

Abstract
Amyloid beta-peptides (Aβ) are known to undergo active transport across the blood-brain barrier, and cerebral amyloid angiopathy has been shown to be a prominent feature in the majority of Alzheimer׳s disease. Quercetin is a natural flavonoid molecule and has been demonstrated to have potent neuroprotective effects, but its protective effect on endothelial cells under Aβ-damaged condition is unclear. In the present study, the protective effects of quercetin on brain microvascular endothelial cells injured by fibrillar Aβ1–40 (fAβ1–40) were observed. The results show that fAβ1–40-induced cytotoxicity in human brain microvascular endothelial cells (hBMECs) can be relieved by quercetin treatment. Quercetin increases cell viability, reduces the release of lactate dehydrogenase, and relieves nuclear condensation. Quercetin also alleviates intracellular reactive oxygen species generation and increases superoxide dismutase activity. Moreover, it strengthens the barrier integrity through the preservation of the transendothelial electrical resistance value, the relief of aggravated permeability, and the increase of characteristic enzyme levels after being exposed to fAβ1–40. In conclusion, quercetin protects hBMECs from fAβ1–40-induced toxicity.
KEY WORDS: Alzheimer׳s disease, Fibrillar Aβ1–40, Human brain microvascular endothelial cells, Quercetin
Graphical abstract
Quercetin increased TEER and permeability efficient (Pe) values of fluorescein sodium and FITC-albumin. From the results of this study, quercetin improved barrier function of hBMECs against fAβ1–40-induced toxicity.

1. Introduction
In the pathogenesis of Alzheimer׳s disease (AD), accumulation and deposition of β-amyloid peptides (Aβ) in the brain plays a vital role. Aβ is cleared from the brain across the blood–brain barrier (BBB)1, 2. In AD patients, destruction of the BBB leads to Aβ clearance disruption, resulting in Aβ plaques and cerebral amyloid angiopathy (CAA)3, 4. The pathology of microvascular CAA is closely related to the cerebrovascular dysfunction that contributes to disease progression2, 5. Among all the cerebral microvasculature comorbidities in AD, cerebrovascular Aβ deposition is the most common pathological finding, presenting in up to 90% of AD patients. Amyloid deposition is observed at sites in the cerebrovasculature, mainly in the leptomeningeal and cortical vessels6, 7. Cerebrovascular amyloidosis is a central factor in AD pathogenesis and supported by increasing evidence.
Brain microvascular endothelial cells (BMECs) contribute to maintenance of brain homeostasis and the formation of the BBB8. In recent studies, it was found that treatment with Aβ could stimulate inflammatory responses, reduce endothelial antioxidants, augment transendothelial migration, and increase the changes in morphology in BMEC monolayers9, 10, 11. Progressive build-up of Aβ in and around vessels induces alteration in the BBB permeability, which chronically limits blood supply and results in deprivation of oxygen and nutrients12, 13. Accordingly, these changes trigger a secondary cascade of metabolic events involving generation of free radicals, oxidative stress, and release of proteases, such as γ-glutamyl transpeptidase (γ-GT) and alkaline phosphatase (ALP)13, 14, 15. Therefore, BMECs are considered as a potential target in AD.
Quercetin (3,3′,4′,5,7-pentahydroxyflavone, Fig. 1), a natural flavonoid compound, is found in common foods, such as tea, berries and apples. It exerts numerous beneficial effects on human health, including anti-inflammation, anti-ischemia, cardiovascular protection and neuroprotective effects16, 17, 18, 19. Quercetin has been reported to pass through the BBB and slow down the progression of degenerative diseases. Additionally, quercetin was shown to reduce the maturation of the amyloid precursor protein in human SH-SY5Y neuroblastoma cells19, 20. Quercetin significantly decreased the ratio of insoluble Aβ in a mouse model of AD21. Furthermore, studies show that quercetin acts as a novel neuroprotectant by mitigating the increased levels of reactive oxygen species (ROS) that can accelerate the progress of AD16, 17, 19.
Figure 1.
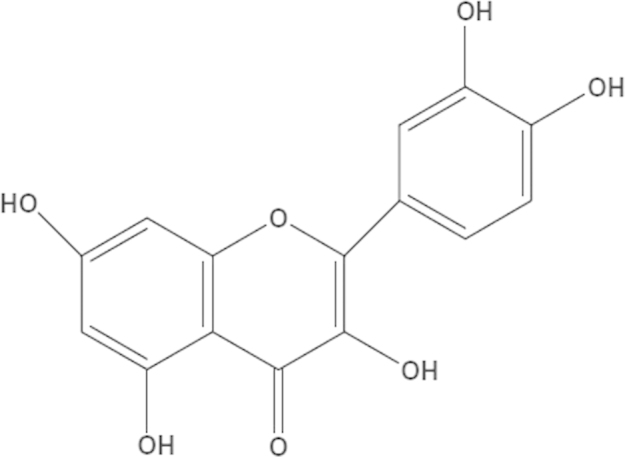
Chemical structure of quercetin.
Considering the protective properties of quercetin and its therapeutic potential, we speculated that quercetin might have a protective effect against Aβ-induced BBB disruption. To test our hypothesis, we employed an in vitro BBB model of human BMECs (hBMECs). Our study assessed the effects of fibrillar Aβ1–40 (fAβ1–40) exposure on hBMECs with respect to changes in cell viability, oxidative stress and the barrier function. Finally, we investigated the protective effect of quercetin on hBMECs against fAβ1–40-induced toxicity.
2. Materials and methods
2.1. Cell culture and treatment
The hBMECs were purchased from ScienCell Research Laboratories (Carlsbad, USA). The hBMECs were cultured in endothelial cell complete medium (ScienCell Research Laboratories, Carlsbad, USA). The cells were incubated at 37 °C in an atmosphere consisting of 95% air and 5% CO2 in a humidified incubator until reaching 80% confluency.
Synthetic Aβ1–40 was purchased from Sangon Biotech Company (Shanghai, China) and dissolved in water to make a stock solution of 0.1 mmol/L to foster the fibrillization state. Quercetin (purity >98%) was purchased from Huike Botanical Development Company (Shanxi, China). It was first dissolved in DMSO at 100 mmol/L, and then diluted in endothelial cell medium at 30.0 μmol/L, 3.0 μmol/L and 0.3 μmol/L. Different concentrations of quercetin were added to the cells at the start of fAβ1–40 injury, and incubated with/without fAβ1–40 for 24 h. The cells were randomly divided into eight groups: (1) control group; control groups in the presence of quercetin at (2) 0.3 μmol/L, (3) 3.0 μmol/L, (4) 30.0 μmol/L for 24 h; (5) fAβ1–40 group treated with 20 μmol/L fAβ1–40 for 24 h; fAβ1–40 groups treated with quercetin (6) 0.3 μmol/L, (7) 3.0 μmol/L, (8) 30.0 μmol/L for 24 h.
2.2. Cell viability assay
MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)2H-tetrazolium, inner salt] assay (Promega, Madison, USA) was used for evaluating cell viability. After being treated with quercetin and fAβ1–40 as described above, the medium was discarded and replaced with 100 μL of MTS solution according to the manufacturer׳s protocol. Then the absorbance of samples at 490 nm was read using a SpectraMax Plus microplate reader (Molecular Devices Corp, Sunnyvale, USA).
2.3. Lactate dehydrogenase (LDH) release assay
LDH, a soluble cytosolic enzyme present in the most eukaryotic cells, is released into the culture medium upon cell death due to damage to the plasma membrane. The increase in LDH activity in the culture supernatant is proportional to the number of lysed cells. The amount of LDH released by cells was determined using an LDH assay kit (Promega, Madision, USA) according to manufacturer׳s protocol. Briefly, after cells were exposed to fAβ1–40, quercetin or both for 24 h, the supernatant in each well was transferred to a new plate for LDH assay and measured in SpectraMax Plus microplate reader.
2.4. Hoechst 33342 staining assay
Nuclear changes were measured in hBMECs using Hoechst 33342 staining (Dojindo Laboratory, Kumamoto, Japan). After the cells were treated with fAβ1–40, quercetin, or both for 24 h, they were washed in phosphate-buffered solution (PBS), fixed in 4% formaldehyde for 1 h, and stained with 5 μg/L Hoechst 33342 for 30 min. Nuclear change was assessed by the value of average fluorescent intensity measured with a Cellomics ArrayScan VTI HCS Reader (Cellomics Inc, Pittsburgh, USA).
2.5. Measurements of intracellular ROS and superoxide dismutase (SOD)
The intracellular ROS level was measured using 2,7-dichlorofluorescein diacetate (DCFH2-DA, Sigma, St. Louis, USA). After treatment, hBMECs were washed with PBS and incubated with DCFH2-DA at a final concentration of 5 μmol/L for 40 min at 37 °C protected from light. The cells were then washed with PBS to remove the extracellular DCFH2-DA, and the fluorescence intensity was measured with SpectraMax Plus microplate reader at an excitation wavelength of 485 nm and an emission wavelength of 538 nm. The intracellular ROS levels were expressed as fluorescent intensity. After the exposure to fAβ1–40, and quercetin, hBMECs were collected and lysedby sonication (60 W with 0.5 s interval for 10 min). The lysate of hBMECs was centrifuged at 10,000×g for 15 min and the supernatant fraction was used to measure the SOD activity using WST-1 based SOD inhibition kit (Dojindo Laboratory, Kumamoto, Japan) according to the manufacturer׳s protocols. The absorbance of the endpoint reaction at 440 nm was measured. Percent inhibition of samples was calculated by the following equation:
| (1) |
where A1, A2, A3 and AS were the absorbances of uninhibited test, blank sample, blank reagent and sample, respectively.
2.6. Transendothelial electrical resistance (TEER)
TEER was measured using an EVOM resistance meter (World Precision Instruments, Sarasota, USA). The extracellular matrix-treated Transwell inserts (Corning Co., Corning, USA) were placed in a 12-well plate containing culture medium and used to measure the background resistance. The resistance measurements of these blank filters were then subtracted from those of filters with cells. TEER values were expressed as Ω·cm cm2 based on culture inserts.
2.7. Transendotheial permeability for sodium fluorescein and FITC-labeled albumin
To measure transendotheial permeability, the extracellular matrix-treated Transwell inserts were placed in a 12-well plate. The flux of sodium fluorescein (MW, 376 Da) and fluorescein isothioyanate (FITC)-labeled albumin (MW, 67 kDa) across endothelial monolayers were determined as previously reported22, 23. The basolateral compartments of 12-wells contained 1.5 mL Ringer solution (118 mmol/L NaCl, 4.8 mmol/L KCl, 2.5 mmol/L CaCl2, 1.2 mmol/L MgSO4, 5.5 mmol/L glucose, 20 mmol/L Hepes, pH 7.4). In the apical chamber, culture medium was replaced by 500 μL Ringer׳s solution containing 10 μg/mL sodium fluorescein and 165 μg/mL FITC-BSA. The inserts were transferred to a new well containing Ringer׳s solution at 20 min, 40 min and 60 min. FITC and fluorescein levels were measured by microplate reader (Molecular Devices Corp, Sunnyvale, USA) with emission at 488 nm, excitation at 535 nm and emission at 525 nm, excitation at 440 nm. Flux across cell-free and cell-containing inserts were both measured. Transport was expressed as μL of donor (luminal) compartment volume from which the tracer is completely cleared. The transendothelial permeability coefficient (Pe) was calculated as previously described23, 24. FITC-BSA concentrations in the basolateral chamber, [BSA]basolateral, were determined from a calibration curve of the fluorescence signal, which was linearly dependent upon BSA concentration.
Permeability, P, was calculated according to a linear approximation of Fick׳s Law:
| (2) |
where S is the surface area of the transwell insert and [BSA]0,apical and [BSA]0,basolateral are the initial concentration of FITC-BSA in the top and bottom chamber, respectively. The flux, J, was calculated as the product of the volume of the basolateral chamber times the rate of increase in basolateral FITC-BSA concentration determined from the linear region of [BSA]basolateral versus time plot. Permeability was measured for each experimental group of endothelial monolayers, as well as for membrane supports in the absence of monolayers. Diffusional Pe were calculated by correction for the permeability of the membrane support in series with the monolayer:
| (3) |
PeS divided by the surface area (1 cm2 for Transwell-12) generated the endothelial permeability coefficient (Pe, 10–3 cm/min).
2.8. Analysis of levels of γ-glutamyl transpeptidase (γ-GT) and alkaline phosphatase (ALP)
The levels of γ-GT and ALP were measured in the cells using the commercially available assay kits. The kits were purchased from Jiancheng Chemical Factory (Nanjing, China). hBMECs were collected by scrapping and low-speed centrifugation (1000 rpm, 10 min) and measured according to manufacturer׳s protocols.
2.9. Statistical analysis
Data were expressed as mean±SEM and analyzed by one-way analysis of variance (ANOVA) with LSD post-hoc correction used for multiple comparisons in our experiment using Graph Pad Prism version 4.0 (GraphPad Inc., La Jolla, USA). Values of P<0.05 are considered statistically significant and F values are given.
3. Results
3.1. Quercetin increased cell viability against fAβ1–40-induced toxicity in hBMECs
In this study, the protective effects of quercetin on hBMECs against fAβ1–40-induced toxicity were investigated in three cytotoxicity assays. Firstly, we examined the effect of quercetin on cell viability of hBMECs subjected to fAβ1–40 in the MTS assay. Cell viability was found to be significantly decreased in the presence of 20 μmol/L fAβ1–40 (Fig. 2A, P<0.001). Quercetin at 0.3 μmol/L, 3.0 μmol/L and 30.0 μmol/L dose-dependently increased cell viability (Fig. 2A, P<0.05, P<0.001, P<0.001). Quercetin did not show obvious effects on cell viability of hBMECs without fAβ1–40 injury at any of the evaluated concentrations.
Figure 2.

Quercetin increased cell viability against fAβ1–40-induced toxicity in hBMECs. (A) Quercetin increased cell viability as evaluated by MTS assay. n=8, F (7, 56)=9.541, P<0.001. (B) Quercetin decreased the levels of extracellular LDH released from hBMECs against fAβ1–40-induced toxicity. n=6, F (7, 40)=24.144, P<0.001. (C) The representative nuclear images stained with Hoechst 33342. (D) Quercetin inhibited the nuclear mean fluorescence intensity in hBMECs against fAβ1–40-induced toxicity. n=4, F (7, 24)=11.761, P<0.001. Date are expressed as mean±SEM, ***P<0.001 versus control group, #P<0.05, ##P <0.01, ###P<0.001 versus fAβ1–40 group.
The cytoprotective effects of quercetin were also demonstrated by detecting LDH leakage caused by fAβ1–40. Treatment with 20 μmol/L fAβ1–40 led to significant enzyme leakage from hBMECs (Fig. 2B, P<0.001). However, when hBMECs were treated with quercetin at 0.3 μmol/L, 3.0 μmol/L and 30.0 μmol/L in the presence of fAβ1–40, the intensity of fluorescence based on the release of LDH in hBMECs decreased significantly (Fig. 2B, P<0.001). No obvious effects of quercetin on the release of LDH from cells without fAβ1–40 at 0.3 μmol/L, 3.0 μmol/L and 30.0 μmol/L was found.
Similar effects were seen in the Hoechst 33342 staining assay. Treatment with fAβ1–40 induced nuclear changes, indicating heterogeneous intensities and chromatin condensation in the nuclei of the fAβ1–40-injured hBMECs (Fig. 2C). These cytotoxic effects were alleviated both in nuclear morphological condensation and fluorescence intensity through treatment with quercetin at 0.3 μmol/L, 3.0 μmol/L and 30.0 μmol/L in a dose-dependent manner (Fig. 2D, P<0.05, P<0.001, P<0.001). Quercetin at the same concentrations did not damage the nuclei of control cells.
Based on the above results, quercetin at 0.3 μmol/L, 3.0 μmol/L and 30.0 μmol/L is found to significantly increase the cell viability, decrease LDH leakage, and ameliorate the nuclear injury induced by fAβ1–40. Moreover, quercetin at the same concentrations did not show any cytotoxicity. Summing up the above, we selected concentrations ranging from 0.3 μmol/L to 30.0 μmol/L for further investigation.
3.2. Quercetin affected fAβ1–40-induced oxidative stress in hBMECs
Oxidative stress plays an important role in Aβ-induced cytotoxicity. Therefore, we examined the effect of quercetin on fAβ1–40-induced oxidative imbalance in hBMECs. The intracellular ROS level of fAβ1–40-treated cells showed a remarkable increase (Fig. 3A, P<0.001), while quercetin scavenged ROS generation to protect cells at the concentrations of 0.3 μmol/L, 3.0 μmol/L and 30.0 μmol/L (Fig. 3A, P<0.01, P<0.001, P<0.001) in the presence of fAβ1–40. The results also revealed that fAβ1–40 treatment induced the redox imbalance through decreasing SOD activity (Fig. 3B, P<0.001). However, when the cells were treated with quercetin at 0.3 μmol/L, 3.0 μmol/L and 30.0 μmol/L, SOD activity was remarkably increased in a dose-dependent manner (Fig. 3B, P<0.05, P<0.01, P<0.01).
Figure 3.

Quercetin affected fAβ1–40-induced oxidative stress in hBMECs. (A) Quercetin decreased the generation of ROS in hBMECs in the presence of fAβ1–40. n=6, F (4, 25)=45.616, P<0.001. (B) Quercetin improved the SOD activity in hBMECs against fAβ1–40-induced toxicity. n=4, F (4, 15)=12.539, P<0.001. Date are expressed as mean±SEM, ***P<0.001 versus control group, #P<0.05, ##P<0.01, ###P<0.001 versus fAβ1–40 group.
3.3. Quercetin improved barrier function of hBMECs against fAβ1–40-induced toxicity
The BBB is a specialized barrier that maintains the integrity of brain by restricting permeability across the brain endothelium. In this study, the barrier integrity of BMECs and paracellular permeability were determined by the measurement of TEER and the flux of fluorescein sodium and FITC-albumin. As shown in Fig. 4A, the values of TEER in fAβ1–40-treated cells were significantly reduced (Fig. 4A, P<0.001). Treatment with quercetin significantly attenuated the reduction of TEER after being exposed to fAβ1–40 at 3.0 μmol/L and 30.0 μmol/L (Fig. 4A, P<0.01, P<0.001).
Figure 4.

Quercetin improved barrier function of hBMECs against fAβ1–40-induced toxicity. (A) Quercetin at concentrations of 3.0 μmol/L and 30.0 μmol/L decreased TEER. n=4, F (4, 15)=28.676, P<0.001. (B) Quercetin at concentration of 30.0 μmol/L decreased transendotheial permeability for sodium fluorescein. n=4, F (4, 15)=14.654, P<0.001. (C) Quercetin at concentrations of 3.0 μmol/L and 30 μmol/L decreased FITC-albumin. n=4, F (4, 15)=13.999, P<0.001. Date are expressed as mean±SEM, **P<0.01, ***P<0.001 versus control group, #P<0.05, ##P<0.01, ###P<0.001 versus fAβ1–40 group.
Permeability and paracellular diffusion across confluent hBMEC monolayers was then evaluated by measuring the permeability coefficient of the permeability markers, fluorescein sodium and FITC-albumin. In the fAβ1–40-treated groups, the Pe values were both increased compared with the control group (Fig. 4B and C, P<0.001, P<0.01). After co-treatment with fAβ1–40 and quercetin for 24 h, the Pe values of fluorescein sodium and FITC-albumin were both increased in the presence of 30.0 μmol/L quercetin (P<0.05); meanwhile, the Pe value of FITC-albumin was increased at 3.0 μmol/L quercetin as well (P<0.05).
3.4. Quercetin modulated the levels of γ-GT and ALP in fAβ1–40-treated hBMECs
γ-GT and ALP are known to be abundant in cerebral microvessels, and the levels of these two enzymes are involved in barrier formation. On the basis of the above results, we determined the effect of the modulation of the levels of γ-GT and ALP in fAβ1–40-treated hBMECs. Levels of γ-GT and ALP were significantly decreased in hBMECs after being exposed to fAβ1–40 (Fig. 5, P<0.01, P<0.001), whereas quercetin reversed both changes at 3.0 μmol/L and 30.0 μmol/L (Fig. 5, P<0.01, P<0.01, P<0.001, P<0.001). Moreover, quercetin at 0.3 μmol/L significantly increased ALP in presence of fAβ1–40, but not γ-GT.
Figure 5.

Quercetin modulated levels of γ-GT and ALP in fAβ1–40-treated hBMECs. Date are expressed as mean±SEM, n=4. γ-GT: F (4, 15)=10.067, P<0.001; ALP: F (4, 15)=28.919, P<0.001. **P<0.01, ***P<0.001 versus control group, ##P<0.01, ###P<0.001 versus fAβ1–40.
4. Discussion
The present study clarifies the beneficial effects of quercetin on AD-associated microvascular endothelial pathology. The present findings indicate that quercetin can protect hBMECs from fAβ1–40-induced toxicity. In these effects, quercetin increases cell viability, reduces the amount of LDH release, relieves nuclear condensation, decreases oxidative stress, and strengthens the barrier integrity and functions through improving TEER, ameliorating transendotheial permeability and modulating barrier enzyme levels. These results suggest that quercetin protected endothelial cells against fAβ1–40-induced endothelial dysfunction.
Recent advances in neuroscience research have contributed to the emerging concept that the structural and functional integrity of the central nervous system (CNS) depends on the coordinated activity of the neurovascular unit. BMECs, known to be essential for brain homeostasis, provide a dynamic interface between circulating blood and the brain parenchyma1. Injuries to the endothelial cells of blood vessels triggered by inflammation and other insults (like Aβ) can result in endothelial death, which contributes to the pathogenesis of diverse neurological disorders25. Accordingly, results from experimental animal models and clinical studies suggest that degenerative disorders are exacerbated, if not initiated, by brain microvasculature injury. Therefore, effective therapeutic strategies to protect neuronal functions, to preserve the integrity of the BBB, and to ensure brain repair should also be targeted toward the brain microvasculature.
Herein, we firstly evidenced that fAβ1–40 treatment causes significant cell injury in the hBMEC experimental system26. The toxic effect of fAβ1–40 at 20 μmol/L was achieved in hBMECs, involving decreasing cell viability, increasing LDH release, inducing nuclear injury, overproducing intracellular ROS, reducing SOD activity, and breaking the barrier function.
Appropriate administrative conditions for quercetin were determined using both control and fAβ1–40-injured hBMECs. Quercetin at the determined optimal concentrations of 0.3 μmol/L, 3.0 μmol/L and 30.0 μmol/L was found to significantly increase the viability of cells injured by fAβ1–40. In line with the increased viability, a decrease in LDH release and relief of nuclear injury also indicated the protective effects of quercetin in this process. In addition, treatment of control cells with quercetin showed no statistically significant differences, indicating that quercetin has no toxicity under basal conditions.
Excessive generation of ROS within endothelial cells in response to Aβ leads to oxidative stress and cellular injury27, 28, 29. In vitro studies have revealed that oxidative stress results in dysfunction of the endothelial cell, destroying the integrity of the vascular barrier and leading to increased endothelial permeability, mitochondrial dysfunction, chronic inflammatory processes and amyloid deposition in blood vessels, which are involved in the imbalance of endothelial transductions during the pathogenesis of Alzheimer׳s deficits30, 31, 32. Neuroprotective interventions for AD should be effective in reducing the severity of oxidative injury and maintaining the integrity of the BBB.
In this study an obvious imbalance was detected after treatment with fAβ1–42 alone, involving a marked increase in ROS generation and a reduction of SOD activity. Treatment with quercetin attenuated fAβ1–42-induced ROS generation and protected SOD activity in hBMECs. Quercetin is a phenolic compound isolated from plants, and has been thought to be effective in scavenging free radicals33. Previous studies indicated that quercetin can act as an antioxidant to protect the skin from oxidative stress induced by ultraviolet rays34, 35. Here, quercetin produced a significant antioxidant effect in hBMECs treated with fAβ1–40. We hypothesize that quercetin might exert cytoprotective effects by suppressing oxidative stress induced by Aβ18. Accumulating evidence suggests that treatment with hydrogen peroxide disturbs the permeability barrier of epithelial cells through disruption of tight junction proteins36, 37. As hydrogen peroxide is a member of ROS, we suggest that quercetin protects hBMECs, at least in part, by reducing the overproduction of ROS and improving anti-oxidant efficacy.
The restrictive nature of BMECs for forming the BBB is due to tight junctions among adjacent endothelial cells. The BBB allows for the regulation of ion flux and paracellular diffusion through the development of high transendothelial electrical resistance and tight barrier properties38, 39, 40. The microvascular barrier function was determined to be suitable for subsequent experiments, as shown by the changes of TEER value, permeability property, and characteristic enzymatic activity.
TEER value is an important indicator of the barrier tightness of interendothelial tight junctions. In this study, the reduction of the TEER value could be markedly attenuated by treatment with quercetin at higher concentrations, indicating that the preservation of monolayer integrity may contribute to its protective effects against fAβ1–40-induced damage. In addition, quercetin treatment at the highest concentrations attenuated apical-to-basolateral diffusion of both fluorescein sodium and FITC-albumin. Although increased permeability can allow deleterious molecules into the brain and aggravate the endothelial injury, it also may help therapeutic agents cross the BBB. A relevant effect of quercetin on the BBB function has been reported41, and the mechanisms attributed to the Aβ25–35-induced toxicity in mice have been suggested to result from suppression of MAPK transduction. In combination with our studies, it provides evidence for the explanation that quercetin can cross the BBB and has molecular effects in the brain.
γ-GT and ALP are the important membranous components of the BBB as well, known to be abundant on the apical surface of microvascular endothelial cells41. The activities of these enzymes can be altered in situations such as oxidative stress42, inflammation43 and hypoxia44. The levels of γ-GT and ALP changed consistently with TEER value and barrier permeability when subjected to fAβ1–40, and they could be preserved with the quercetin treatment in a dose-dependent manner at 3.0 μmol/L and 30.0 μmol/L. These effects are consistent with the preservation of monolayer integrity based on TEER detection and transendotheial permeability. Thus, we extrapolate that quercetin is capable of protecting the barrier integrity and function of microvascular endothelial monolayers treated with fAβ1–40.
5. Conclusions
In summary, we have presented novel evidence to show that quercetin effectively prevented fAβ1–40-induced hBMEC damage, which was characterized by reduced cell viability, increased LDH leakage and aggravated nuclear condensation. Furthermore, we found that quercetin showed sufficient activity in regulating the redox imbalance and strengthening barrier integrity through the preservation of permeability and characteristic enzymatic activities in the fAβ1–40-treated hBMECs. To our knowledge, this is the first direct evidence for an effect of quercetin on brain microvascular endothelial cells. Our results provide a rational basis for the therapeutic application of this compound in the prevention/treatment of AD.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Nos. 81373388, 81473374 and 81102830).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.

Contributor Information
Rui Liu, Email: liurui@imm.ac.cn.
Tiantai Zhang, Email: ttzhang@imm.ac.cn.
References
- 1.Kalaria R.N. The blood-brain barrier and cerebral microcirculation in Alzheimer disease. Cerebrovasc Brain Metab Rev. 1992;4:226–260. [PubMed] [Google Scholar]
- 2.Ghiso J., Fossati S., Rostagno A. Amyloidosis associated with cerebral amyloid angiopathy: cell signaling pathways elicited in cerebral endothelial cells. J Alzheimers Dis. 2014;42 Suppl. 3:S167–S176. doi: 10.3233/JAD-140027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parham C., Auckland L., Rachwal J., Clarke D., Bix G. Perlecan domain V inhibits amyloid-β induced brain endothelial cell toxicity and restores angiogenic function. J Alzheimers Dis. 2014;38:415–423. doi: 10.3233/JAD-130683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sagare A.P., Bell R.D., Zlokovic B.V. Neurovascular defects and faulty amyloid-β vascular clearance in Alzheimer׳s disease. J Alzheimers Dis. 2013;33 Suppl. 1:S87–S100. doi: 10.3233/JAD-2012-129037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu R., Meng F.R., Zhang L., Liu A.L., Qin H.L., Lan X. Luteolin isolated from the medicinal plant Elsholtzia rugulosa (Labiatae) prevents copper-mediated toxicity in β-amyloid precursor protein Swedish mutation overexpressing SH-SY5Y cells. Molecules. 2011;16:2084–2096. doi: 10.3390/molecules16032084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grammas P., Yamada M., Zlokovic B. The cerebromicrovasculature: a key player in the pathogenesis of Alzheimer׳s disease. J Alzheimers Dis. 2002;4:217–223. doi: 10.3233/jad-2002-4311. [DOI] [PubMed] [Google Scholar]
- 7.Grinberg L.T., Thal D.R. Vascular pathology in the aged human brain. Acta neuropathol. 2010;119:277–290. doi: 10.1007/s00401-010-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deane R., Zlokovic B.V. Role of the blood–brain barrier in the pathogenesis of Alzheimer׳s disease. Curr Alzheimer Res. 2007;4:191–197. doi: 10.2174/156720507780362245. [DOI] [PubMed] [Google Scholar]
- 9.Deane R., Du Yan S., Submamaryan R.K., LaRue B., Jovanovic S., Hogg E. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 10.Liu R., Wu C.X., Zhou D., Yang F., Tian S., Zhang L. Pinocembrin protects against β-amyloid-induced toxicity in neurons through inhibiting receptor for advanced glycation end products (RAGE)-independent signaling pathways and regulating mitochondrion-mediated apoptosis. BMC Med. 2012;10:105. doi: 10.1186/1741-7015-10-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan S.D., Chen X., Fu J., Chen M., Zhu H., Roher A. RAGE and amyloid-β peptide neurotoxicity in Alzheimer׳s disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 12.Morris A.W., Carare R.O., Schreiber S., Hawkes C.A. The cerebrovascular basement membrane: role in the clearance of β-amyloid and cerebral amyloid angiopathy. Front Aging Neurosci. 2014;6:251. doi: 10.3389/fnagi.2014.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fossati S., Ghiso J., Rostagno A. Insights into caspase-mediated apoptotic pathways induced by amyloid-β in cerebral microvascular endothelial cells. Neurodegener Dis. 2012;10:324–328. doi: 10.1159/000332821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee J.K., Jin H.K., Park M.H., Kim B.R., Lee P.H., Nakauchi H. Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer׳s disease. J Exp Med. 2014;211:1551–1570. doi: 10.1084/jem.20132451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.More S.S., Vince R. Potential of a γ-glutamyl-transpeptidase-stable glutathione analogue against amyloid-β toxicity. ACS Chem Neurosci. 2012;3:204–210. doi: 10.1021/cn200113z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ansari M.A., Abdul H.M., Joshi G., Opii W.O., Butterfield D.A. Protective effect of quercetin in primary neurons against Aβ(1−42): relevance to Alzheimer׳s disease. J Nutr Biochem. 2009;20:269–275. doi: 10.1016/j.jnutbio.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi R.C., Zhu J.T., Leung K.W., Chu G.K., Xie H.Q., Chen V.P. A flavonol glycoside, isolated from roots of Panax notoginseng, reduces amyloid-beta-induced neurotoxicity in cultured neurons: signaling transduction and drug development for Alzheimer׳s disease. J Alzheimers Dis. 2010;19:795–811. doi: 10.3233/JAD-2010-1293. [DOI] [PubMed] [Google Scholar]
- 18.Jiménez-Aliaga K., Bermejo-Bescós P., Benedí J., Martín-Aragón S. Quercetin and rutin exhibit antiamyloidogenic and fibril-disaggregating effects in vitro and potent antioxidant activity in APPswe cells. Life Sci. 2011;89:939–945. doi: 10.1016/j.lfs.2011.09.023. [DOI] [PubMed] [Google Scholar]
- 19.Liu R., Zhang T.T., Zhou D., Bai X.Y., Zhou W.L., Huang C. Quercetin protects against the Aβ(25–35)-induced amnesic injury: involvement of inactivation of rage-mediated pathway and conservation of the NVU. Neuropharmacology. 2013;67:419–431. doi: 10.1016/j.neuropharm.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 20.Huebbe P., Wagner A.E., Boesch-Saadatmandi C., Sellmer F., Wolffram S., Rimbach G. Effect of dietary quercetin on brain quercetin levels and the expression of antioxidant and Alzheimer׳s disease relevant genes in mice. Pharmacol Res. 2010;61:242–246. doi: 10.1016/j.phrs.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 21.Tongjaroenbuangam W., Ruksee N., Chantiratikul P., Pakdeenarong N., Kongbuntad W., Govitrapong P. Neuroprotective effects of quercetin, rutin and okra (Abelmoschus esculentus Linn.) in dexamethasone-treated mice. Neurochem Int. 2011;59:677–685. doi: 10.1016/j.neuint.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 22.Veszelka S., Pásztói M., Farkas A.E., Krizbai I., Ngo T.K., Niwa M. Pentosan polysulfate protects brain endothelial cells against bacterial lipopolysaccharide-induced damages. Neurochem Int. 2007;50:219–228. doi: 10.1016/j.neuint.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Zhao L., Hou L., Sun H.J., Yan X., Sun X.F., Li J.G. Apigenin isolated from the medicinal plant Elsholtzia rugulosa prevents β-amyloid 25–35-induces toxicity in rat cerebral microvascular endothelial cells. Molecules. 2011;16:4005–4019. [Google Scholar]
- 24.Vargo M.A., Voss O.H., Poustka F., Cardounel A.J., Grotewold E., Doseff A.I. Apigenin-induced-apoptosis is mediated by the activation of PKC δ and caspases in leukemia cells. Biochem Pharmacol. 2006;72:681–692. doi: 10.1016/j.bcp.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 25.Deli M.A., Veszelka S., Csiszár B., Tóth A., Kittel A., Csete M. Protection of the blood-brain barrier by pentosan against amyloid-β-induced toxicity. J Alzheimers Dis. 2010;22:777–794. doi: 10.3233/JAD-2010-100759. [DOI] [PubMed] [Google Scholar]
- 26.Liu R., Li J.Z., Song J.K., Sun J.L., Li Y.J., Zhou S.B. Pinocembrin protects human brain microvascular endothelial cells against fibrillar amyloid-β(1–40) injury by suppressing the MAPK/NF-κB inflammatory pathways. BioMed Res Int. 2014;2014:470393. doi: 10.1155/2014/470393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng J., Qi X.L., Guan Z.Z., Yan X.M., Huang Y., Wang Y.L. Pretreatment of SH-SY5Y cells with dicaffeoylquinic acids attenuates the reduced expression of nicotinic receptors, elevated level of oxidative stress and enhanced apoptosis caused by β-amyloid peptide. J Pharm Pharmacol. 2013;65:1736–1744. doi: 10.1111/jphp.12096. [DOI] [PubMed] [Google Scholar]
- 28.Ashabi G., Ahmadiani A., Abdi A., Abraki S.B., Khodagholi F. Time course study of Aβ formation and neurite outgrowth disruption in differentiated human neuroblastoma cells exposed to H2O2: protective role of autophagy. Toxicol In Vitro. 2013;27:1780–1788. doi: 10.1016/j.tiv.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Carrano A., Hoozemans J.J.M., van der Vies S.M., Rozemuller A.J.M., van Horssen J., de Vries H.E. Amyloid beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal. 2011;15:1167–1178. doi: 10.1089/ars.2011.3895. [DOI] [PubMed] [Google Scholar]
- 30.Strazielle N., Ghersi-Egea J.F., Ghiso J., Dehouck M.P., Frangione B., Patlak C. In vitro evidence that β-amyloid peptide 1–40 diffuses across the blood-brain barrier and affects its permeability. J Neuropathol Exp Neurol. 2000;59:29–38. doi: 10.1093/jnen/59.1.29. [DOI] [PubMed] [Google Scholar]
- 31.Bell R.D., Zlokovic B.V. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer׳s disease. Acta Neuropathol. 2009;118:103–113. doi: 10.1007/s00401-009-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thal D.R., Griffin W.S., de Vos R.A., Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer׳s disease. Acta Neuropathol. 2008;115:599–609. doi: 10.1007/s00401-008-0366-2. [DOI] [PubMed] [Google Scholar]
- 33.Song Y., Liu J., Zhang F., Zhang J., Shi T., Zeng Z. Antioxidant effect of quercetin against acute spinal cord injury in rats and its correlation with the p38MAPK/iNOS signaling pathway. Life Sci. 2013;92:1215–1221. doi: 10.1016/j.lfs.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 34.Liu D., Hu H., Lin Z., Chen D., Zhu Y., Hou S. Quercetin deformable liposome: preparation and efficacy against ultraviolet B induced skin damages in vitro and in vivo. J Photochem Photobiol B. 2013;127:8–17. doi: 10.1016/j.jphotobiol.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 35.Yin Y., Li W., Son Y.O., Sun L., Lu J., Kim D. Quercitrin protects skin from UVB-induced oxidative damage. Toxicol Appl Pharmacol. 2013;269:89–99. doi: 10.1016/j.taap.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suganya N., Bhakkiyalakshmi E., Suriyanarayanan S., Paulmurugan R., Ramkumar K.M. Quercetin ameliorates tunicamycin-induced endoplasmic reticulum stress in endothelial cells. Cell Prolif. 2014;47:231–240. doi: 10.1111/cpr.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nekohashi M., Ogawa M., Ogihara T., Nakazawa K., Kato H., Misaka T. Luteolin and quercetin affect the cholesterol absorption mediated by epithelial cholesterol transporter niemann-pick c1-like 1 in caco-2 cells and rats. PLoS One. 2014;9:e97901. doi: 10.1371/journal.pone.0097901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seok S.M., Kim J.M., Park T.Y., Baik E.J., Lee S.H. Fructose-1,6-bisphosphate ameliorates lipopolysaccharide-induced dysfunction of blood-brain barrier. Arch Pharm Res. 2013;36:1149–1159. doi: 10.1007/s12272-013-0129-z. [DOI] [PubMed] [Google Scholar]
- 39.Fischer S., Renz D., Schaper W., Karliczek G.F. In vitro effects of fentanyl, methohexital, and thiopental on brain endothelial permeability. Anesthesiology. 1995;82:451–458. doi: 10.1097/00000542-199502000-00015. [DOI] [PubMed] [Google Scholar]
- 40.Gloor S.M., Wachtel M., Bolliger M.F., Ishihara H., Landmann R., Frei K. Molecular and cellular permeability control at the blood-brain barrier. Brain Res Brain Res Rev. 2001;36:258–264. doi: 10.1016/s0165-0173(01)00102-3. [DOI] [PubMed] [Google Scholar]
- 41.Lawrenson R., Williams T., Farmer R. Clinical information for research: the use of general practice databases. J Public Health Med. 1999;21:299–304. doi: 10.1093/pubmed/21.3.299. [DOI] [PubMed] [Google Scholar]
- 42.Kugelman A., Choy H.A., Liu R., Shi M.M., Gozal E., Forman H.J. gamma-Glutamyl transpeptidase is increased by oxidative stress in rat alveolar L2 epithelial cells. Am J Respir Cell Mol Biol. 1994;11:586–592. doi: 10.1165/ajrcmb.11.5.7946387. [DOI] [PubMed] [Google Scholar]
- 43.Das M., Singh S.V., Mukhtar H., Awasthi Y.C. Differential inhibition of rat and human glutathione S-transferase isoenzymes by plant phenols. Biochem Biophys Res Commun. 1986;141:1170–1176. doi: 10.1016/s0006-291x(86)80167-x. [DOI] [PubMed] [Google Scholar]
- 44.Yu S., Iwatsuki H., Ichinohe N., Mori F., Shoumura K. ‘In vivo perfusion Turnbull׳s reaction’ for Fe (II) histochemistry in non-anoxic/non-ischemic and anoxic/ischemic cat brains. Neurosci Lett. 2001;308:79–82. doi: 10.1016/s0304-3940(01)01944-9. [DOI] [PubMed] [Google Scholar]