

Abstract
Abdominal aortic aneurysm (AAA) is an inflammatory vascular disorder with high mortality. Accumulating evidence shows that toll-like receptor 2 (TLR2) plays a critical role in the regulation of wound-repairing process after tissue injury. We wondered if TLR2 signaling contributed to the pathogenesis of AAA and that targeting TLR2 would attenuate AAA development and progression. In this study, enhanced expression of TLR2 and its ligands were observed in human AAA tissue. Neutralization of TLR2 protected against AAA development and caused established AAA to regress in mouse models of AAA. In addition, TLR2-deficient mice also failed to develop AAA. The prophylactic and therapeutic effects of blocking TLR2 were accompanied by a significant resolution of inflammation and vascular remodeling, as indicated by the decreased expression or activity of MMP-2/9, α-SMA, inflammatory cytokines, and transcription factors NF-κB, AP-1 and STAT1/3 in AAA tissue. Mechanistically, blocking TLR2 decreased the expression and interaction of TLR2 and several endogenous ligands, which diminished chronic inflammation and vascular remodeling in the vascular tissue of AAA. Our studies indicate that the interactions between TLR2 and its endogenous ligands contribute to the pathogenesis of AAA and that targeting TLR2 offers great potential toward the development of therapeutic agents against AAA.
Abbreviations: AAA, abdominal aortic aneurysm; AP-1, activator protein-1; bip, binding immunoglobulin protein; Ang II, angiotensin II; DAMP, damage associated molecular pattern; DHE, dihydroethidium; HMGB1, high mobility group B-1; HSP, heat shock protein; IOD, integrated optical density; MCP-1, monocyte chemoattractant protein-1; MMP, matrix metalloproteinase; NF-κB, nuclear factor kappa B; PAMP, pathogen-associated molecular pattern; PRRs, pattern recognition receptors; RAMPs, resolution-associated molecular patterns; ROS, reactive oxygen species; α-SMA, α-smooth muscle actin; STAT1/3, signal transducer and activator of transcription 1/3; Th2, type 2 T help; TLR, toll-like receptor; VVG, Verhoeff van Gieson; WT, wide-type
KEY WORDS: Abdominal aortic aneurysm, DAMPs, Vascular remodeling, TLR2, Immune microenvironment
Graphical abstract
The interactions between toll-like receptor 2 (TLR2) and its endogenous ligands contribute to the pathogenesis of abdominal aortic aneurysm (AAA) and targeting TLR2 offers great potential toward the development of therapeutic agents against AAA.
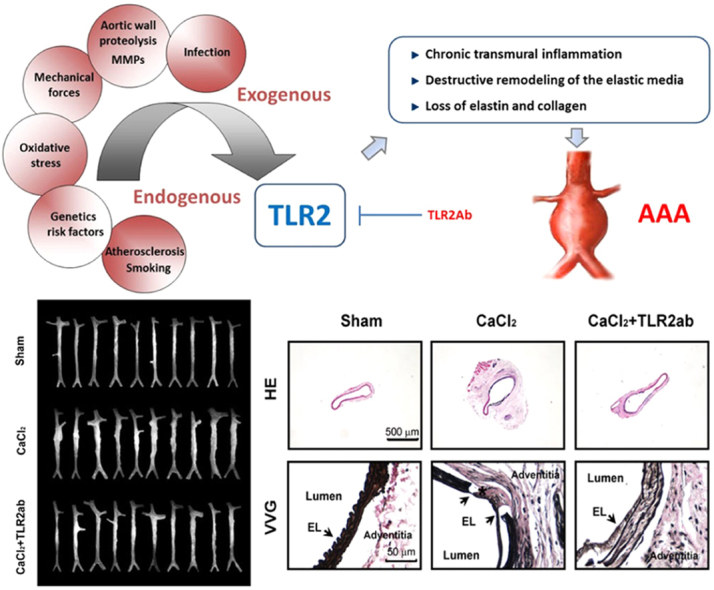
1. Introduction
Abdominal aortic aneurysm (AAA), which is characterized by chronic transmural inflammation and destructive remodeling of the vascular wall, results in dilation, triggers clinical complications, and eventually ruptures to lead to high morbidity and mortality1. Although the pathogenesis of AAA formation and progression remains poorly understood, chronic inflammation, enzymatic degradation of elastin and collagen, and malignant vascular remodeling constitute the prominent pathological changes associated with AAA2. Currently, the primary therapeutic strategies are surgical procedures, especially for large AAAs, including replacement of the damaged aorta with an artificial vascular graft or conducting endovascular stent graft repair. However, considering the risk of endoleaks, graft displacement, postoperative survival rates of the endovascular stent and the high risk of open repair, surgery must be restricted to within narrow limits3. Therefore, more effective therapeutic alternatives for the management of AAAs are necessary.
Recent work suggests that innate and adaptive immune responses participate in the pathogenesis of AAA4, 5. Vascular inflammation may result from a wide variety of insults, including infection, oxidative stress and chemotherapeutic agents. Persistent or repeated insulting stimuli activate and sustain the tissue wound-healing process in blood vessels. The host response to these insults is intricately directed toward recognizing pathogen-associated molecular patterns (PAMPs), which are produced by pathogenic organisms, or damage-associated molecular patterns (DAMPs), which are released from damaged tissues through an interaction with pattern recognition receptors (PRRs) on immune and residual cells. These endogenous DAMPs, including high-mobility group box 1 (HMGB1), heat shock proteins (HSPs), S100 family, uric acid and adenosine, can activate immune responses and participate in the development and progression of vascular diseases6, 7. Interestingly, these endogenous factors may also act as resolution-associated molecular patterns (RAMPs) to promote the resolution of inflammation and injured tissues8, 9. Activation of PRRs by DAMPs also initiates and shapes adaptive immune responses by activating antigen-presenting cells and recruitment of T and B cell subsets10. For instance, Shimizu et al.11 found that there was a shift toward type 2 T help (Th2) cell responses in human AAAs compared with stenotic atheromas, while the balance of Th1/Th2 cytokines has an important regulatory role in modulating matrix remodeling, which has been implicated in the pathogenesis of AAAs and arteriosclerosis.
Pryshchep et al.12 reported that the vessel-specific risk for inflammatory vasculopathies is partly because of distinct toll-like receptor (TLR) profiles and subsequent wall-infiltrating T cell responses. TLR2 is ubiquitously present in the aorta, and enhanced TLR2 expression has been described in human atherosclerotic lesions13. Moreover, TLR2 activation plays a central role in the regulation of inflammation after chronic tissue injury, which leads to tissue fibrosis in many organs, including the aorta14. Indeed, the underlying mechanisms of aneurysm involve a complex vascular remodeling. We recently found that targeting TLR2 prompts the resolution of chronic inflammation and protects against doxorubicin-induced cardiovascular remodeling and dysfunction15. We thus hypothesized that TLR2 signaling might involve in the pathogenesis of AAA. Our studies demonstrate that TLR2 activity critically participates in the pathogenesis of AAA and that targeting TLR2 signaling may have therapeutic potential against AAA formation and progression.
2. Materials and methods
2.1. Acquisition of human aortic samples
Human aortic aneurysm and control tissues were obtained from AAA patients who received an open surgical repair process. For immunofluorescence analysis, frozen sections (8 μm) of aortic samples were incubated with anti-human TLR2 (eBioscience Inc.) and HMGB1 (Sigma-Aldrich), followed by Alexa Fluor 488- and 647-conjugated IgG. For ex vivo organ cultures of human aortic specimens, fresh tissues were divided into 1 mm segments and cultured in DMEM with 1% BSA in a 24-well plate as described previously16. The conditioned media was collected after the tissue was treated with or without 20 μg/mL of TLR2ab for 48−96 h. The activities of matrix metalloproteinase-2/9 (MMP-2/9) were determined by gelatin zymography with an equal volume of conditioned media. All protocols using human specimens were approved by the Institutional Review Board of Chinese Academy of Medical Sciences and Peking Union Medical College. The study conforms to the principles outlined in the Declaration of Helsinki.
2.2. Animals and materials
Male C57BL/6 mice (6–8 weeks) were purchased from Vital River Inc. (Beijing, China). Tlr2−/− mice (C57BL/6 background and backcrossed more than 9 times to the C57BL/6 background), Apoe−/− mice and corresponding wide-type (WT) mice were obtained from Jackson Laboratories (Bar Harbor, USA). TLR2-neutralizing mAb was purchased from R&D System Inc. (Minneapolis, USA). Angiotensin (Ang) II was obtained from Sigma-Aldrich (St. Louis, USA). ALZET osmotic pumps were purchased from DURECT Corporation (Cupertino, USA).
2.3. Generation of AAA models
A CaCl2-induced mouse AAA model was generated by periaortic application of 0.5 mol/L CaCl2 according to the procedures described previously17, 18. Each mouse was anesthetized with sodium pentobarbital (45 mg/kg, i.p.) before surgically exposing the abdominal aorta and inferior vena cava. For the prevention study, mice were randomly divided into each group (n=12–15). In the sham group, the aorta was treated with saline instead of CaCl2. As positive controls, mice were treated with doxycycline (30 mg/kg, p.o.) daily for 6 weeks. Mice were treated with either TLR2-neutralizing antibody (TLR2ab) or isotype-IgG (200 μg/kg, i.v.) on days 1, 3 and 7 after CaCl2 application and then weekly for 6 weeks. Mice (n=10/group) were randomly assigned to receive ultrasonography. For the regression study, AAA was generated by periaortic administration of 0.5 mol/L of CaCl2 (n=15) or saline (n=11). Abdominal aortic ultrasonography was performed 6 weeks after CaCl2 administration to confirm AAA formation. Mice with well-established AAA were randomly treated with TLR2ab (200 μg/kg, i.v.) or saline for additional 6 weeks. In the end of study, mice (n=8/group) were randomly assigned to receive ultrasonography.
To generate an Ang II-induced AAA model, six-month-old Apoe−/− male mice were administered saline or Ang II (1000 ng/kg/min) via Alzet osmotic minipumps (Durect Corporation, Cupertino, USA) for 28 days to induce suprarenal aneurysms as described previously19. Briefly, mice were anaesthetized intraperitoneally using sodium pentobarbital (45 mg/kg), and the pumps were placed into the subcutaneous space of the mice through a small incision in the back of the neck.
2.4. Morphometric analysis
Mice were anesthetized with sodium pentobarbital (40–50 mg/kg, i.p.) and the aortas were immediately perfused with saline or 4% paraformaldehyde for half an hour20. The external diameter was measured based on the images of fixed-aortas and lumen diameter was examined by measuring the lumen perimeters of cross sections of paraffin-embedded aortas using Image-Pro Plus software (Media Cybernetics).
2.5. Ultrasonography
Mice were anesthetized with sodium pentobarbital (40–50 mg/kg, i.p.). The ultrasound imaging system Visual Sonics Vevo 770 (Visual Sonics, Canada) was used to perform B- and M-mode imaging of the abdominal aorta. The abdominal aorta was located using anatomical landmarks and Doppler signals were used to confirm the identification of the abdominal aorta. A longitudinal image of the abdominal aorta between the left renal arteries and the iliac bifurcation was acquired and recorded for at least 8–10 cardiac cycle in B-mode and M-mode. To get excellent reproducibility, the maximal diameter and other parameters were measured, rescanned and remeasured at least five different locations by two individuals blinded to the groups.
2.6. Histology and immunohistochemistry
Formaldehyde-fixed paraffin sections (3 μm) were stained for general structure (hematoxylin and eosin, H&E), elastin (Verhoeff van Gieson, VVG), collagen I/III (picrosirius red), macrophages (CD11b), T cells (CD4), myofibroblasts (α-smooth muscle actin, α-SMA) and cytokines. Picrosirius-stained sections were imaged by ordinary polychromatic and polarized light microscopes using identical shutter conditions21. All images were acquired using the same exposure time and loaded into the Image-Pro Plus software for analysis. The positive staining was highlighted red color by the same threshold (hue, saturation and brightness). The region of the labeled red were measured and analyzed in a blinded way. Data were obtained from 5–8 serial cross-sections, 8 aortas per treatment, and presented as the integrated optical density (IOD). For each section, data were presented as averages from at least 8 non-overlapping images. Species- and isotype-matched IgG was used as a negative control.
2.7. Immunofluorescence
Frozen sections (8 μm) were fixed in cold formaldehyde for 5 min, rinsed twice in PBS (pH 7.4), and blocked with 3% BSA, processed as described22. The sections were incubated with indicated primary and secondary antibodies and observed under an E2000U confocal microscope or fluorescent microscopy and evaluated using Image-Pro Plus software.
2.8. ROS analysis
To evaluate ROS production in aortic tissues from mice treated with or without Ang II, the abdominal aortas were harvested, snap-frozen and embedded in optimal cutting temperature compound. The sections (10 μm) were incubated with dihydroethidine hydrochloride (5 μmol/L) (Molecular Probes, USA) at 37 °C for 30 min and covered with DAPI solution. ROS content (red fluorescence) was detected with confocal microscopy.
2.9. Western blot
The aorta was homogenized and centrifuged at 14,000×g at 4 °C 20 min to collect protein extract as previously described23.
2.10. Gelatin zymography
Aortic protein was extracted as described and 20 μg proteins were separated on 12% SDS-PAGE gels containing 1 mg/mL gelatin under nonreducing conditions. After electrophoresis, the gel was soaked in renaturing buffer and equilibrated in developing buffer. The gel was incubated in fresh developing buffer overnight at 37 °C. Then the gels were stained with 0.5% Coomassie blue R250 and destained in 25% methanol/20% acetic acid.
2.11. Statistics
Data are expressed as mean±SD and analyzed with SPSS software version 11.0 (SPSS Inc.). For statistical analysis, comparisons between groups were performed using one-way ANOVA with the LSD test or a nonparametric Mann-Whitney U test if the data were not normally distributed. A Kaplan-Meier analysis summarized the survival rate. Statistical significance was accepted at P<0.05.
3. Results
3.1. Human AAA vascular tissue over-expresses TLR2 and TLR2 ligands
Although TLR2 has been linked with several forms of cardiovascular disease, no direct evidence suggests a role for TLR2 in the development of AAA. We examined the expression of TLR2 and its endogenous ligands in human AAA tissue obtained from patients who received an open surgery repair. Detailed descriptions of these patients including age, gender, and average aortic aneurysm diameters et al. were shown in Table S1 in Supporting information. Human AAA tissue manifested the characteristic morphologic changes, such as local elastin degeneration, accumulation of necrotic cells, intra-aortic bleeding in vascular tissue (Fig. 1A). Notably, the expression of TLR2 and its endogenous ligands, including HMGB1, HSP70 and S100A8, were up-regulated in human aneurysm tissue compared to the control aortic tissue (Fig. 1B−D). Moreover, TLR2 and HMGB1 were found to co-localize in adventitia and outer media (Fig. 1E), suggesting a potential interaction between TLR2 and HMGB1. Indeed, blocking TLR2 attenuated the expression or activation of inflammatory factors NF-κB and HMGB1 in human AAA tissue and the activity of secreted MMP-2/9 in conditioned media of cultured human AAA tissue (Fig. 1F and G). These data indicate that TLR2 signaling may play a crucial role in the pathogenesis of human AAA.
Figure 1.

TLR2 and its endogenous ligands are up-regulated in human AAA. (A) Representative H&E staining of human AAA or control abdominal aortic tissue. (B and C) The expression of TLR2, HMGB1, HSP70 and S100A8 was detected by Western blot (n=5/group). Data are means±SD. #P<0.05, ##P<0.01, ###P<0.001, compared to control group. (D) The expression of TLR2 was detected by immunofluorescence (TLR2, FITC, green; DNA, DAPI, blue). (E) The colocalization of TLR2 and HMGB1 (TLR2, Alexa 488, green; HMGB1, Alexa 647, red; and DNA, DAPI, blue). (F and G) Blocking TLR2 attenuated the activity and expression of NF-κBp65 and HMGB1 detected by Western blot (F), and the MMP-2/9 activity in conditioned media in vitro cultured human AAA tissue detected by gelatin zymography (G). Representative images of Western blot or gelatin zymography are shown with quantitative analysis (n=5/group).
3.2. Targeting TLR2 protected mice from developing CaCl2- induced AAA
We recently found that TLR2 activity is critically involved in the regulation of the injury repair process after acute tissue injury23, 24. We suspected whether antagonizing TLR2 would protect animals from CaCl2-induced AAA. We found that blocking TLR2 activity using a TLR2ab attenuated the dilation of aneurysm compared to vehicle control (1.14±0.31 versus 1.69±0.49; P<0.01; Fig. 2A) or with isotype-IgG (1.14±0.31 versus 1.59±0.38; P<0.01; Fig. 2A). In addition, doxycycline, a non-specific MMP inhibitor, as a positive control25, showed a similar protective role as TLR2ab compared to vehicle control (1.15±0.43 versus 1.69±0.49; P<0.01; Fig. 2A). The data of maximum external diameter from ultrasound also confirmed these results (Fig. 2B, data not shown). The CaCl2-damaged aorta was characterized by low elastin content and flattening and fragmentation of naturally wavy elastic lamellae. TLR2ab and doxycycline treated mice relatively preserved elastic laminar integrity and waviness (Fig. 2C and D). Using the morphometry of H&E staining of cross sections, we found that CaCl2 induced a time-dependent increase in the internal aortic diameters (data not shown), while the expansion of internal aortic diameter was significantly reduced after 6 weeks of treatment with TLR2ab (0.46±0.06 versus 0.57±0.05; P<0.01; Fig. 2E). These data suggest that blocking TLR2 protects against the development of CaCl2-induced AAA.
Figure 2.

Targeting TLR2 protects mice from the development of AAA. The CaCl2-induced AAA mice were immediately treated with the indicated agents for 6 weeks. (A) A representative photograph of aortas showed that doxycycline and TLR2ab decreased the maximal external aortic diameters (n=12–15/group). (B) A representative ultrasound image of aortas (arrows indicate the aorta lumen), and histological studies of AAA. (C) H&E stains of cross sections of aorta (top), local lesions (middle) and VVG stains for elastin (bottom). (D) Quantitative elastin content analysis is shown as the percentage of the surface area occupied by elastic fibers stained with VVG. (E) Internal aortic diameter was reduced by TLR2ab, as indicated by morphometry using H&E staining of cross sections of the aorta (middle panel of C). Arrowheads indicate elastic lamella (n=8/group). Data are means±SD. ##P<0.01, ###P<0.001, compared to sham group. ⁎P<0.05, ⁎⁎P<0.01, compared to CaCl2 group.
Excess expression and activation of MMP-2/9 has been found to contribute to the destructive remodeling of extracellular matrix in AAA18. Targeting TLR2 attenuated the expression (Fig. 3A) and activation (Fig. 3B) of MMP-2/9. After tissue injury, a wound-healing response was activated; the fate of recruited/activated fibroblasts in injured tissues may ultimately determine whether normal healing or end-stage fibrosis occurs. Indeed, the expression of α-SMA, a marker for myofibroblasts26, was significantly increased in a time-dependent manner. The α-SMA-positive cells migrated to and surrounded the lesion sites within two weeks and stayed there for 12 weeks after CaCl2 application (Fig. 3C) with collagen I/III accumulation. However, targeting TLR2 reduced α-SMA-positive cells in adventitia (data not shown), α-SMA expression (Fig. 3D), and protected against deposition of collagen I/III (Fig. 3E). These observations indicate that blocking TLR2 attenuates vascular fibrosis and remodeling in CaCl2-induced AAA.
Figure 3.

Targeting TLR2 reduces AAA lesions in vascular tissue of CaCl2-induced AAA. (A and B) Blocking TLR2 attenuated the expression (A) and activities of MMP-2/9 (B) in the CaCl2-injured vascular tissue (n=6). (C and D) CaCl2 stimulated a time-dependent increase in α-SMA expression in vascular tissue (n=5/time point/group) and targeting TLR2 decreased the expression of α-SMA (n=6). (E) Targeting TLR2 reduced the content of collagen I/III in aneurismal tissue stained by picrosirius red (n=8). Data are means±SD. #P<0.05, ##P<0.01, ###P<0.001, compared to sham group. ⁎P<0.05, ⁎⁎P<0.01, ⁎⁎⁎P<0.001, compared to CaCl2 group.
3.3. Targeting TLR2 ameliorated CaCl2-induced AAA
To determine the potential therapeutic significance of TLR2 in AAA development, the mice with established AAA were treated with or without TLR2ab for 6 weeks. We found that therapeutic antagonism against TLR2 resulted in a decreased external aortic diameter of AAA compared to the vehicle control, as detected by morphometry (1.08±0.232 versus 1.38±0.375; P<0.01; Fig. 4A and C) and by ultrasonography (data not shown), while the expansion of the internal aortic diameters of AAA was also inhibited compared to vehicle control (0.51±0.101 versus 0.61±0.048; Fig. 4C), indicating that blocking TLR2 attenuated the luminal expansion. Furthermore, elastic laminar integrity and waviness were preserved in TLR2ab-treated mice (Fig. 4E). These data suggested that therapeutic blocking of TLR2 inhibits elastin degeneration and attenuates aneurismal remodeling, leading to a regression of established AAA.
Figure 4.

Targeting TLR2 results in the regression of established AAA, and TLR2 deficiency protected mice from CaCl2-induced AAA. (A) AAA animals were treated with TLR2ab for six weeks. Representative photographs of aortas from sham-treated mice and mice treated with saline or TLR2ab (n=10/group). (B) The CaCl2-induced AAA was generated in WT or Tlr2−/− mice. Representative photographs of aortas from sham- and CaCl2-treated WT mice and Tlr2−/− mice (n=4/group). (C) The morphometric analysis was conducted for the maximal external and internal diameters of aortas from sham or AAA mice treated with indicated agents (n=10/group). (D) Tlr2−/− mice showed a reduction in the maximal external and internal diameters of aortas compared to WT mice (n=8/group). (E and F) Histological analysis of cross sections of aorta was performed with VVG and H&E staining. The elastin content was evaluated as the percentage of elastic fibrous area stained with VVG. Arrowheads indicate elastic lamella. Data are means±SD. ##P<0.01, ###P<0.001, compared to sham group. ⁎P<0.05, ⁎⁎P<0.01, compared to CaCl2 group.
To validate the crucial roles of TLR2 in AAA development, Tlr2−/− and WT mice were subjected to CaCl2-induced AAA. We found that TLR2 deficiency prevented CaCl2-induced dilation of the aorta compared to WT mice, as illustrated in Fig. 4B and D (0.95±0.12 versus 1.33±0.27; P<0.05). Compared to WT mice, Tlr2−/− mice showed a much smaller internal aortic diameter, as measured by the lumen perimeters of aortic cross sections (0.45±0.11 versus 0.58±0.05; P<0.05; Fig. 4D). Histological study with VVG staining revealed that a deficiency of TLR2 protected from aortic dilation and elastin degradation in media and exhibited a significant increase in the elastin content of aortas compared to that from WT animals (Fig. 4F).
3.4. Targeting of TLR2 reduced the expression of TLR2 and TLR2 ligands in vascular tissue
Periaortic application of CaCl2 resulted in a time-dependent increase of TLR2+ cells in AAA and targeting TLR2 reduced the accumulation of TLR2+ cells in aortic tissues (Fig. 5A). Furthermore, TLR2+ cells co-localized with infiltrating CD11b+ macrophages in adventitia of the injured aortas, and the expression of TLR2 on CD11b+ macrophages was up-regulated after CaCl2 application (Fig. 5B). The HMGB1 bound to TLR2 in the adventitia and outer media of aortic walls from AAA aortas (Fig. 5C), but was not detected in the control aorta (data not shown), indicating that this endogenous TLR2 ligand interacted with TLR2. CaCl2-induced vascular injury resulted in a time-dependent increase of HMGB1 expression (Fig. 5D), and blocking TLR2-suppressed HMGB1 expression (Fig. 5E). Additionally, the expression of HSP70 and S100A8 was increased, and targeting of TLR2 reduced CaCl2-stimulated expression of these DAMPs in aneurismal tissue measured by immunostaining (Fig. S1A and C in Supporting information) and Western blot (Fig. S1B and D).
Figure 5.

Targeting TLR2 attenuates expression of TLR2 and endogenous ligands in CaCl2-injured vascular tissue. (A) A time-dependent increase of TLR2+ cells was found in CaCl2-injured vascular tissue (n=5/time point/group). Targeting TLR2 reduced TLR2+ cells in adventitia (6 weeks, n=8; 12 weeks, n=10). (B) TLR2+ cells colocalized with CD11b+ macrophages in adventitia. DNA (top left, DAPI, blue); TLR2 (top right, green); CD11b (bottom left, red); merge of TLR2, CD11b and DNA (bottom right); yellow arrowheads indicate TLR2+/CD11b+/adventitial macrophages. (C) Colocalization (yellow) of HMGB1 (red) and TLR2 (green) was detected by immunofluorescence (arrowheads). (D) A time-dependent increase in HMGB1 expression was found in CaCl2-injured vascular tissue (n=5/time point/group). (E) Blocking TLR2 attenuated HMGB1 expression (n=6). Data are means±SD. #P<0.05, ##P<0.01, ###P<0.001, compared to sham group. ⁎P<0.05, ⁎⁎⁎P<0.001, compared to CaCl2 group.
3.5. Blocking TLR2 reduced the infiltration of inflammatory cells and cytokines in vascular tissue
CaCl2 stimulated an increased CD11b+ macrophage and CD4+ T cell infiltration in aortic tissue, mainly in the outer aortic wall (Fig. S2A and B). Moreover, the levels of MCP-1, TGF-β1, IL-10, IL-6, IFN-γ and IL-17 A (Figs. S2C, D and S3A) were increased. The phosphorylation of transcription factors, including NF-κBp65, AP-1 (c-Jun, Fig. S3B), and STAT1/3 (Fig. S3C), were up-regulated in CaCl2-damaged aortic tissue. However, systemic administration of TLR2ab reduced the infiltration of inflammatory cells and the expression of cytokines, as well as the activation of these transcription factors. These data suggest that TLR2 signaling has a crucial role in chronic inflammation in CaCl2-induced AAA.
3.6. Targeting TLR2 inhibited the Ang II-induced AAA formation
To determine the relationship between TLR2 activation and AAA pathogenesis, an established Ang II-induced AAA model was generated in Apoe−/− mice. We found that a four-week infusion of Ang II in Apoe−/− mouse resulted in the formation of suprarenal aortic aneurysm (Fig. 6A). Blocking TLR2 significantly not only decreased animal death from ruptured aortic aneurysm compared to the untreated AAA animals (2/16 versus 10/24, P<0.03; Fig. 6B), but reduced the incidence rate of AAA (7/16 versus 20/24, P<0.01), including type I-IV AAA, as indicated previously (Fig. 6C). There was also a decrease in the maximal aortic diameter in TLR2ab-treated mice compared to the mice infused with Ang II alone (1.53±0.35 versus 2.16±0.39, P<0.01; Fig. 6D). Furthermore, blocking TLR2 reduced the activity of MMP-2 in the aortic tissues collected from the thoracic aorta, suprarenal and infrarenal aorta and MMP-9 in all selected aortic parts (Fig. 6E). Infusion of Apoe−/− mice with Ang II enhanced the aortic lumen and wall thickness, resulted in the degradation of elastin and deposition of extracellular matrix (Fig. 6F), and promoted thrombus and inflammatory infiltration in the aortic wall, as reported27. However, TLR2 blocking protected from the destruction of elastic media and inhibited the aortic expansion. Importantly, antagonism of TLR2 activity significantly reduced the Ang II-induced ROS production in vascular tissue (Fig. 6F). Moreover, blocking TLR2 activity attenuated inflammatory responses (Fig. S4), inhibited the phosphorylation of c-Jun N-terminal kinase (JNK), NF-κBp65, AP-1 and STAT1/3 in Ang II-induced AAA (Fig. S5). These data demonstrate that the therapeutic efficacy of TLR2 blocking against the formation of AAA is independent of the animal models.
Figure 6.

Targeting TLR2 protects against Ang II-induced AAA formation. AAA was generated by infusion of Ang II (1000 ng/kg/min) in Apoe−/− mouse. (A) Typical suprarenal aneurysms were induced by Ang II (arrows). (B) Targeting TLR2 reduced animal death from Ang II-induced aneurysm rupture. Survival rate was analyzed by Kaplan-Meier. (C) Targeting TLR2 reduced the incidence of AAA. (D) Maximal abdominal aortic diameter in Apoe−/− mice with Ang II infusion. (E) MMP-2/9 activities were evaluated with gelatin zymography in aortic arch (a), thoracic (t), suprarenal (s) and infrarenal aorta (i) from the Apoe−/− mice infused with Ang II. The qualitative analysis was summarized from four separate experiments. (F) Representative H&E staining, VVG, picrosirius red and dihydroethidium (DHE) for detecting ROS in the aortic tissue. Data are means±SD. #P<0.05, ##P<0.01, compared to sham group. ⁎P<0.05, ⁎⁎P<0.01, compared to Ang II group.
4. Discussion
Our studies indicate that the innate immune receptor TLR2 plays an essential role in the development and progression of AAA. Our finding is supported by the following facts. First, the expression of TLR2 and its endogenous ligands were up-regulated in human aneurysm compared to control aortic tissue. Moreover, an enhanced interaction between TLR2 and its ligand has been identified in human AAA tissue, and blocking TLR2 in vitro attenuated TLR2-mediated expression or activation of inflammatory factors and MMP-2/9 in AAA vascular tissue. Second, functionally or genetically blocking TLR2 attenuates AAA formation and causes established AAA regression through attenuating the CaCl2- or Ang II-enhanced MMP-2/9 activation and vascular remodeling. Third, CaCl2- or Ang II-induced vascular injury and inflammatory response promoted the expression of TLR2 and several endogenous TLR2 ligands in aortic tissue. Blocking TLR2 decreased the expression and interaction of TLR2 with its endogenous TLR2 ligands, and thus, interrupted the positive feedback loop between TLR2 activation and inflammatory response. Fourth, blocking TLR2 attenuated the recruitment of CD11b+ macrophages and CD4+ T cells, expression of inflammatory factors, and activation of transcription factors NF-κB, AP-1 and STAT1/3. Indeed, TLRs expressed on or in human blood vessels have been found to mediate selective immune responses and impose vessel-specific risk for inflammatory vascular disorders12; the interactions between oxidized lipoproteins and TLRs promote the development of atherosclerosis through the initiation of a sterile inflammatory response28. Aoyama et al.29 report recently that TLR2 plays a fundamental role in periodontal bacteria-accelerated AAA development. These authors and we obtained similar results regarding the role of TLR2 signaling in the regulation of AAA development by using different animal models of AAA. Moreover, because our work demonstrates that targeting TLR2 by an anti-TLR2 antibody induces the regression of AAA, TLR2 may be a potential target for the development of therapeutics against AAA.
Two in vivo AAA models were utilized for studying the role of TLR2 activity in the development and progression of AAA. Periarterial application of CaCl2 or infusion of Ang II is a convenient and reliable model for induction of AAA in mice30. Underlying mechanisms for the models are mainly due to the accumulation of intracellular calcium and free radicals after exposure to a high concentration of calcium or Ang II, which are crucial for initiating inflammation to cause severe vascular injury and tissue necrosis31. The injured or necrotic vascular tissue liberates several intracellular factors functioning as DAMPs to stimulate PRRs, such as TLR2, on the immune and residue cells to lead to a chronic inflammation in vascular tissue. This DAMP/TLR2 interaction–induced chronic inflammation is crucial in maintaining a persistent activation of MMPs, extracellular matrix degradation and fibrosis, apoptosis of vascular smooth muscle, and vascular remodeling in AAA24, 32. According to Matzinger’s ‘danger’ mode, the crucial event controlling the initiation of inflammation by Ang II or calcium was not infection, but the production of danger signals from stressed, damaged, and/or dying cells in the local tissue33. Indeed, recent studies clearly indicate that the endogenous triggers are equally effective at activating a reparative and protective response, including the activation of the immune system through interaction with PRRs such as TLRs34. Satoh et al.35 demonstrated that Ang II increases the expression of the TLR ligand cyclophilin A in smooth muscle and that cyclophilin A mediates Ang II-induced AAA formation by inducing ROS accumulation, recruiting inflammatory cells and activating MMP-2. An increase in the elastin content of aortic tissue in AAA mice treated with an anti-TLR2 antibody indicate that blocking TLR2 signaling attenuates the CaCl2-induced elastin degradation by inhibiting inflammation and particularly, MMP-2/9 enzyme activity, and also promotes a regenerative and scarless repair in AAA vascular tissue36. It needs pointing out that several DAMPs released from damaged tissues including small HSPs and binding immunoglobulin protein (bip) display anti-inflammatory response or favor the resolution of inflammation that are named as RAMPs8, 9. However, because the study regarding how these DAMPs can act as the RAMPs is in its infancy, further experiments are necessary to explore how these RAMPs interact with what PPR to negatively regulate inflammation and promote immunological homeostasis.
Blocking of TLR2 provides a tremendous protective effect against animal death from the ruptured aortic aneurysm caused by Ang II. These results clearly indicate that chronic blockade of TLR2 activity diminishes Ang II-induced inflammation, reverses a massive destruction of elastin and promotes the reconstruction of the aneurysm wall. However, because inhibition of matrix degradation seems insufficient to result in the regression of AAA, other mechanisms may involve the reconstruction of the aneurysm wall by blocking TLR2. Indeed, TLR2 blocking attenuates inflammation and tissue fibrosis23, while less fibrosis before surgery may enable faster regression after surgery in patients with aortic stenosis37. Using TLR2- and TLR4-neutralizing antibodies, we have recently demonstrated that blocking TLR2 attenuates the development of bleomycin- and silica-induced pulmonary fibrosis, but blocking TLR4 promotes the development of pulmonary fibrosis23, 38. We also found that blocking TLR2 attenuates the tumor metastasis, but blocking TLR4 promotes tumor metastasis24, 39. Similarly, blocking TLR2 prompts but blocking TLR4 attenuates the resolution of chronic inflammation and protects against or aggravates doxorubicin-induced cardiovascular remodeling and dysfunction15. These observations indicate that TLR2 or TLR4 signaling has a differentiate regulation in the pathogenesis of tissue fibrosis, cancers and cardiomyopathy. Interestingly, we have found that targeting TLR4 can attenuate the progression of AAA in CaCl2-induced AAA through a similar mechanism to current study (unpublished observation).
However, Owens et al.40 reported recently that MyD88 deficiency attenuates Ang II-induced abdominal aortic aneurysm formation independent of signaling through toll-like receptors 2 and 4. This finding seems conflicting with the finding in the current study. Nevertheless, the AAA model used by these authors was induced by a deficiency of adaptor molecule MyD88 in leukocytes whereas AAA model used by current study was induced by a deficiency of either TLR2 or TLR4 in whole animal. Therefore, these different observations reminder us how TLR2 activity interferes with the capacity of vascular repair and regeneration and how blocking TLR2 promotes the reconstruction of aneurysm wall from the injured AAA are still open and need further investigation.
In summary, our study may have significant clinical implications. In contrast to several therapeutic agents, such as inhibitors of MMP and anti-inflammatory agents16, 41, which can promote a regression of AAA, targeting TLR2, which is a membrane receptor and contributes to multiple aspects in AAA development, will probably provide greater therapeutic benefit and easy access.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81030056 and 81400286). Dr. Xiaowei Zhang is supported by a grant from Basic Research Program of Institute of Materia Medica (No. 2014RC04). We are indebted to Dr. Koichi Yoshimura of Yamaguchi University School of Medicine of Japan for his expert advice in the preparation of CaCl2-induced AAA models.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.apsb.2015.03.007.
Appendix A. Supplementary materials
Supplementary data
References
- 1.Wassef M, Upchurch GR Jr., Kuivaniemi H, Thompson RW, Tilson MD., 3rd Challenges and opportunities in abdominal aortic aneurysm research. J Vasc Surg. 2007;45:192–198. doi: 10.1016/j.jvs.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Diehm N, Dick F, Schaffner T, Schmidli J, Kalka C, Di Santo S. Novel insight into the pathobiology of abdominal aortic aneurysm and potential future treatment concepts. Prog Cardiovasc Dis. 2007;50:209–217. doi: 10.1016/j.pcad.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Baxter BT, Terrin MC, Dalman RL. Medical management of small abdominal aortic aneurysms. Circulation. 2008;117:1883–1889. doi: 10.1161/CIRCULATIONAHA.107.735274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuivaniemi H, Platsoucas CD, Tilson MD 3rd. Aortic aneurysms: an immune disease with a strong genetic component. Circulation. 2008;117:242–252. doi: 10.1161/CIRCULATIONAHA.107.690982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jagadesham VP, Scott DJ, Carding SR. Abdominal aortic aneurysms: an autoimmune disease? Trends Mol Med. 2008;14:522–529. doi: 10.1016/j.molmed.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 7.Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in Apoe-/- mice. J Clin Invest. 2008;118:183–194. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shields AM, Panayi GS, Corrigall VM. Resolution-associated molecular patterns (RAMP): RAMParts defending immunological homeostasis? Clin Exp Immunol. 2011;165:292–300. doi: 10.1111/j.1365-2249.2011.04433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shields AM, Thompson SJ, Panayi GS, Corrigall VM. Pro-resolution immunological networks: binding immunoglobulin protein and other resolution-associated molecular patterns. Rheumatology. 2012;51:780–788. doi: 10.1093/rheumatology/ker412. [DOI] [PubMed] [Google Scholar]
- 10.Hou B, Reizis B, Defranco AL. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29:272–282. doi: 10.1016/j.immuni.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimizu K, Shichiri M, Libby P, Lee RT, Mitchell RN. Th2-predominant inflammation and blockade of IFN-γ signaling induce aneurysms in allografted aortas. J Clin Invest. 2004;114:300–308. doi: 10.1172/JCI19855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pryshchep O, Ma-Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel-specific toll-like receptor profiles in human medium and large arteries. Circulation. 2008;118:1276–1284. doi: 10.1161/CIRCULATIONAHA.108.789172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michelsen KS, Doherty TM, Shah PK, Arditi M. TLR signaling: an emerging bridge from innate immunity to atherogenesis. J Immunol. 2004;173:5901–5907. doi: 10.4049/jimmunol.173.10.5901. [DOI] [PubMed] [Google Scholar]
- 14.Shishido T, Nozaki N, Takahashi H, Arimoto T, Niizeki T, Koyama Y. Central role of endogenous toll-like receptor-2 activation in regulating inflammation, reactive oxygen species production, and subsequent neointimal formation after vascular injury. Biochem Biophys Res Commun. 2006;345:1446–1453. doi: 10.1016/j.bbrc.2006.05.056. [DOI] [PubMed] [Google Scholar]
- 15.Ma Y, Zhang X, Bao H, Mi S, Cai W, Yan H. Toll-like receptor (TLR) 2 and TLR4 differentially regulate doxorubicin induced cardiomyopathy in mice. PLoS One. 2012;7:e40763. doi: 10.1371/journal.pone.0040763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshimura K, Aoki H, Ikeda Y, Fujii K, Akiyama N, Furutani A. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11:1330–1338. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 17.Chiou AC, Chiu B, Pearce WH. Murine aortic aneurysm produced by periarterial application of calcium chloride. J Surg Res. 2001;99:371–376. doi: 10.1006/jsre.2001.6207. [DOI] [PubMed] [Google Scholar]
- 18.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korshunov VA, Berk BC. Flow-induced vascular remodeling in the mouse: a model for carotid intima-media thickening. Arterioscler Thromb Vasc Biol. 2003;23:2185–2191. doi: 10.1161/01.ATV.0000103120.06092.14. [DOI] [PubMed] [Google Scholar]
- 21.Bevilacqua R, Benassi CA, Largajolli R, Veronese FM. Psychoactive butyrophenones: binding to human and bovine serum albumin. Pharmacol Res Commun. 1979;11:447–454. doi: 10.1016/s0031-6989(79)80008-9. [DOI] [PubMed] [Google Scholar]
- 22.Liu YY, Cai WF, Yang HZ, Cui B, Chen ZR, Liu HZ. Bacillus Calmette-Guérin and TLR4 agonist prevent cardiovascular hypertrophy and fibrosis by regulating immune microenvironment. J Immunol. 2008;180:7349–7357. doi: 10.4049/jimmunol.180.11.7349. [DOI] [PubMed] [Google Scholar]
- 23.Yang HZ, Cui B, Liu HZ, Chen ZR, Yan HM, Hua F. Targeting TLR2 attenuates pulmonary inflammation and fibrosis by reversion of suppressive immune microenvironment. J Immunol. 2009;182:692–702. doi: 10.4049/jimmunol.182.1.692. [DOI] [PubMed] [Google Scholar]
- 24.Yang HZ, Cui B, Liu HZ, Mi S, Yan J, Yan HM. Blocking TLR2 activity attenuates pulmonary metastases of tumor. PLoS One. 2009;4:e6520. doi: 10.1371/journal.pone.0006520. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Mosorin M, Juvonen J, Biancari F, Satta J, Surcel HM, Leinonen M. Use of doxycycline to decrease the growth rate of abdominal aortic aneurysms: a randomized, double-blind, placebo-controlled pilot study. J Vasc Surg. 2001;34:606–610. doi: 10.1067/mva.2001.117891. [DOI] [PubMed] [Google Scholar]
- 26.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saraff K, Babamusta F, Cassis LA, Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:1621–1626. doi: 10.1161/01.ATV.0000085631.76095.64. [DOI] [PubMed] [Google Scholar]
- 28.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by toll-like receptor 2. J Clin Invest. 2005;115:3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aoyama N, Suzuki J, Ogawa M, Watanabe R, Kobayashi N, Hanatani T. Toll-like eeceptor-2 plays a fundamental role in periodontal bacteria-accelerated abdominal aortic aneurysms. Circ J. 2013;77:1565–1573. doi: 10.1253/circj.cj-12-1191. [DOI] [PubMed] [Google Scholar]
- 30.Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2004;24:429–434. doi: 10.1161/01.ATV.0000118013.72016.ea. [DOI] [PubMed] [Google Scholar]
- 31.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weintraub NL. Understanding abdominal aortic aneurysm. N Engl J Med. 2009;361:1114–1116. doi: 10.1056/NEJMcibr0905244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DAMPs Bianchi ME. PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 35.Satoh K, Nigro P, Matoba T, O’Dell MR, Cui Z, Shi X. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II–induced aortic aneurysms. Nat Med. 2009;15:649–656. doi: 10.1038/nm.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Redd MJ, Cooper L, Wood W, Stramer B, Martin P. Wound healing and inflammation: embryos reveal the way to perfect repair. Philos Trans R Soc Lond B Biol Sci. 2004;359:777–784. doi: 10.1098/rstb.2004.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petrov G, Regitz-Zagrosek V, Lehmkuhl E, Krabatsch T, Dunkel A, Dandel M. Regression of myocardial hypertrophy after aortic valve replacement: faster in women? Circulation. 2010;122:S23–S28. doi: 10.1161/CIRCULATIONAHA.109.927764. [DOI] [PubMed] [Google Scholar]
- 38.Yang HZ, Wang JP, Mi S, Liu HZ, Cui B, Yan HM. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol. 2012;180:275–292. doi: 10.1016/j.ajpath.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 39.Yan J, Wang ZY, Yang HZ, Liu HZ, Mi S, Lv XX. Timing is critical for an effective anti-metastatic immunotherapy: the decisive role of IFNγ/STAT1-mediated activation of autophagy. PLoS One. 2011;6:e24705. doi: 10.1371/journal.pone.0024705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Owens AP, 3 rd, Rateri DL, Howatt DA, Moore KJ, Tobias PS, Curtiss LK. MyD88 deficiency attenuates angiotensin II–induced abdominal aortic aneurysm formation independent of signaling through toll-like receptors 2 and 4. Arterioscler Thromb Vasc Biol. 2011;31:2813–2819. doi: 10.1161/ATVBAHA.111.238642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyake T, Aoki M, Masaki H, Kawasaki T, Oishi M, Kataoka K. Regression of abdominal aortic aneurysms by simultaneous inhibition of nuclear factor κB and Ets in a rabbit model. Circ Res. 2007;101:1175–1184. doi: 10.1161/CIRCRESAHA.107.148668. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data