1) were scanned from 4000 cm−1 to 400 cm−1 and recorded.
2.2.2. Preparation of SLNs
In a preliminary laboratory study, various factors like ratio of drug to lipid (1:2), surfactant concentration (Tween 80, 1.5% w/v), ratio of chloroform to ethanol (1:1, 2.5% v/v) (as the solvent for drug and lipid), homogenization time (30 min), stirring time (2 h), stirring speed (2500 rpm), and sonication time (5 min) were fixed and their effect on particle size and entrapment efficiency was determined. Factors like ratio of drug to lipid, surfactant concentration, stirring speed and stirring time were further optimized in this study. All of the experiments were performed in triplicate and the averages were considered as the response. Table 1 shows the composition of various batches.
Table 1.
Composition of various batches of HP-SLNs.
| Formulation | Variable |
||||
|---|---|---|---|---|---|
| Drug (mg) | Lipid (mg) | Surfactant (% w/v) | Stirring speed (rpm) | Stirring time (h) | |
| OH1 | 50 | 100 | 1.5 | 3000 | 2 |
| OH2 | 43.75 | 87.5 | 1.5 | 3000 | 2 |
| OH3 | 43.75 | 87.5 | 1.625 | 3000 | 2 |
| OH4 | 50 | 100 | 1.625 | 3000 | 2 |
| OPH | 50 | 100 | 1.625 | 3000 | 2.5 |
Drug-loaded SLNs were prepared by the modified solvent emulsification–diffusion technique19, 23, 24, 25. Accurately weighed lipid (87.5–100 mg) was dissolved in a 2.5 mL mixture of ethanol and chloroform (1:1) as the internal oil phase. Drug (43.75–50 mg, ratio of drug to lipid 1:2) was dispersed in the above solution. This organic phase was then added drop by drop into a homogenizer tube containing 22.5 mL of an aqueous solution of Tween 80 (1.5–1.625% w/v) as the external aqueous phase and homogenized for 30 min at 3000 rpm (Remi Instruments Pvt. Ltd., India) to form a primary emulsion (o/w). The above primary emulsion was poured into 75 mL of ice-cold water (2–3 °C) containing surfactant (1.5–1.625% w/v) and stirred to extract the organic solvent into the continuous phase and for proper solidification of SLNs. The stirring was continued for 2–2.5 h at 3000 rpm to disperse the SLNs. The SLN dispersion was sonicated for 5 min (1 cycle, 100% amplitude, Bandelin sonoplus, Germany) to produce SLN dispersions of uniform size. The dispersion was then centrifuged at 18,000 rpm for 20 min (Remi Instruments Pvt., Ltd., India) to separate the solid lipid material containing the drug and washed with deionized water several time to ensure the complete removal of organic solvent. This was then redispersed in 1.5–1.625% (w/v) of an aqueous surfactant mixture of Tween 80 and sonicated for 5 min to obtain the SLNs. The SLN dispersions were lyophilized in the presence of 5% (w/v) mannitol as a cryoprotectant.
2.2.3. Particle size, zeta potential and morphology study
Average particle size, polydispersity index (PDI) and zeta potential were measured by photon correlation spectroscopy (PCS; Zetasizer, HAS 3000; Malvern Instruments, Malvern, UK). The analysis was performed at 25 °C with an angle of detection 90°26.
Surface morphology of optimized SLNs (OPH) was determined by using transmission electron microscopy (TEM, Philips CM 10, Holland). To perform the TEM observation, SLN dispersions (approx. 10 µL) were dropped on a 300 mesh copper grid coated with carbon film and allowed to sit for 10 min until air-dried. After complete drying the sample was stained with 2% w/v phosphotungstic acid solution with several replications and dried at room temperature. Digital micrograph and Soft Imaging Viewer software were used to perform the image capture and analysis.
2.2.4. Drug loading and drug entrapment efficiency determination
A fixed quantity of HP-SLN dispersions (10 mL) was fractionated (Remi Instruments, Pvt. Ltd., India) at 18,000 rpm for 20 min at 20 °C23. The supernatant fraction was analyzed spectrophotometrically at λmax of 247.5 nm (Shimadzu 1800, Japan) for determination of unencapsulated drug27, 28. The drug loading (%) and drug entrapment efficiency (%) were calculated by using the following equations21, 23:
| (1) |
| (2) |
where Wt is the total weight of drug used, Ws is the weight of drug in the supernatant after centrifugation and WL is the weight of the lipid used in preparing the SLNs.
2.2.5. X-ray diffractometric analysis
X-ray powder diffraction patterns of drug, lipid and optimized HP-SLNs were measured by X-ray diffractometer (PANanalytical, X’Pert Pro model, Germany) equipped with an X-ray generator. Analysis was performed on the samples using Cu Kα radiation (45 kV, 40 mA) and scanned from 5° to 90° (2θ). The scanning rate is 2°/min using an automatic divergence slit assembly and a proportional detector. Samples were scanned at 25 °C 29.
2.2.6. Differential scanning calorimetry
The thermograms of drug, lipid and optimized HP-SLNs were recorded with a DSC (Pyris 6 DSC Perkin Elmer, CT, USA) under an inert atmosphere which was maintained by purging with nitrogen. Sample (5 mg) was loaded into an aluminum pan and sealed tightly. An empty aluminum pan was used as a reference. Samples were heated at a scanning rate of 10 °C/min over a temperature range between 40–230 °C and the thermograms were recorded30.
2.2.7. In vitro drug release and release kinetics study
Drug release from HP-SLNs was determined using a dialysis bag diffusion technique using a dialysis membrane (Himedia, molecular weight cut off 12,000–14,000 D)31. An accurately weighed amount of HP-SLN containing the drug equivalent to 10 mg was transferred to a dialysis bag and sealed. The sealed bag was then suspended in a beaker containing 100 mL of phosphate buffer pH 7.4 and stirred at a constant speed at 37±0.5 °C. Aliquots were withdrawn at predetermined time intervals up to 24 h from receiver compartment (beaker) and replaced with an equal volume of fresh medium to maintain a ‘sink’ condition. The samples were analyzed spectrophotometrically at λmax of 247.5 nm (Shimadzu 1800, Japan). In vitro release data was fitted to zero order, first order and Higuchi release models32, 33. The percent cumulative amount of drug release versus time was plotted for zero kinetics, log percent drug remaining versus time was plotted for first order kinetics and percent cumulative amount of drug release versus square root of time was plotted for the Higuchi release model and the correlation coefficient was determined for each model. Initial equations for various models are given below:
| (3) |
| (4) |
| (5) |
where X is the amount of drug released, K is the release rate constant, and t is time. To construct the plot for zero order and the Higuchi model, percent cumulative drug release determined, and for the first order model, log percent drug remaining was determined.
2.2.8. Stability study
The stability study was carried out to determine the effect of the presence of formulation additives on the stability of drug and also to determine the physical stability of the prepared formulation under conditions of storage temperature and relative humidity34.
The optimized HP-SLNs were subjected to stability studies and the studies were performed in triplicate. The storage conditions used for stability testing were 4±2 °C (refrigerator), 25±2 °C/60±5% RH, and 40±2 °C/75±5% in a stability chamber (Hicon instruments, N. Delhi). The sample was withdrawn after a period of 0, 1, 3 and 6 months and the effect on particles size, PDI, zeta potential, entrapment efficiency and loading capacity was determined.
2.2.9. Pharmacokinetics study
A pharmacokinetic study was performed with male albino Wistar rats (adult/weighing 200–250 g). A protocol for animal studies was approved by Institutional animal ethical committee and project number was 03.
The animals were kept under standard laboratory conditions, temperature of 22±3 °C and relative humidity of 30%–70%. The animals were housed in polypropylene cages, 6 animals per cage with free access to standard laboratory diet and water ad libitum. Rats were divided into three groups: group A, positive control for intravenous (i.v.) drug administration (HP sol.); group B, positive control for intranasal (i.n.) drug administration (HP sol.); and group C, intranasal (i.n.) formulation administration (HP-SLNs). Each group was further divided into 8 subgroups on time basis as 0.167, 0.5, 1, 2, 4, 6, 8 and 24 h, with each group receiving drug at time 0 and then analyzed at the indicated time. Each subgroup contained 6 animals. The dose for rats was calculated based on the body weight and surface area ratio of the rat35. Surface area ratio calculated for a 200 g rat relevant to a 70 kg human is 56. The equation is
| (6) |
where x is rat dose per 200 g and the dose of HP for human is 10 mg. x=0.179 mg for 200 g of rat on this basis; the dose required for rat of 1 kg is 0.893 mg.
Drug solution (positive control), containing 0.179 mg (for 200 g of rat) of HP (equivalent to 0.89 mg/kg body weight), was injected through the tail vein (10 µL) of one group of Wistar rats. Similarly, drug solution and drug formulation (HP-SLNs) containing 0.179 mg of HP were administered in each nostril in the other two groups with the help of micropipette (10–100 µL) with 0.1 mm internal diameter at the delivery site. The rats were anaesthetized prior to nasal administration by pentobarbital sodium (35–50 mg/kg, i.p.) and held firmly from the back in a slanted position during nasal administration.
The rats were killed humanely by an overdose of pentobarbital sodium at the designated time intervals (0.167, 0.5, 1, 2, 4, 6, 8 and 24 h) and the blood was collected using cardiac puncture in EDTA-coated Eppendorf tubes. The blood was fractionated at 4000 rpm for 20 min and aliquots of the supernatant separated and stored at −21 °C until drug analysis was carried out using HPLC36.
At the same interval of blood collection, the rats were sacrificed to remove the brain. Brain was rinsed twice with normal saline, made free from adhering tissue/fluid and weighed. Cold normal saline solution was added (brain weight: normal saline, 1:5) to the brain and homogenized. The homogenate was centrifuged at 4000 rpm for 20 min with the temperature of 4 °C, and aliquots of the supernatant were separated and stored at –21 °C until drug analysis was carried out using HPLC37, 38.
Chromatographic separation was achieved with a Cosmosil C18 column (250 mm×4.6 mm, particle size 5 µm). The mobile phase consisted of 100 mmol/L potassium dihydrogen phosphate–acetonitrile–TEA (10:90:0.1, v/v/v) and the pH was adjusted with o-phosphoric acid to 3.5. The mobile phase was sonicated for 15 min and filtered through a 0.22 μm membrane filter before use. The flow rate of the mobile phase was maintained at 2 mL/min and eluents were monitored at 230 nm. Twenty μL of sample was injected using an HPLC injector. All determinations were performed at ambient temperature for a run time of 5 min.
The extraction of HP from plasma and brain samples was carried out using the liquid–liquid extraction (LLE) technique. Plasma sample and homogenized brain tissue (0.5 mL) were mixed with 100 µL loratidine (100 ng/mL) as an internal standard39, 40. The mixture was shaken with 2 mL isopropyl alcohol for 2 min on a vortex mixer and then fractionated at 4000 rpm for 10 min until a clear organic layer was separated. The organic layer was evaporated to dryness under nitrogen gas at room temperature. Dried sample was then reconstituted with 0.3 mL mobile phase and evaluated by HPLC for the presence of HP.
Plasma concentration–time profiles of HP after i.n. and i.v. delivery were evaluated with pharmacokinetic software (PK Functions for Microsoft Excel, Pharsight Corporation, Mountain View, CA, USA). Various pharmacokinetic parameters such as Cmax, Tmax, AUC0–∞, AUMC0–∞, elimination rate constant and MRT were calculated. The percent brain targeting efficiency (DTE%) and nose-to-brain direct transport percentage (DTP%) were calculated with the help of the following formulae41.
| (7) |
| (8) |
whereF=(AUC0–24,brain,i.v./AUC0–24,blood,i.v.)×AUC0–24,blood,i.n., AUC0–24, brain, i.n. is the area under the curve of brain following i.n. administration, AUC0–24, brain, i.v. is the area under the curve of brain following i.v. administration, AUC0–24 blood, i.v. is the area under the curve of blood following i.v. administration, and AUC0–24 blood, i.n. is the area under the curve of blood following i.n. administration.
2.2.10. Statistical analysis
All data are reported as mean±standard deviation (SD). Differences between the groups were tested using Student׳s t-test and differences among more than two groups were compared using ANOVA (analysis of variance). A P-value <0.05 was considered statistically significant.
3. Results and discussion
3.1. Excipients selection
Excipients used for formulation development should be pharmaceutically acceptable, non-irritating, and non-sensitizing in nature. They should be generally regarded as safe. For the SLNs development, selection of a suitable lipid and surfactant is important. Solubility of drug in the lipid is a determinant of encapsulation efficiency. It is expected that high lipid solubility will result in high encapsulation efficiency42.
Solubility studies indicated that in comparison of GMS, Compritol 888 ATO, precirol ATO 5, stearic acid and palmitic acid, GMS effectively solubilised the HP (Table 2). The solubilising potential, coupled with already reported biocompatibility and acceptability of GMS for nose to brain and parenteral route delivery favours its selection for the present study23.
Table 2.
Solubility of drug in various lipids.
| Lipid name | Melting point of lipid (°C) | Amount of lipid requireda |
|---|---|---|
| Glyceryl monostearate | 59 | 47.66±0.95 |
| Compritol 888 ATO | 70 | 49.51±0.83 |
| Precirol ATO 5 | 56 | 55.34±2.24 |
| Stearic acid | 69 | 82.89±2.10 |
| Palmitic acid | 63 | 142.37±2.06 |
Data are expressed as mean±SD, n=3.
The FTIR spectra over the range of 400–4000 cm−1 for HP, GMS and physical mixture of HP and GMS are shown in Fig. 1. Important peaks at 3120 cm−1 appeared the characteristic peak of OH stretch, at 2951 cm−1 aromatic CH group stretch, at 2911 cm−1 aliphatic CH2 stretch, at 1676 cm−1 and 1587 cm−1 corresponded to C O carbonyl stretching and a substituted aromatic ring, respectively, at 1138 cm−1 CH deformation of F substituted aromatic ring, and at 995 cm−1 is due to —Cl substituted aromatic ring43. These characteristic peaks of HP were also observed in the FTIR spectrum of physical mixture of HP and GMS without any distinct shifts. This fact verified that no chemical interaction between the drug and the polymer had occurred.
Figure 1.

IR spectra of HP (a), GMS (b) and physical mixture of HP and GMS (c).
On the basis of drug lipid solubility and drug lipid compatibility study, GMS was selected as the lipid. The criterion for surfactant selection was its hydrophilic–lipophilic balance (HLB) value that can emulsify lipid and form the stable microemulsion in an acceptable concentration. Tween 80 with HLB of 15 was selected as the surfactant.
3.2. Evaluation of HP-SLNs
3.2.1. Particle size, zeta potential and morphology study
The concentration of the surfactant (Tween 80) was optimized in order to obtain a smaller size of SLNs with maximum percent drug entrapment. The concentration of the surfactant was optimized to 1.625% w/v.
The mean particle size of different batches of SLNs ranges from 115.1±2.78 to 156.77±3.95 nm. The particle size of optimized batch (OPH) batch was appreciably lower (115.1±2.78 nm) compared to other batches. This is due to the addition of surfactant to solid lipid nanoparticles that causes the interfacial film to condense and stabilize44. All batches had particles in the nano range which is well evident from the values of PDI. PDI is essentially the ratio of standard deviation to the mean particle size. The value of PDI was acceptable for all batches (Table 3).
Table 3.
Observed responses of various batches.
| Formulation code | Response |
||||
|---|---|---|---|---|---|
| Particle size (nm)a | PDI | Zeta potential (mV) | Entrapment efficiency (%)a | Drug loading (%)a | |
| OH1 | 146.34±3.59 | 0.479 | −13.68 | 69.65±0.89 | 25.82±2.45 |
| OH2 | 156.77±3.95 | 0.453 | −14.56 | 68.35±2.67 | 23.47±0.57 |
| OH3 | 150.25±5.76 | 0.437 | −16.12 | 70.43±2.56 | 23.97±0.95 |
| OH4 | 140.49±2.97 | 0.429 | −15.98 | 70.65±1.78 | 26.01±1.23 |
| OPH | 115.10±2.78 | 0.409 | −16.70 | 71.56±1.56 | 26.35±0.56 |
Data are expressed as mean±SD, n=3.
On increasing the stirring time and stirring speed from 2 to 2.5 h and from 2000 to 3000 rpm (data is not shown), a decrease in the particle size was observed for all the batches; an increase in the percent of drug entrapment was also observed. On a further increase a marginal increase in particle size was observed. Hence, stirring time and stirring speed were optimized to 2.5 h and 3000 rpm, respectively. Similar findings have also been reported by Singh et al.23.
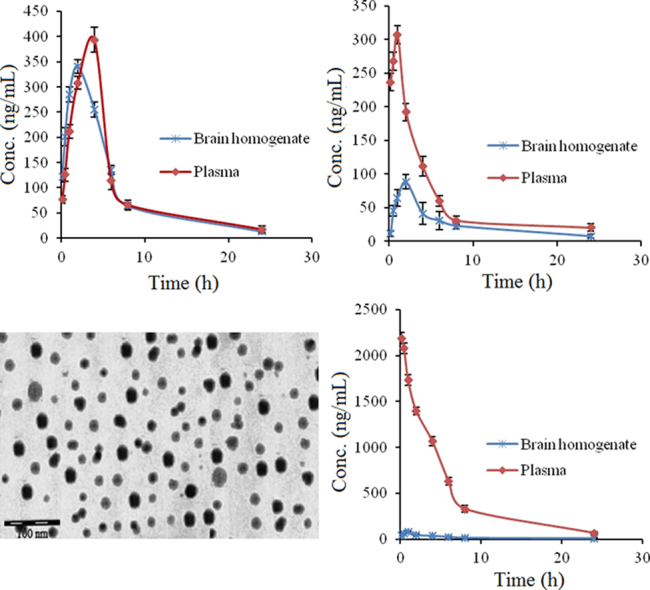
Percent drug loading and percent drug entrapment efficiency of the optimized batches were found to be 26.35±0.56 and 71.56±0.1.56, respectively (Table 3). For the optimized batch, a dense roughly spherical pattern was observed by transmission electron microscopy (Fig. 2). The surface carried negative charges with a zeta potential of −16.7 mV when the drug-to-lipid ratio, surfactant concentration, stirring time, and stirring speed were 1:2, 1.625% (w/v), 2.5 h and 3000 rpm, respectively.
Figure 2.

TEM image of optimized HP-SLNs.
3.2.2. X-ray diffractometric analysis
The X-ray defraction (XRD) pattern of HP showed sharp peaks at 2θ-scattered angles at 15.5° and 20.5°, indicating the highly crystalline nature of the drug. The XRD pattern of GMS showed a peak at about 20°, indicating the crystalline nature of the lipid. The principal peak of HP is absent in SLN XRD spectra. Furthermore, the principal peak of lipid did not shift but had a reduced intensity as compared to free lipid. This may be attributed to the incorporation of HP between the parts of the crystal lattice of the lipid, leading to a change in the crystallinity of the HP-SLNs (Fig. 3).
Figure 3.
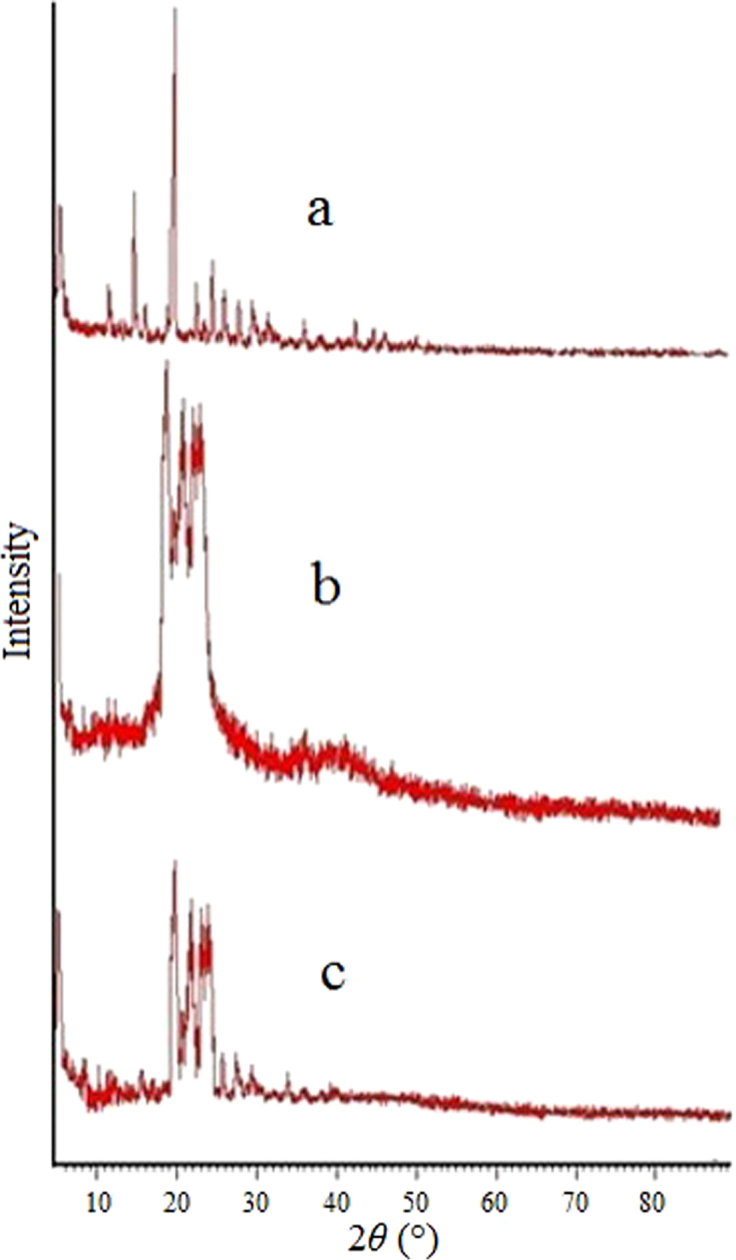
XRD of HP (a), GMS (b) and optimized HP-SLNs (c).
3.2.3. Differential scanning calorimetry
The DSC thermogram of HP showed a melting peak of 151.895 °C while GMS and optimized HP-SLNs showed at 60.376 °C and 55.856 °C, respectively (Fig. 4). The thermogram of SLNs did not show the melting peak of crystalline haloperidol around 151.895 °C, indicating that the drug was completely solubilized inside the lipid matrix and was in the amorphous form. The melting peak of GMS in SLNs was decreased by 4.52 °C, suggesting that GMS in SLNs might be in crystalline form. The decrease in the melting point of SLNs loaded with haloperidol was due to their small particles size that lead to high surface energy, creating an energetically suboptimal state as described by the Thomson equation45.
Figure 4.

DSC thermograms of haloperidol (a), GMS (b) and optimized HP-SLNs (c).
3.2.4. In vitro release and release kinetics studies
For the optimized batch, release from 10.98±2.96% to 94.16±4.78% was obtained as the drug slowly diffused through the lipid core (Fig. 5a). The drug release data was fitted into zero-order, first-order and Higuchi kinetics models22. For the optimized batch, the highest value of the correlation coefficient (R2=0.9941) was observed for Higuchi׳s model, followed by the first-order (R2=0.9894) and zero order (R2=0.8842) models, as shown in Fig. 5.
Figure 5.

In vitro drug release from optimized HP-SLNs (a), zero-order release model (b), first-order release model (c), and Higuchi release model (d).
3.2.5. Stability studies
No significant change was observed in particle size when they were stored at 4±2 °C (refrigerator) and 25±2 °C/60±5% RH, but the size of particles increased when the they were stored at 40±2 °C/75±5% RH due to aggregation (P<0.05, Table 4).
Table 4.
Characteristics of HP-SLNs after a 6-month stability studies at different conditions.
| Temp (°C)/RH(%) | Time (month) | Characteristics parameter |
||||
|---|---|---|---|---|---|---|
| Particle size (nm)a | PDI | Zeta potential (mV)a | Entrapment efficiency (%)a | Drug loading (%)a | ||
| 4±2 | 0 | 112.34±4.73 | 0.424 | −18.6±1.7 | 72.53±2.64 | 26.61±0.85 |
| 1 | 117.65±2.54 | 0.506 | −17.6±1.5 | 71.54±1.20 | 26.35±0.42 | |
| 3 | 115.94±5.82 | 0.332 | −17.3±1.2 | 71.23±1.94 | 26.27±0.63 | |
| 6 | 125.67±2.34 | 0.463 | −16.1±2.6 | 71.92±2.45 | 26.45±0.86 | |
| 25±2/60±5 | 0 | 112.34±4.73 | 0.424 | −18.6±1.7 | 72.53±1.64 | 26.61±0.57 |
| 1 | 112.21±1.04 | 0.556 | −17.3±1.2 | 70.65±1.24 | 26.10±0.53 | |
| 3 | 119.76±5.57 | 0.575 | −16.6±1.3 | 69.78±1.98 | 25.87±0.59 | |
| 6 | 135.34±5.86 | 0.637 | −15.1±2.6 | 71.87±2.34 | 26.44±0.71 | |
| 40±2/75±5 | 0 | 112.34±4.73 | 0.424 | −18.6±1.7 | 72.53±2.64 | 26.61±0.75 |
| 1 | 120.45±2.81 | 0.554 | −14.2±1.9 | 70.39±2.85 | 26.04±0.91 | |
| 3 | 355.35±3.98 | 0.594 | −11.6±2.3 | 69.53±1.27 | 25.80±0.65 | |
| 6 | 1345.93±4.60 | 0.642 | −8.34±3.9 | 70.36±2.54 | 26.03±0.89 | |
PS, Particle size; PDI, polydispersity index.
Data are expressed as mean±SD, n=3; P<0.05 (The measurements obtained at each time point (1, 3 and 6 months) at specific storage condition were compared with measurements obtained at zero month at that storage condition. Comparison of different parameters was also made between different temperatures.)
Zeta potential plays an important role in physical stability. There was no significant change between 4±2 °C (refrigerator) and 25±2 °C/60±5% RH, but zeta potential dropped at 40±2 °C/75±5% RH (P<0.05). This might be due to dissolution of the coating of lipid which leads to aggregation of particles.
3.2.6. Pharmacokinetics studies
The HP concentrations in brain following the i.n. administration of HP-SLNs were found to be significantly higher at all the time points as compared to both HP sol. i.n. and HP sol. i.v. (P<0.05). The haloperidol concentration in plasma following the i.n. of HP-SLNs was found to be significantly lower at all the time points compared to HP sol. i.v. administration (P<0.05, Table 5). Fig. 6 shows the drug concentration versus time profile of HP in blood and brain after (a)HP- SLNs i.n., (b) HP sol. i.n., and (c) HP sol. i.v. Various pharmacokinetic parameters of HP were calculated by using PK Functions for Microsoft Excel (Pharsight Corporation, Mountain View, CA, US) as shown in Table 5. The lower value of Tmax for brain (2 h) as compared to blood (4 h) may be attributed to the preferential nose to brain transport following i.n. administration. The values of Cmax and AUC of brain for HP sol. i.n., HP sol. i.v. and HP- SLNs i.n. were compared, the value of Cmax (329.17±20.89 ng/mL) and AUC0–∞ (2389.17±78.82 ng·h/mL) of HP-SLNs were found to be significantly higher than HP sol. (i.n. and i.v.) because of the direct transport of drug through olfactory route by bypassing the BBB.
Table 5.
Pharmacokinetic parameters of HP after HP-SLNs i.n., HP sol. i.n. and HP sol. i.v. administration to rats in brain and plasma.
| Parameter (unit) | SLNs i.n. |
Drug solution i.n. |
Drug solution i.v. |
|||
|---|---|---|---|---|---|---|
| Brain | Plasma | Brain | Plasma | Brain | Plasma | |
| Cmax (ng/mL) | 329.17±20.89 | 393.5±24.63 | 90.13±6.28 | 306.96±13.47 | 76.95±7.62 | 2190±60.67 |
| Tmax (h) | 2 | 4 | 2 | 1 | 1 | 0.167 |
| AUC0–24 h (ng·h/mL) | 2172.33±60.41 | 2433.05±18.54 | 623.16±8.51 | 1460.71±15.67 | 433.65±15.46 | 11,464.59±150.45 |
| AUC0–∞ (ng·h/mL) | 2389.17±78.82 | 2612.31±40.67 | 683.15±30.17 | 1681.82±32.83 | 500.82±12.78 | 12,017.5±180.87 |
| AUMC0–24 h (ng·h2/mL) | 12,172.67±56.59 | 13,725.21±135.43 | 2881.23±27.08 | 8696.86±124.78 | 2881.31±30.76 | 57,642.09±580.45 |
| AUMC0–∞ (ng·h2/mL) | 15,665.20±25.59 | 19,864.67±256.43 | 7079.16±35.53 | 14,650.31±145.75 | 5199.46±120.67 | 70,374.14±960.87 |
| Ke (h−1) | 0.079±0.0065 | 0.097±0.003 | 0.077±0.005 | 0.11±0.003 | 0.095±0.003 | 0.15±0.007 |
| MRT (h) | 12.60±0.99 | 7.60±0.32 | 9.17±0.45 | 8.9±0.57 | 10.38±0.65 | 5.92±0.57 |
All data are expressed as mean±SD, n=6; P<0.05 (Pharmacokinetics parameters after various route of administration were compared with each other).
Figure 6.

Drug concentration versus time profile of HP in blood and brain after administration of HP-SLNs i.n. (a), HP sol. i.n. (b) and HP sol. i.v. (c).
The value of AUC0–∞ in brain for HP-SLNs i.n. was found to be nearly 4.77 times higher than that of HP sol. i.v., whereas 3.49 times higher than HP sol. i.n. This result reveals that drug uptake into the brain from the nasal mucosa mainly occurs via two different pathways. One is the systemic pathway by which some of the drug is absorbed into the systemic circulation and subsequently reaches the brain by crossing the BBB. The other is the olfactory pathway by which the drug partly travels from the nasal cavity to CSF and/or brain tissue46, 47. The DTE% and DTP% represent the percentage of drug directly transported to the brain via the olfactory pathway. The higher DTE (2362.43%) and DTP (95.77%) were found with HP- SLNs (Table 6). Higher DTE (%) and DTP (%) suggest that HP-SLNs have better brain targeting efficiency. Similar types of results have also been reported by Zhang et al.48.
Table 6.
DTE (%) and DTP (%) of optimized HP-SLNs i.n. and HP sol. i.n.
| Formulation and route of administration | DTE (%) | DTP (%) |
|---|---|---|
| HP-SLNs | 2362.43 | 95.77 |
| HP sol. | 1128.61 | 91.14 |
4. Conclusions
The present research work proposed a lipid nanoparticulate drug delivery system (SLNs) for intranasal delivery of HP. SLNs were prepared by the modified emulsification–diffusion technique and evaluated for particle size, particle size distribution (PDI), zeta potential, entrapment efficiency, in vitro release and stability studies. All measurements were found to be in an acceptable range. In vitro drug release was found to be 94.14±4.78% over 24 h, indicating a controlled and sustained release profile of HP-SLNs. Pharmacokinetics studies were performed on Wistar rats, in which the DTE (%) and DTP (%) are more indicative of direct nose to brain transport bypassing the BBB thereby representing the superiority of HP-SLNs over HP sol. i.n. and i.v. It was concluded that HP-SLNs could be an effective drug delivery system for the treatment of psychiatric conditions like schizophrenia via nose to brain route. However, clinical data is still needed to evaluate the risk/benefit ratio.
Acknowledgments
The authors express deep appreciation and thanks to the management of ITS paramedical college (pharmacy), Muradnagar, Ghaziabad, UP, India for their valuable cooperation in the present research work. The authors gratefully acknowledge the Vamsi labs ltd., Solapur, Maharashtra, India for providing the gift sample of HP. Further, they acknowledge the Advanced Research Instrumentation Facility (ARIF, JNU, New Delhi, India) for XRD facilities.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.

References
- 1.Kingsley J.D., Dou H.Y., Morehead J., Rabinow B., Gendelman H.E., Destache C.J. Nanotechnology: a focus on nanoparticles as a drug delivery system. J Neuroimmune Pharmacol. 2006;1:340–350. doi: 10.1007/s11481-006-9032-4. [DOI] [PubMed] [Google Scholar]
- 2.Falzarano M.S., Passarelli C., Bassi E., Fabris M., Perrone D., Sabatelli P., et al. Biodistribution and molecular studies on orally administered nanoparticle-AON complexes encapsulated with alginate aiming at inducing dystrophin rescue in mdx mice. BioMed Res Int. 2013;2013:527418. doi: 10.1155/2013/527418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sahoo S.K., Labhasetwar V. In: Nanoparticle technology for drug delivery. Gupta R.B., Kompella U.B., editors. Taylor & Francis, NY, USA; New York: 2006. Nanoparticles interface: an important determinant in nanoparticle-mediated drug/gene delivery; pp. 139–154. [Google Scholar]
- 4.Misra A., Ganesh S., Shahiwala A., Shah S.P. Drug delivery to the central nervous system: a review. J Pharm Pharm Sci. 2003;6:252–273. [PubMed] [Google Scholar]
- 5.Alam M.I., Baboota S., Ahuja A., Ali M., Ali J., Sahni J.K., et al. Pharmacoscintigraphic evaluation of potential of lipid nanocarriers for nose-to-brain delivery of antidepressant drug. Int J Pharm. 2014;470:99–106. doi: 10.1016/j.ijpharm.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 6.Illum L. Nasal drug delivery: new developments and strategies. Drug Discov Today. 2002;7:1184–1189. doi: 10.1016/s1359-6446(02)02529-1. [DOI] [PubMed] [Google Scholar]
- 7.Krishnamoorthy R., Mitra A.K. Prodrugs for nasal drug delivery. Adv Drug Deliv Rev. 1998;29:135–146. doi: 10.1016/s0169-409x(97)00065-3. [DOI] [PubMed] [Google Scholar]
- 8.Haque S., Md S., Sahni J.K., Ali J., Baboota S. Development and evaluation of brain targeted intranasal alginate nanoparticles for treatment of depression. J Psychiatr Res. 2014;48:1–12. doi: 10.1016/j.jpsychires.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 9.Benvegnú D.M., Barcelos R.C., Boufleur N., Reckziegel P., Pase C.S., Ourique A.F., et al. Haloperidol-loaded polysorbate-coated polymeric nanocapsules increase its efficacy in the antipsychotic treatment in rats. Eur J Pharm Biopharm. 2011;77:332–336. doi: 10.1016/j.ejpb.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 10.Yasir M., Sara U.V.S. Development and validation of UV spectrophotometric method for the estimation of haloperidol. Br J Pharm Res. 2014;4:1407–1415. [Google Scholar]
- 11.Settle E.C., Ayd F.J. Haloperidol: a quarter century of experience. J Clin Psychiatry. 1983;44:440–448. [PubMed] [Google Scholar]
- 12.Forsman A.O. Individual variability in response to haloperidol. Proc Roy Soc Med. 1976;69 (Suppl 1):S9–S13. [PMC free article] [PubMed] [Google Scholar]
- 13.Vella-Brincat J., Macleod A.D. Haloperidol in palliative care. Palliat Med. 2004;18:195–201. doi: 10.1191/0269216304pm881oa. [DOI] [PubMed] [Google Scholar]
- 14.Chang W.H., Lam Y.W.F., Jann M.W., Chen H. Pharmacokinetics of haloperidol and reduced haloperidol in chinese schizophrenic patients after intravenous and oral administration of haloperidol. Psychopharmacology. 1992;106:517–522. doi: 10.1007/BF02244824. [DOI] [PubMed] [Google Scholar]
- 15.Budhian A., Siegel S.J., Winey K.I. Haloperidol-loaded PLGA nanoparticles: systematic study of particle size and drug content. Int J Pharm. 2007;336:367–375. doi: 10.1016/j.ijpharm.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 16.Hollister L.E. 6th ed. Prentice Hall; London: 1995. Basic and Clinical Pharmacology. [Google Scholar]
- 17.Kaur I.P., Bhandari R., Bhandari S., Kakkar V. Potential of solid lipid nanoparticles in brain targeting. J Control Release. 2008;127:97–109. doi: 10.1016/j.jconrel.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 18.Schwarz C., Mehnert W., Lucks J.S., Muller R.H. Solid lipid nanoparticles (SLN) for controlled drug delivery. I. production, characterization and sterilization. J Control Release. 1994;30:83–96. [Google Scholar]
- 19.Yasir M., Sara U.V.S. Preparation and optimization of haloperidol loaded solid lipid nanoparticles by box-behnken design. J Pharm Res. 2013;7:551–558. [Google Scholar]
- 20.Helgason T., Awad T.S., Kristbergsson K., McClements D.J., Weiss J. Effect of surfactant surface coverage on formation of solid lipid nanoparticles (SLN) J Colloid Interface Sci. 2009;334:75–81. doi: 10.1016/j.jcis.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 21.Kumar R., Yasir M., Saraf S.A., Gaur P.K., Kumar Y., Singh A.P. Glyceryl monostearate based nanoparticles of mefenamic acid: fabrication and in vitro characterization. Drug Invention Today. 2013;5:246–250. [Google Scholar]
- 22.Shah K.A., Date A.A., Joshi M.D., Patravale V.B. Solid lipid nanoparticles (SLN) of tretinoin: potential in topical delivery. Int J Pharm. 2007;345:163–171. doi: 10.1016/j.ijpharm.2007.05.061. [DOI] [PubMed] [Google Scholar]
- 23.Singh A.P., Saraf S.K., Saraf S.A. SLN approach for nose-to-brain delivery of alprazolam. Drug Deliv Transl Res. 2012;2:498–507. doi: 10.1007/s13346-012-0110-2. [DOI] [PubMed] [Google Scholar]
- 24.Serralheiro A., Alves G., Fortuna A., Falcão A. Intranasal administration of carbamazepine to mice: a direct delivery pathway for brain targeting. Eur J Pharm Sci. 2014;60:32–39. doi: 10.1016/j.ejps.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 25.Trotta M., Debernardi F., Caputo O. Preparation of solid lipid nanoparticles by a solvent emulsification-diffusion technique. Int J Pharm. 2003;257:153–160. doi: 10.1016/s0378-5173(03)00135-2. [DOI] [PubMed] [Google Scholar]
- 26.Jores K., Mehnert W., Drechsler M., Bunjes H., Johann C., Mäder K. Investigations on the structure of solid lipid nanoparticles (SLN) and oil-loaded solid lipid nanoparticles by photon correlation spectroscopy, field-flow fractionation and transmission electron microscopy. J Control Release. 2004;95:217–227. doi: 10.1016/j.jconrel.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 27.Venkateswarlu V., Manjunath K. Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J Control Release. 2004;95:627–638. doi: 10.1016/j.jconrel.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 28.Varshosaz J., Tabbakhian M., Mohammadi M.Y. Formulation and optimization of solid lipid nanoparticles of buspirone HCl for enhancement of its oral bioavailability. J Liposome Res. 2010;20:286–296. doi: 10.3109/08982100903443065. [DOI] [PubMed] [Google Scholar]
- 29.Tamjidi F, Shahedi M, Varshosaz J, Nasirpour A. Design and characterization of astaxanthin-loaded nanostructured lipid carriers. Innov Food Sci Emerg Technol2 July 2014. Available from: 10.1016/j.ifset.2014.06.012, in press [DOI]
- 30.Chadha R., Bhandari S. Drug-excipient compatibility screening—role of thermoanalytical and spectroscopic techniques. J Pharm Biomed Anal. 2014;87:82–97. doi: 10.1016/j.jpba.2013.06.016. [DOI] [PubMed] [Google Scholar]
- 31.Chen D.B., Yang T.Z., Lu W.L., Zhang Q. In vitro and in vivo study of two types of long-circulating solid lipid nanoparticles containing paclitaxel. Chem Pharm Bullo. 2001;49:1444–1447. doi: 10.1248/cpb.49.1444. [DOI] [PubMed] [Google Scholar]
- 32.Korsmeyer R.W., Gurny R., Doelker E., Buri P., Peppas N.A. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15:25–35. doi: 10.1002/jps.2600721021. [DOI] [PubMed] [Google Scholar]
- 33.Higuchi T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52:1145–1149. doi: 10.1002/jps.2600521210. [DOI] [PubMed] [Google Scholar]
- 34.Souto E.B., Wissing S.A., Barbosa C.M., Müller R.H. Development of a controlled release formulation based on SLN and NLC for topical clotrimazole delivery. Int J Pharm. 2004;278:71–77. doi: 10.1016/j.ijpharm.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 35.Ghosh M.N. 3rd ed. Kolkata: Hilton & Company, West Bengal, India; 2005. Fundamentals of experimental pharmacology. [Google Scholar]
- 36.Kumar M., Misra A., Babbar A.K., Mishra A.K., Mishra P., Pathak K. Intranasal nanoemulsion based brain targeting drug delivery system of risperidone. Int J Pharm. 2008;358:285–291. doi: 10.1016/j.ijpharm.2008.03.029. [DOI] [PubMed] [Google Scholar]
- 37.Fazil M., Md S., Haque S., Kumar M., Baboota S., Sahni J.K., et al. Development and evaluation of rivastigmine loaded chitosan nanoparticles for brain targeting. Eur J Pharm Sci. 2012;47:6–15. doi: 10.1016/j.ejps.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 38.Haque S., Md S., Fazil M., Kumar M., Sahni J.K., Ali J., et al. Venlafaxine loaded chitosan NPs for brain targeting: pharmacokinetic and pharmacodynamic evaluation. Carbohydr Polym. 2012;89:72–79. doi: 10.1016/j.carbpol.2012.02.051. [DOI] [PubMed] [Google Scholar]
- 39.Jain T., Bhandari A., Ram V., Sharma S., Chaudhary R.K., Parakh M. High-performance liquid chromatographic method with diode array detection for quantification of haloperidol levels in schizophrenic patients during Routine clinical practice. J Bioanal Biomed. 2011;3:8–12. [Google Scholar]
- 40.Rahman N., Khatoon A., Rahman H. Studies on the development of spectrophotometric method for the determination of haloperidol in pharmaceutical preparation. Quím Nova. 2012;35:392–397. [Google Scholar]
- 41.Md S., Khan R.A., Mustafa G., Chuttani K., Baboota S., Sahni J.K., et al. Bromocriptine loaded chitosan nanoparticles intended for direct nose to brain delivery: pharmacodynamic, pharmacokinetic and scintigraphy study in mice model. Eur J Pharm Sci. 2013;48:393–405. doi: 10.1016/j.ejps.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 42.Müller R.H., Mäder K., Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery—a review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–177. doi: 10.1016/s0939-6411(00)00087-4. [DOI] [PubMed] [Google Scholar]
- 43.Janicki C.A., Ko C.T. In: Florey K., editor. 9th. Academic Press; New York: 1991. Haperidol; pp. 342–344. (Analytical profile of drug substances). [Google Scholar]
- 44.Sanjula B., Shah F.M., Javed A., Alka A. Effect of poloxamer 188 on lymphatic uptake of carvedilol-loaded solid lipid nanoparticles for bioavailability enhancement. J Drug Target. 2009;17:249–256. doi: 10.1080/10611860902718672. [DOI] [PubMed] [Google Scholar]
- 45.Bunjes H., Unruh T. Characterization of lipid nanoparticles by differential scanning calorimetry, X-ray and neutron scattering. Adv Drug Deliv Rev. 2007;59:379–402. doi: 10.1016/j.addr.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 46.Khan S., Patil K., Bobade N., Yeole P., Gaikwad R. Formulation of intranasal mucoadhesive temperature-mediated in situ gel containing ropinirole and evaluation of brain targeting efficiency in rats. J Drug Target. 2010;18:223–234. doi: 10.3109/10611860903386938. [DOI] [PubMed] [Google Scholar]
- 47.Al-Ghananeem A.M., Saeed H., Florence R., Yokel R.A., Malkawi A.H. Intranasal drug delivery of didanosine-loaded chitosan nanoparticles for brain targeting: an attractive route against infections caused by aids viruses. J Drug Target. 2010;18:381–388. doi: 10.3109/10611860903483396. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Q.Z., Jiang X.G., Jiang W.M., Lu W., Su L.N., Shi Z.Q. Preparation of nimodipine-loaded microemulsion for intranasal delivery and evaluation on the targeting efficiency to the brain. Int J Pharm. 2004;275:85–96. doi: 10.1016/j.ijpharm.2004.01.039. [DOI] [PubMed] [Google Scholar]