

Summary
The human deafness-pigmentation syndromes Waardenburg syndrome type 2a and Tietz syndrome are characterized by profound deafness but only partial cutaneous pigmentary abnormalities. Both syndromes are caused by mutations in MITF. To illuminate differences between cutaneous and otic melanocytes in these syndromes, their development and survival in heterozygous Microphthalmia-White (MitfMi‐wh/+) mice were studied and hearing function of these mice characterized. MitfMi-wh/+ mice have a profound hearing deficit, characterized by elevated ABR thresholds, reduced distortion product otoacoustic emissions, absent endocochlear potential, loss of outer hair cells, and stria vascularis abnormalities. MitfMi-wh/+ embryos have fewer melanoblasts during embryonic development than their wild-type littermates. Although cochlear melanocytes are present at birth, they disappear from the MitfMi-wh/+ cochlea between P1 and P7. These findings may provide insight into the mechanism of melanocyte and hearing loss in human deafness-pigmentation syndromes such as Waardenburg syndrome and Tietz syndrome, and illustrate differences between otic and follicular melanocytes.
Keywords: cochlea, development, melanocyte, intermediate cell, deafness
INTRODUCTION
The human syndromes Waardenburg syndrome (WS) type 2a (WS2a) and Tietz syndrome are dominantly-inherited deafness-pigmentation syndromes caused by mutations in MITF (Amiel et al., 1998, Tassabehji et al., 1994, Hughes et al., 1994, Smith et al., 2000). WS was first described as a syndrome combining pigmentary defects of the iris and of the hair of the head with congenital deafness and various facial abnormalities (Waardenburg, 1951). Patients with the WS2a subtype of WS2 have identified mutations in the gene MITF, comprising approximately 15% of all patients with the WS2 phenotype (Read and Newton, 1997). Tietz syndrome is a related dominant syndrome featuring deafness and generalized skin hypopigmentation (Tietz, 1963). The two Tietz syndrome pedigrees reported to date contain distinct mutations in the basic, DNA-binding region of the MITF gene (Amiel et al., 1998, Smith et al., 2000). Hence, MITF activity appears to be important both for proper pigmentation and for hearing function in humans. The presence of neural crest-derived melanocytes in the inner ear (Steel and Barkway, 1989, Steel et al., 1987, Hilding and Ginzberg, 1977) substantiates their role in hearing.
Mouse coat color mutants have been invaluable both for defining the genetic requirements of melanocyte development and for demonstrating the importance of melanocytes in the development of the auditory system. Experimental evidence supporting the importance of melanocytes for hearing function was obtained from an analysis of the murine coat color mutant viable dominant spotting (KitW‐v). The absence of melanocytes from the stria vascularis in KitW‐v/W‐v mice, together with the loss of endocochlear potential from the substantial majority of these mice, both strengthened the basis for the claim that strial melanocytes are derived from the neural crest and established the requirement for melanocytes in the maintenance of normal structure and function of the stria vascularis (Steel and Barkway, 1989). The signaling pathway that has been described linking activation of KIT with the enhanced phosphorylation, ubiquitination, and degradation of MITF (Hemesath et al., 1998, Wu et al., 2000) may account for the similarities between the pigmentary and hearing phenotypes observed in individuals with deafness-pigmentation syndromes attributed to mutations in KIT or MITF.
However, one perplexing problem in the field of human deafness-pigmentation syndromes is the disparity between the cutaneous pigmentation phenotype, which can be minimal, and the prevalence of deafness, which is often high. The extent of white spotting in WS2 is often minimal, limited to a white forelock, the iris, and/or small areas of the skin. In contrast, the reported incidence of congenital deafness in WS2 patients is 77% (Liu et al., 1995). Likewise, Tietz syndrome patients have a limited cutaneous hypopigmentation phenotype. However, they are uniformly deaf (Smith et al., 2000, Amiel et al., 1998, Tietz, 1963). One possibility accounting for these disparities is that the mutation in MITF affects the intrinsic survival capabilities of cutaneous melanocytes differently from that of otic melanocytes, even though both sets of melanocytes are of neural crest origin. Another possibility is that cutaneous and otic melanocytes remain equivalent in terms of their intrinsic properties and sensitivity to MITF dosage or dysfunction, but that the follicular environment in the skin and the substantially different environment of the cochlear lateral wall support the survival of MITF-deficient melanocytes in quite different ways.
In this report, we address the difference between the mild pigmentation phenotype and often severe hearing phenotype in WS2 and Tietz by analyzing melanocyte development and survival in the MitfMi-wh/+ mouse. Most mice with mutations in Mitf do not have a visible heterozygous phenotype, with homozygosity for a mutant Mitf allele required for a pigmentary aberration in the coat. However, a small set of Mitf mutants with heterozygous phenotypes, MitfMi‐or/+ and MitfMi-wh/+, have mild pigmentary phenotypes, rendering such mutants appropriate both phenotypically and genetically as models for these human conditions. Here we show that the cochlear structural abnormality previously described in MitfMi-wh/+ mice (Deol, 1970, Deol, 1967) is accompanied by a functional hearing deficit, similar to that described previously with KitW‐v/W‐v mice (Steel and Barkway, 1989). Although a limited number of melanoblasts migrate to and are incorporated into the MitfMi‐wh/+ stria vascularis during development, they fail to survive in the cochlear environment during early post-natal life. These findings provide additional information to implicate MITF as an important melanocyte survival factor (McGill et al., 2002) in the cochlear environment, and may account for the difference in severity between the cutaneous and cochlear pigmentary phenotypes in certain congenital disorders of pigmentation.
RESULTS
Auditory Brainstem Responses, Otoacoustic Emissions, and Endocochlear Potentials of MitfMi-wh/+ mice
C57BL6-MitfMi-wh/+ mice, in addition to possessing altered cochlear structure, have a grayish-brown coat color and ventral white spot which distinguish them from wild-type littermates and C57BL6-MitfMi-wh/Mi-wh homozygotes at an early age (Supplementary Figure 1). The work of Deol (Deol, 1970, Deol, 1967) established that cochlear structural abnormality was a feature of certain homozygous Mitf mutants as well as C57BL6-MitfMi-wh/+ mice. Specifically, the stria vascularis was described to be “abnormal in its entirety” (Deol, 1970), with “severe dedifferentiation and cellular migrations” noted in the cochlear duct and saccule in most ears (Deol, 1970). However, a formal assessment of hearing function was not performed. To determine the hearing function of MitfMi-wh/+ mice, we first measured the auditory brainstem responses (ABRs) as a function of age. ABRs were measured on C57BL6-MitfMi-wh/+ mice and their wild-type littermates at P18, P28, and P45. At P18, only 1/2 MitfMi-wh/+ mice tested exhibited a detectable ABR which was limited to the 4 kHz and 12kHz frequencies, with no measurable ABRs detected at frequencies < 4 kHz or at 24 kHz. 5/5 MitfMi-wh/+ mice tested at P28 demonstrated a measurable ABR at frequencies ranging from 4 kHz through 24 kHz, but the threshold for response was higher for MitfMi‐wh/+ mice throughout the frequency range tested. For example, the mean threshold shift for P28 MitfMi‐wh/+ mice at 12 kHz was 70 dB. At P45, no measurable ABRs were detected in 2/2 MitfMi‐wh/+ mice at ranges from 1 kHz through 24 kHz (Table 1, Figure 1A). Representative waveforms are shown in Figure 1B. At P28, the mean ABRs for the wild-type and MitfMi‐wh/+ mice differed significantly (Figure 1A and legend to Figure 1). Despite the significant threshold shift between wild-type and MitfMi‐wh/+ ABRs, the MitfMi‐wh/+ mice exhibited a Preyer reflex. Together, these results suggest that MitfMi‐wh/+ mice in the P18-45 age range have a profound sensorineural hearing deficit.
Table 1.
ABR thresholds of P18, P28, and P45 C57BL6-MitfMi-wh/+ (Mu) mice and C57BL6-+/+ (Wt) littermates at various frequencies (kHz).
| ABR thresholds | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | Genotype | Sex | 1kHz | 2kHz | 4kHz | 8kHz | 12kHz | 16kHz | 24kHz |
| H1-p28 | Wt | F | * | * | 35 | * | 20 | * | 33 |
| H3-p28 | Wt | F | * | * | 32 | 29 | 15 | 35 | 40 |
| H5-p28 | Wt | F | * | * | 41 | * | 30 | * | 35 |
| H7-p28 | Wt | F | * | * | 33 | 33 | 19 | 34 | 38 |
| H2-p28 | Mu | M | * | * | 100 | 92 | 96 | 95 | NR |
| H4-p28 | Mu | F | * | * | 95 | 92 | 85 | 90 | 104 |
| H6-p28 | Mu | M | * | * | 90 | * | 87 | * | 104 |
| H8-p28 | Mu | F | * | * | 98 | * | 94 | * | 104 |
| H9-p18 | Wt | F | NR | NR | 66 | * | 36 | * | 45 |
| H10-p18 | Mu | M | NR | NR | NR | * | NR | * | * |
| H11-p18 | Wt | M | 96 | 84 | 49 | * | 20 | * | 50 |
| H12-p18 | Mu | F | NR | NR | 95 | * | 90 | * | NR |
| H13-p18 | Wt | M | 94 | 80 | 40 | * | 26 | * | 47 |
| H10-p45 | Mu | M | NR | NR | NR | * | NR | * | NR |
| H11-p45 | Wt | M | 95 | 75 | 45 | * | 25 | * | 45 |
| H12-p45 | Mu | F | NR | NR | NR | * | NR | * | NR |
| H13-p45 | Wt | M | NR | 95 | 60 | * | 25 | * | 45 |
NR = No response observed at 105 dB SPL
* = Not recorded
Figure 1.
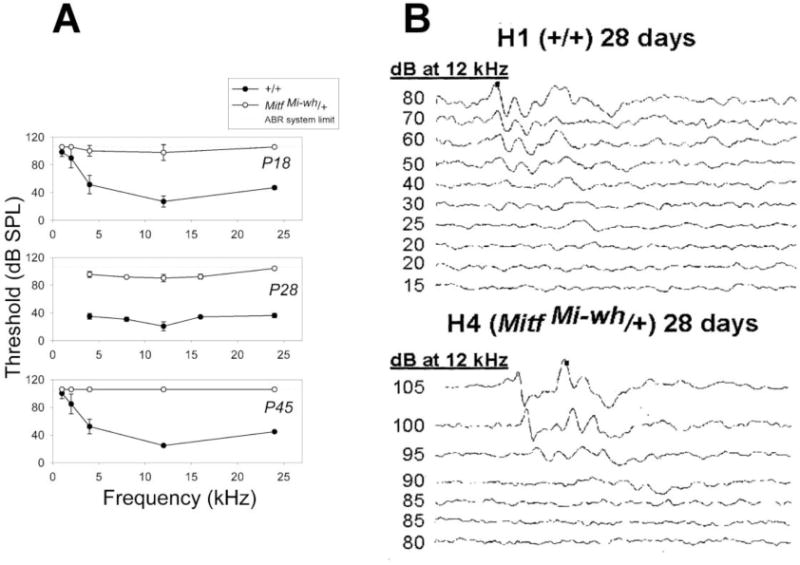
Auditory brainstem responses of +/+ and MitfMi-wh/+ mice. (A) Post-natal day 18 (P18) MitfMi‐wh/+ mice (○) exhibited a 63dB ABR threshold shift compared to wild-type (+/+) littermates (●) at 12 KHz frequency. Similarly, P28 MitfMi‐wh/+ mice exhibited a 70 dB ABR threshold shift compared to wild-type littermates at 12 KHz frequency. P45 MitfMi‐wh/+ mice had an absence of ABR waveforms, with an ABR threshold shift of >80 dB. Error bars signify standard deviation from the mean. A value of 105 dB SPL was used by convention for graphing and calculation purposes when no response was detected. P < 0.05 for P28 comparisons of MitfMi-wh/+ and wild-type responses at 8 kHz and 16 kHz. For comparison of responses at 4 kHz, 12 kHz, and 24 kHz, P < 1 × 106, 1 × 105, and 1 × 104 respectively by two-tailed Student’s t test. (B) Comparison of ABR waveforms of +/+ (H1, P28) and MitfMi‐wh/+ (H4, P28) littermates at 12 kHz. ABR waveforms are no longer evident at 20 dB in the wild-type mouse, but disappear at 85 dB in the MitfMi‐wh/+ littermate.
To provide information on outer hair cell function in MitfMi-wh/+ mice, distortion product otoacoustic emissions (DPOAE) were obtained on one MitfMi‐wh/+ P28 mouse and compared to that of 2 wild-type littermates. An example DPOAE from one wild-type is shown in Figure 2. The amplitude of the DPOAE (8.7 kHz, F2/F1=1.2, L1 = L2 – 10 dB) exceeded the noise floor from 50–80 dB SPL. In the MitfMi-wh/+ mouse, the DPOAE amplitude did not exceed the noise floor.
Figure 2.
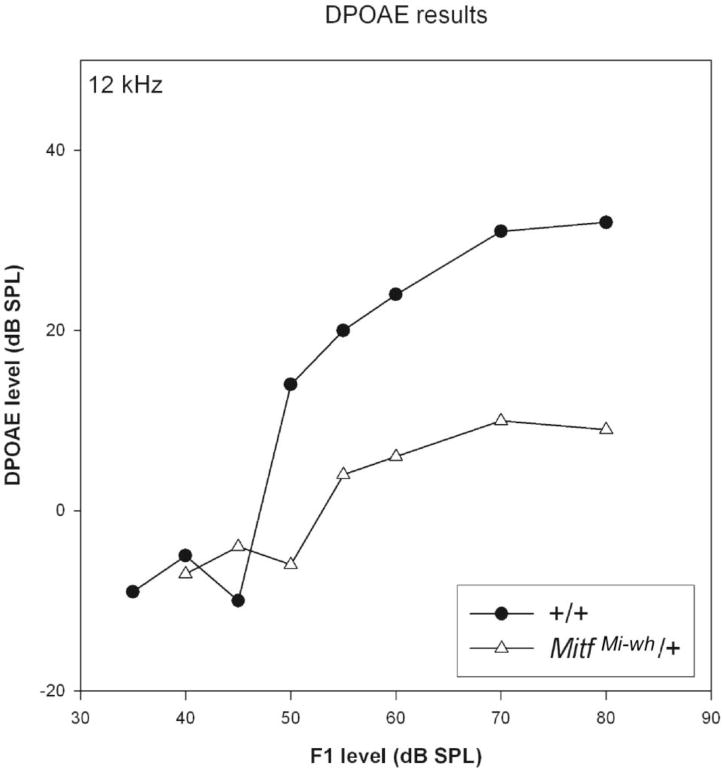
Distortion product otoacoustic emissions (DPOAE) for post-natal 29 (P29) wild-type (A) and MitfMi-wh /+ littermates (B). At 12kHz (geometric mean of the primaries), the 2F1 – F2 levels recorded from wild-type mouse are about 20–30 dB higher than from MitfMi-wh/+ littermate.
Endocochlear potentials (EPs) are absent in KitW-v/W-v mice (Steel and Barkway, 1989), which lack both coat pigmentation and strial intermediate cells. The ABR and DPOAE abnormalities we found in MitfMi‐wh/+ mice prompted us to evaluate the EP in these mice as well. EP measurements were obtained from 2 wild-type and 3 MitfMi‐wh/+ mice representing 2 litters at ages P34 and P36. While EP values were >100 mV in the 2 wild-type mice examined, repeated measurements on the 3 MitfMi‐wh/+ mice revealed the absence of an EP (Table 2, Supplementary Figure 2).
Table 2.
Endocochlear potentials of wild-type and MitfMi-wh/+ littermates.
| Mouse age | Genotype | EP (mV) |
|---|---|---|
| P34 | C57BL6-+/+ | 110 |
| P36 | C57BL6-+/+ | 130 |
| P34 | C57BL6-MitfMi-wh/+ | 0 |
| P34 | C57BL6-MitfMi-wh/+ | 0 |
| P36 | C57BL6-MitfMi-wh/+ | 0 |
Stria vascularis structure, cochlear hair cell integrity, and strial melanocyte distribution in MitfMi‐wh/+ mice
To determine the structure of the stria vascularis of MitfMi‐wh/+ mice at ages before and after the P34 and P36 timepoints where EP was absent, histologic sections were examined from cochleas of MitfMi‐wh/+ mice and their wild-type littermates at P28 and P37. The striae vascularis of wild-type mice at these stages (Figure 3A, Supplementary Figure 3) show condensed marginal cell nuclei present in a thin cell layer adjacent to the scala media, exhibiting extensive interdigitations that obscure the boundaries between cell layers. In contrast, the striae vascularis of MitfMi‐wh/+ mice exhibit enlarged marginal cell nuclei with prominent nuclear markings. Most prominently, the MitfMi‐wh/+ stria vascularis shows a sharp boundary between the marginal cell layer and basal cell layer that differs from the extensively interdigitated boundary present in the wild-type stria vascularis (Figure 3B, Supplementary Figure 3).
Figure 3.
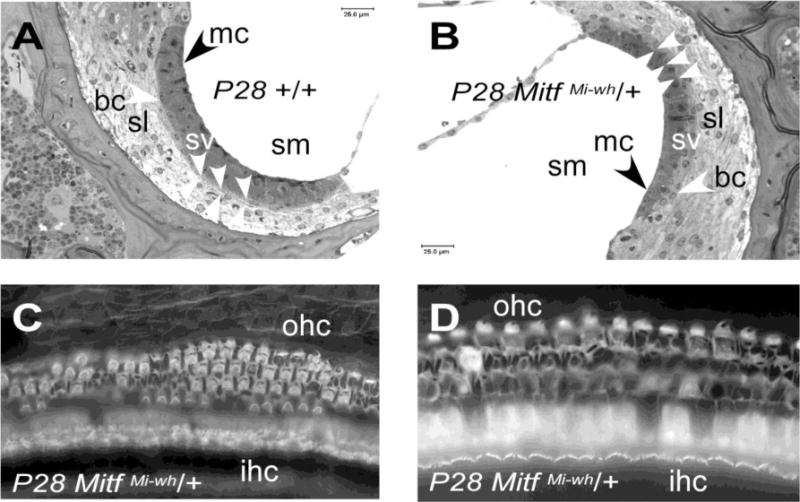
Morphology of +/+ and MitfMi-wh/+ stria vascularis and cochlear hair cells. Examples from P28 MitfMi-wh/+ mouse (B) and +/+ littermate (A) are shown. (A) Stria vascularis of P28 wild-type mouse. Arrowheads point to the condensed marginal cell (mc) nuclei and basal cell (bc) nuclei of the stria vascularis, with grouped arrowheads defining the boundary between the stria vascularis (sv) and the spiral ligament (sl). The boundaries between cell layers in the stria vascularis are obscured by faint interdigitations. (B) Stria vascularis of P28 MitfMi‐wh/+ mouse. In this panel, the grouped arrowheads delineate the sharp boundary present between the marginal and basal cell layers. The marginal cell (mc) nuclei are rounded rather than condensed. (C) Apex of P28 MitfMi‐wh/+ cochlea. Whole-mount phalloidin staining reveals no inner hair cell (IHC) loss, with sporadic loss of outer hair cells (OHCs). (D) Whole-mount phalloidin staining of base of P28 MitfMi‐wh/+ cochlea shows similar OHC loss with preservation of IHC layer. Other symbols: sm, scala media.
Whole-mount phalloidin staining of the P28 MitfMi‐wh/+ organ of Corti was performed to determine whether loss of cochlear hair cells correlated with degeneration of the stria vascularis and loss of auditory function. Inner hair cells were normal at P28 in all (n=5) MitfMi-wh/+ mice examined. In contrast, there was extensive loss of outer hair cells (OHCs) at P28. OHC loss increased progressively from apex (Figure 3C) to base (Figure 3D). Quantitative cytocochlear analysis revealed that OHC loss was 10.0(±4.1)% at approximately midway between the apex and base, increasing to 37.5(±33.9)% midway between the midpoint and the base, and reaching 76.1(±16.5)% loss at the cochlear base (Table 3, Supplementary Figure 4). The difference between the mean percent OHC loss at the midpoint and the base was highly significant (p<0.001, two-tailed t-test with unequal variances). Because of the role of OHCs as cochlear amplifiers (Kachar et al., 1986, Raphael and Altschuler, 2003, Fettiplace and Hackney, 2006, Ashmore, 2008), the selective loss of these OHCs, at least at P28, is the likely explanation of the marked difference in ABR threshold observed between wild-type and MitfMi-wh/+ mice.
Table 3.
Outer hair cell loss in MitfMi-wh/+ cochleas as a function of distance from apex.
| at 50% | at 75% | at 100% | ||||
|---|---|---|---|---|---|---|
| Mouse | %OHCs* | %dist | %OHCs** | %dist | %OHCs*** | %dist |
| Mitf2 | 10.9 | 48.4 | 66.8 | 79.8 | 86.1 | 100 |
| Mitf7 | 9.9 | 51 | 8.6 | 70.1 | 73.7 | 100 |
| Mitf8 | 11.2 | 48.5 | 6.1 | 72.7 | 48.8 | 100 |
| Mitf11 | 3.5 | 46.8 | 26.2 | 74.3 | 81.9 | 100 |
| Mitf14 | 14.7 | 50 | 79.8 | 76.9 | 90.2 | 100 |
| Average | 10.0 | 48.9 | 37.5 | 74.8 | 76.1 | 100.0 |
| Stdev | 4.1 | 1.6 | 33.9 | 3.7 | 16.5 | 0.0 |
The percentage of OHCs absent at the closest point to the 50% point between the apex and base was determined.
The percentage of OHCs absent at the closest point to the 75% point between the apex and base was determined.
The percentage of OHCs absent at the base was determined.
The studies previously described demonstrate hearing loss, greatest at higher frequencies, associated with degeneration of the stria vascularis and progressive loss of OHCs approaching the cochlear base. To determine whether melanocyte loss or dysfunction determines the cochlear phenotype in MitfMi‐wh/+ mice, we analyzed cochlear melanocyte development and survival in MitfMi‐wh/+ embryos and post-natal mice. Embryonic analysis of another dominant-negative Mitf heterozygote, Mitfmi/+, has revealed that fewer melanoblasts are present in the heterozygote relative to wild-type embryos at similar developmental stages (Hornyak et al., 2001). MitfMi-wh/+ mice were intercrossed with Dct-lacZ transgenic mice (Hornyak et al., 2001) to generate embryos for analysis. At E12.5, wild-type embryos contained numerous lacZ‐expressing melanoblasts in the otic vesicle (OV) region (Figure 4A). In contrast, MitfMi-wh/+ embryonic littermates contained significantly fewer lacZ-positive melanoblasts in the OV region (Figure 4B). Like Mitfmi/+ mice, MitfMi-wh/+ mice have fewer melanoblasts in the cephalic region at E10.5 and E12.5 (Figure 4C).
Figure 4.
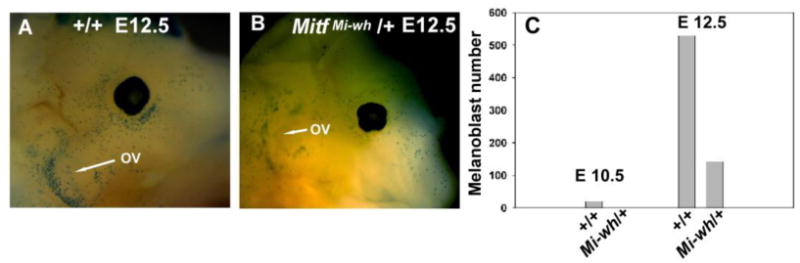
Embryonic melanoblasts in cephalic and otic vesicle regions of Dct‐lacZ transgenic +/+ and MitfMi-wh/+ embryos. (A) At E12.5, numerous Dct-lacZ+ melanoblasts are present in the periorbital region, throughout the cephalic region, and in the region of the otic vesicle (ov) in this wild-type transgenic embryo. (B) Fewer melanoblasts in the cephalic and otic vesicle (ov) region of E12.5 MitfMi‐wh/+ transgenic embryonic littermate. (C) Quantification of Dct-lacZ melanoblast numbers in cephalic region of MitfMi-wh/+ and +/+ embryonic littermates at E10.5 and E12.5. Melanoblasts were counted as described in Methods. P < 0.10 by Student’s two-tailed t test for difference between MitfMi-wh/+ and +/+ embryos at E12.5.
To investigate the presence and distribution of melanocytes in the post-natal cochlea, Dct-lacZ transgenic mice were also used. In wild-type cochleas, lacZ-expressing melanocytes describe an approximate one and three-quarters turn spiral from the cochlear base through the apex (Figure 5, Supplementary Figure 5). Compared to the wild-type cochlea, the P1 MitfMi‐wh/+ cochlea contains fewer lacZ-positive melanocytes than the wild-type cochlea. These melanocytes appear to be localized at the central stria (Supplementary Figure 6). P7 MitfMi‐wh/+ cochleas contain either very few (Figure 6A) or no (Figure 5, Supplementary Figure 5) lacZ-positive melanocytes. P14 and P28 MitfMi-wh/+ cochleas contain no lacZ-positive melanocytes (Figure 5, Supplementary Figure 5, Supplementary Figure 6).
Figure 5.
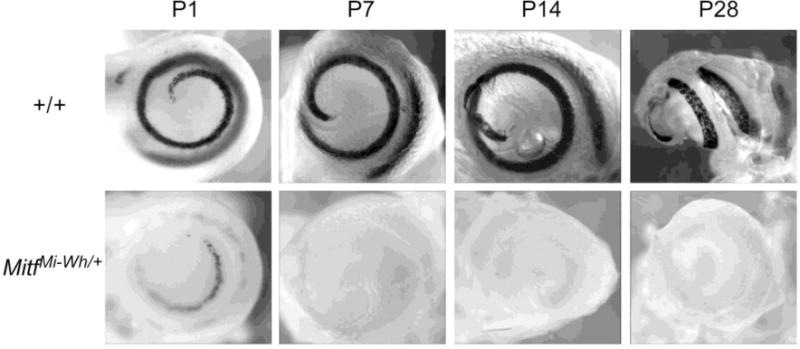
Stria vascularis melanocytes in neonatal +/+ and MitfMi-wh/+ mice. +/+ and MitfMi‐wh/Mi-wh mice were intercrossed with Dct-lacZ transgenic mice to permit visualization of cochlear melanocytes after staining for β-galactosidase activity. Wild-type cochlear melanocytes are present in a continuous, 1 3/4 turn configuration in whole-mount P1, P7, P14, and P28 cochleas (top row). MitfMi-wh/+ cochlear melanocytes (bottom row) are more sparse and distributed intermittently along the stria vascularis at P1, yet still present in a similar distribution as in the wild-type cochlea. In most P7cochleas and in older (P14 and P28) cochleas, MitfMi-wh/+ melanocytes (left) are no longer visible.
Figure 6.
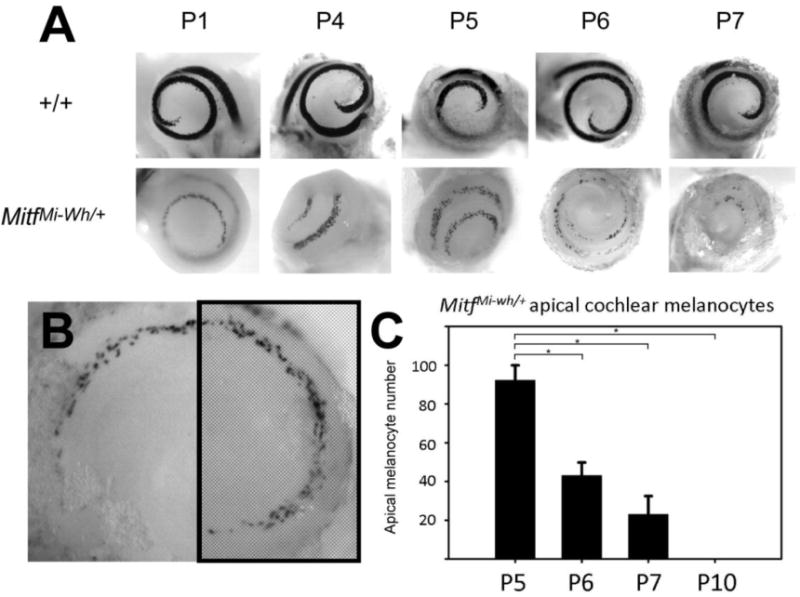
Selective melanocyte loss from early post-natal MitfMi-wh/+ cochleas. (A) Representative X-gal stained Dct-lacZ cochleas from P1-P7 +/+ and MitfMi-wh/+ mice show selective loss of cochlear melanocytes through post-natal day 7. (B) Representation of apical first half-turn used for counting MitfMi-wh/+ cochlear melanocytes. (C) Quantification of cochlear MitfMi-wh/+ melanocytes. Individual melanocytes in the area depicted by cross-hatching in (B) were counted. Progressive loss of cochlear melanocytes is observed from P5-P10. * indicates p < 0.005 for comparison. Error bars represent standard deviation from the mean.
The striking loss of melanocytes in the early post-natal cochleas of MitfMi-wh/+ mice prompted us to investigate whether the loss of melanocytes was progressive or abrupt. To determine this, Dct-lacZ-expressing cochleas were obtained at intermediate time points between P1 and P7 (Figure 6A). Dct-lacZ melanocytes were quantified in the first apical half-turn (Figure 6B) to characterize melanocyte loss. The results of this analysis (Figure 6C) suggest that MitfMi‐wh/+ cochlear melanocyte loss is progressive rather than abrupt, with a decrease in melanocyte number over the P5–P7 stages leading to a total absence by P10.
Discussion
Here we show that hearing dysfunction in MitfMi-wh/+ mice is due to an absence of melanocytes from the post-natal cochlea. We show that the absence of cochlear melanocytes in MitfMi-wh/+ mice following the immediate post-natal period results from a deficiency in melanocyte survival, not in melanocyte migration to the cochlea during embryogenesis. We also show that these mice have selective OHC loss, providing an explanation for the elevated ABR threshold, with the maintenance of a Preyer reflex, observed at P18, P28, and P45. Similar to KitW‐v/W-v mice (Steel et al., 1987, Steel and Barkway, 1989) and KitW‐s/W-s rats (Araki et al., 2002), MitfMi‐wh/+ mice lack strial melanocytes, a positive endocochlear potential, and feature structural abnormalities of the stria vascularis including distorted marginal and basal cell structure and loss of interdigitations. In contrast to these mice, the MitfMi-wh/+ mutant is largely pigmented at the ages we made our findings, and retains its pigment throughout adult life. The disparity between the cochlear and cutaneous pigmentary phenotypes suggests either that intrinsic differences between otic and cutaneous melanocytes account for their differential survival or that the cochlear and follicular environments differ in supporting the survival of melanocytes partially deficient in Mitf.
The generalized lightening of the coat color and ventral white belly spot of the MitfMi-wh/+ mouse results from a heterozygous, Ile212Asn mutation in the basic, DNA-binding region of Mitf (Steingrímsson et al., 1994). Since coat color dilution in mice, because of the follicular localization of their melanocytes, is analogous to hypopigmentation in humans, the pigmentary characteristics of the MitfMi-wh/+ mouse combine both the hypopigmentation described in Tietz syndrome patients as well as the localized spotting of Waardenburg syndrome. Our demonstration of the hearing abnormality present in these mice reinforces the notion of their similarity to these human syndromes, since Tietz syndrome patients are invariably deaf (Amiel et al., 1998, Smith et al., 2000) and approximately 75% of Waardenburg syndromes are deaf (Read and Newton, 1997). The two Tietz syndrome mutations in the MITF gene are also located in the basic region of the gene, whose basic region sequence is 100% conserved between mice and humans (Tachibana et al., 1994). One is an Asn210Lys substitution (Smith et al., 2000) and the other a delArg217 mutation, identical to the mutation responsible for another Mitf mutant allele, Mitfmi. However, the Mitfmi/+ mouse does not have coat color dilution, and has a relatively mild phenotype with small ventral white belly spots and no apparent cochlear abnormality (Deol, 1970). This difference in phenotype associated with an identical mutation in humans and mice indicates that the activity of MITF/Mitf is not exactly the same in each species. Patients with the WS2 phenotype and mutations in the basic region have also been described (Pingault et al., 2010, Chen et al., 2010).
The unique phenotypic characteristics of the MitfMi-wh/+ mouse may not be due exclusively to the specific mutation, but to interactions between mutant and wild-type splice isoforms of the Mitf-M protein, the transcribed form specific to the neural crest-derived melanocyte (Amae et al., 1998). Mitf-M, which dimerizes in vitro (Hemesath et al., 1994), is expressed in two distinct isoforms, (+) Mitf-M and (−) Mitf-M, distinguished by the presence or absence of a 6-amino acid stretch immediately N-terminal to the DNA-binding basic region domain (Hodgkinson et al., 1993, Steingrímsson et al., 1994). In the context of the Mi-wh mutation, the (+) isoform was found to exhibit an enhanced ability to stabilize DNA binding in a heterodimeric complex with the transcription factor TFE3 (Hemesath et al., 1994), an effect that was specific for this mutant form of the protein and not for the mi mutant. Mitf possesses an anti-proliferative effect, mediated by the (+) Mitf isoform (Bismuth et al., 2005), due to its induction of the p21CIP1 cyclin-dependent kinase inhibitor (Carreira et al., 2005) and p16INK4A (Loercher et al., 2005). Since a diminished number of melanoblasts are also found in Mitfmi/+ embryos (Hornyak et al., 2001), it is unlikely that preferential activity or stabilization of (+) isoform-containing heterodimers in MitfMi-wh/+ melanocytes contributes to the diminished number of melanoblasts noted during embryonic development. Perhaps isoform-specific regulatory activites, or neomorphic activities potentially attributable to the MitfMi-wh protein (Steingrímsson, 2010) contribute to the apparent, compromised environment-dependent survival that appears to be quite specific for heterozygotes. It is also possible that there is exacerbated loss of both cochlear and follicular melanocytes during the neonatal time period, with differential loss in the cochlea resulting in complete melanocyte absence with resulting hearing dysfunction, but sufficient retention in the hair follicle to establish a melanocyte stem cell population for recurrent hair pigmentation throughout successive hair cycles.
In summary, we have demonstrated that the profound hearing deficit exhibited by MitfMi‐wh/+ mice is due to the absence of melanocytes in the mature stria vascularis. Although any loss of cochlear melanocytes from humans with Waardenburg and Tietz syndromes may be highly variable and occur on a distinct time course, our findings nonetheless suggest strategies, including the delivery of growth factors to the cochlear environment or the introduction of melanocyte stem cells (Nishimura et al., 2002), that could be devised to maintain the presence of melanocytes in patients with these disorders to preserve cochlear structure and some degree of auditory function.
methods
Characterization, breeding, and analysis of embryonic and adult mice
C57BL/6-MitfMi-wh/ Mi-wh homozygotes were obtained from the colony of Dr. Lynn Lamoureux, Texas A&M College of Veterinary Medicine, and used to establish a colony for these experiments maintained on the C57BL/6 background by serial backcross. C57BL/6-MitfMi‐wh/+ progeny or their C57BL/6-+/+ littermates were used for determination of the auditory brainstem response (ABR), endocochlear potential (EP), distortion product otoacoustic emissions (DPOAE), and for histological and immunohistochemical studies. Tg(Dct-lacZ) transgenic mice (Hornyak et al., 2001) maintained on the CD1 background were intercrossed with C57BL6‐MitfMi-wh/+ or C57BL6-MitfMi-wh/ Mi-wh mice to obtain Tg(Dct-lacZ); MitfMi-wh/+ embryos or neonates for analysis of melanocyte presence in the cochlea and numbers in the cephalic region. Cephalic melanoblasts were counted as previously described (Hornyak et al., 2001). Animal care, handling, and procedures were approved by the Institutional Animal Care and Use Committees of the Henry Ford Health System, The University of Michigan, and the National Cancer Institute.
Noon of the day a vaginal plug was found was defined as E0.5, and embryos were harvested at various time points thereafter. Embryos and mice were genotyped either by inference from parental genotypes, by observation of neonatal skin or coat color, or, in select cases, by using PCR to amplify Mitf exon 7 from DNA purified from yolk sacs or tails. The primers utilized were:
Forward: 5′-GGC CGT TGT AGA ATG AAG TG-3′
Reverse: 5′-CCT GCC TCA AGC CCC AAG CTC-3′.
Amplicons were sequenced for MitfMi-wh mutation detection using sequencing primer MITFSEQF 5′- TCA TAA ATG AGG AGA CTC CAA AGG -3′. For detecting the presence of the Dct-lacZ transgene, PCR was used with primer pairs for detecting lacZ and RAPSYN as previously described (Hanley and Merlie, 1991).
Whole-mount staining and melanocyte quantification of embryos and cochleas
Embryos were microdissected and fixed in 0.4% paraformaldehyde (PFA) for periods of 2–4 hours, depending on their developmental stages, and washed in phosphate-buffered saline (PBS), pH 7.4. Embryos were stained in X-Gal buffer (2mM MgCl2, 0.02% NP-40, 2mM K3Fe(CN)6, 2mM K4Fe(CN)6 containing 0.05% X-Gal dissolved in dimethylformamide). After the reaction, the samples were washed and post-fixed in 4% PFA. For MitfMi-wh/+ embryos, cephalic melanoblasts were counted as previously described (Hornyak et al., 2001). To quantify post-natal cochlear melanocytes, cells from X-gal-stained photomicrographs were counted in the first apical half-turn as depicted in Figure 6B.
ABR measurement
Animals were anesthetized (mice: ketamine 65 mg/kg, xylazine 3.5 mg/kg, and acepromazine 2mg/kg). Body temperature was maintained through the use of water circulating heating pads and heat lamps. Additional anesthetic (ketamine and xylazine) was administered if needed to maintain anesthesia depth sufficient to insure immobilization and relaxation. ABRs were recorded in an electrically and acoustically shielded chamber (Acoustic Systems, Austin, TX, USA). Needle electrodes (Grass E2 platinum) were placed at vertex (active) and the test ear (reference) and contralateral ear (ground) pinnae. Tucker Davis Technologies (TDT) System II hardware and SigGen/BioSig software (TDT, Alachua, FL USA) were used to present the stimulus and record responses. Tones were delivered through a Beyer driver (Beyer Dynamic Inc., Farmingdale, NY, USA; Aluminum-shielded enclosure made in house), with the speculum placed just inside the tragus. Stimulus presentation was 15 millisecond tone bursts, with 1 millisecond rise/fall times, presented 10 per second. Up to 1024 responses were averaged for each stimulus level. Responses were collected for stimulus levels in 10 dB steps at higher stimulus levels, with additional 5 dB steps near threshold. Thresholds were interpolated between the lowest stimulus level where a response was observed, and 5 dB lower, where no response was observed.
DPOAE measurement
Animals were anesthetized as described above. The primary tones, F1 and F2, were set at a ratio of F2/F1 = 1.2. The intensity of F1 (L1) was varied in 5 or 10 dB steps, with the intensity of F2 (L2) held at 10 dB quieter than L1. The DPOAE was measured at 2F1 – F2. Tones were presented via two Beyer drivers (Beyer Dynamic Inc., Farmingdale, NY, USA; Aluminum-shielded enclosure made in house) connected through an Etymotic microphone (ER 10B+, Etymotic Research, Inc., Elk Grove Village, IL, USA). TDT System II hardware and SigGen/BioSig software were used to present the stimuli and record responses.
Recording of the EP
Animals were anesthetized as described above, and given a dose of glycopyrrolate (0.2 mg/kg). They were placed into a headholder. The external pinna was removed and soft tissue dissected away from the bulla. The ossicles and tympanic membrane were removed and some of the bulla wall was removed to allow clear visualization of the cochlea. A small opening was made in the otic capsule over the stria vascularis for penetration of a glass micropipette (filled with 1.5 M KCl) into scala media. The micropipette was inserted with a hydraulic microdrive. The electrode signals were amplified by a capacity-compensated dc preamplifier, and recorded using TDT hardware and “chart recorder” software written in-house. Animals were sacrificed at the end of the recording.
Supplementary Material
Supplementary Figure 1. Coat color phenotypes of MitfMi‐wh mutant mice. MitfMi‐wh/Mi-wh homozygotes (B) have a white coat due to absence of melanocytes from hair follicles. MitfMi-wh/+ heterozygotes have an intermediate phenotype characterized by a diluted, gray-brown coat (B) on a C57BL/6 background (B) along with an occasionally dorsal white spot (B) and, invariably, a ventral white depigmented spot or patch (A).
Supplementary Figure 2. Representative EP measurement of +/+ and MitfMi-wh/+ mice. Repeated advancing and withdrawal of the electrode revealed the expected EP of ~130 mV in a wild-type mouse at P36 (top panel). Similar measurement attempts with an age-matched MitfMi-wh/+ mouse showed absence of an endocochlear potential (bottom panel).
Supplementary Figure 3. Cochlear and hair cell morphology in +/+ and MitfMi-wh/+ mice. (A) Stria vascularis of P37 wild-type mice. Structure of P37 normal stria vascularis is similar to P28 wild-type stria vascularis in Figure 3, with condensed marginal cell (mc) nuclei and strial interdigitations. Grouped arrowheads designate the boundary between the stria vascularis (sv) and the spiral ligament (sl). (B) Stria vascularis of P37 MitfMi‐wh/+ mice. P37 MitfMi-wh/+ stria vascularis is thinner than wild-type P37 stria vascularis. A sharp, rather than interdigitated, boundary remains between the marginal and basal cell (bc) layers of the MitfMi-wh/+ stria vascularis (grouped arrowheads). Marginal cell (mc) nuclei are open, rather than condensed, in the MitfMi-wh/+ stria vascularis. (C) Detail of P28 MitfMi-wh/+ apical IHC morphology. (D) Detail of P28 MitfMi-wh/+ apical OHC morphology.
Supplementary Figure 4. Representative cytocochleograms of a MitfMi-wh/+ mouse at P28. The percentage of missing inner hair cells (IHC) or outer hair cells (OHC1-3 for separate layers, OHC combined) as a function of distance from the apex is indicated. (A) The percentage of missing cells in all hair cell categories as a function of distance from the apex is indicated. (B) The percentage of missing inner hair cells and outer hair cells (average) as a function of distance from the apex is indicated. (C) The percentage of missing cells in all hair cell categories as a function of fractional distance from the apex is indicated. (D) The percentage of missing inner hair cells and outer hair cells (average) as a function of fractional distance from the apex is indicated.
Supplementary Figure 5. Additional examples of X-gal-stained, Dct-lacZ wild-type and MitfMi-wh/+ cochleas at P1, P7, P14, and P28, depicting progressive and persistent loss of cochlear melanocytes in MitfMi-wh/+ post-natal mice.
Supplementary Figure 6. Cryosections of X-gal-stained, Dct-lacZ wild-type and MitfMi-wh/+ cochleas at P1, P7, P14, and P28, demonstrating the localization of Dct-lacZ-expressing melanocytes in the stria vascularis and their progressive loss from MitfMi-wh/+ striae.
Significance.
Melanocytes in the skin and hair follicle reveal their molecular and cellular secrets through the pigmentary phenotypes readily appreciated by even the casual observer. The characteristics of pigment cells in other organs, such as the inner ear, are not as immediately accessible. Studying human deafness-pigmentation syndromes, and their murine counterparts, provides an opportunity to compare and contrast cutaneous and otic melanocyte populations to understand their functions better. Here we correlate the auditory capability and cochlear melanocyte presence of the heterozygous Microphthalmia-White mouse, revealing a selective survival deficit of these melanocytes that provides insight into human Waardenburg and Tietz syndromes.
Acknowledgments
The technical assistance of Lisa Kabara is appreciated. We acknowledge Lynn Lamoreux for originally providing Mitf mutant mice. This work was supported by NIH R01-AR047951 (to T.J.H.) and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- AMAE S, FUSE N, YASUMOTO K, SATO S, YAJIMA I, YAMAMOTO H, UDONO T, DURLU YK, TAMAI M, TAKAHASHI K, SHIBAHARA S. Identification of a novel isoform of microphthalmia-associated transcription factor that is enriched in retinal pigment epithelium. Biochem Biophys Res Commun. 1998;247:710–5. doi: 10.1006/bbrc.1998.8838. [DOI] [PubMed] [Google Scholar]
- AMIEL J, WATKIN PM, TASSABEHJI M, READ AP, WINTER RM. Mutation of the MITF gene in albinism-deafness syndrome (Tietz syndrome) Clin Dysmorphol. 1998;7:17–20. [PubMed] [Google Scholar]
- ARAKI S, MIZUTA K, TAKESHITA T, MORITA H, MINETA H, HOSHINO T. Degeneration of the stria vascularis during development in melanocyte-deficient mutant rats (Ws/Ws rats) Eur Arch Otorhinolaryngol. 2002;259:309–15. doi: 10.1007/s00405-002-0480-z. [DOI] [PubMed] [Google Scholar]
- ASHMORE J. Cochlear Outer Hair Cell Motility. Physiol Rev. 2008;88:173–210. doi: 10.1152/physrev.00044.2006. [DOI] [PubMed] [Google Scholar]
- BISMUTH K, MARIC D, ARNHEITER H. MITF and cell proliferation: the role of alternative splice forms. Pigment Cell Res. 2005;18:349–59. doi: 10.1111/j.1600-0749.2005.00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARREIRA S, GOODALL J, AKSAN I, LA ROCCA SA, GALIBERT MD, DENAT L, LARUE L, GODING CR. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature. 2005;433:764–9. doi: 10.1038/nature03269. [DOI] [PubMed] [Google Scholar]
- CHEN H, JIANG L, XIE Z, MEI L, HE C, HU Z, XIA K, FENG Y. Novel mutations of PAX3, MITF, and SOX10 genes in Chinese patients with type I or type II Waardenburg syndrome. Biochem Biophys Res Commun. 2010;397:70–4. doi: 10.1016/j.bbrc.2010.05.066. [DOI] [PubMed] [Google Scholar]
- DEOL M. The relationship between abnormalities of pigmentation and of the inner ear. Proc Roy Soc Lond A. 1970;175:201–217. doi: 10.1098/rspb.1970.0019. [DOI] [PubMed] [Google Scholar]
- DEOL MS. The neural crest and the acoustic ganglion. J Embryol Exp Morphol. 1967;17:533–41. [PubMed] [Google Scholar]
- FETTIPLACE R, HACKNEY CM. The sensory and motor roles of auditory hair cells. Nat Rev Neurosci. 2006;7:19–29. doi: 10.1038/nrn1828. [DOI] [PubMed] [Google Scholar]
- HANLEY T, MERLIE JP. Transgene detection in unpurified mouse tail DNA by polymerase chain reaction. Biotechniques. 1991;10:56. [PubMed] [Google Scholar]
- HEMESATH TJ, PRICE ER, TAKEMOTO C, BADALIAN T, FISHER DE. MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature. 1998;391:298–301. doi: 10.1038/34681. [DOI] [PubMed] [Google Scholar]
- HEMESATH TJ, STEINGRÍMSSON E, MCGILL G, HANSEN MJ, VAUGHT J, HODGKINSON CA, ARNHEITER H, COPELAND NG, JENKINS NA, FISHER DE. microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994;8:2770–2780. doi: 10.1101/gad.8.22.2770. [DOI] [PubMed] [Google Scholar]
- HILDING DA, GINZBERG RD. Pigmentation of the stria vascularis. The contribution of neural crest melanocytes. Acta Otolaryngol. 1977;84:24–37. doi: 10.3109/00016487709123939. [DOI] [PubMed] [Google Scholar]
- HODGKINSON CA, MOORE KJ, NAKAYAMA A, STEINGRÍMSSON E, COPELAND NG, JENKINS NA, ARNHEITER H. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell. 1993;74:395–404. doi: 10.1016/0092-8674(93)90429-t. [DOI] [PubMed] [Google Scholar]
- HORNYAK TJ, HAYES DH, CHIU LY, ZIFF EB. Transcription factors in melanocyte development: distinct roles for Pax-3 and Mitf. Mech Dev. 2001;101:47–59. doi: 10.1016/s0925-4773(00)00569-4. [DOI] [PubMed] [Google Scholar]
- HUGHES AE, NEWTON VE, LIU XZ, READ AP. A gene for Waardenburg syndrome type 2 maps close to the human homologue of the microphthalmia gene at chromosome 3p12-p14.1. Nat Genet. 1994;7:509–12. doi: 10.1038/ng0894-509. [DOI] [PubMed] [Google Scholar]
- KACHAR B, BROWNELL WE, ALTSCHULER R, FEX J. Electrokinetic shape changes of cochlear outer hair cells. Nature. 1986;322:365–368. doi: 10.1038/322365a0. [DOI] [PubMed] [Google Scholar]
- LIU XZ, NEWTON VE, READ AP. Waardenburg syndrome type II: phenotypic findings and diagnostic criteria. Am J Med Genet. 1995;55:95–100. doi: 10.1002/ajmg.1320550123. [DOI] [PubMed] [Google Scholar]
- LOERCHER AE, TANK EM, DELSTON RB, HARBOUR JW. MITF links differentiation with cell cycle arrest in melanocytes by transcriptional activation of INK4A. J Cell Biol. 2005;168:35–40. doi: 10.1083/jcb.200410115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCGILL GG, HORSTMANN M, WIDLUND HR, DU J, MOTYCKOVA G, NISHIMURA EK, LIN YL, RAMASWAMY S, AVERY W, DING HF, JORDAN SA, JACKSON IJ, KORSMEYER SJ, GOLUB TR, FISHER DE. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell. 2002;109:707–18. doi: 10.1016/s0092-8674(02)00762-6. [DOI] [PubMed] [Google Scholar]
- NISHIMURA EK, JORDAN SA, OSHIMA H, YOSHIDA H, OSAWA M, MORIYAMA M, JACKSON IJ, BARRANDON Y, MIYACHI Y, NISHIKAWA S. Dominant role of the niche in melanocyte stem-cell fate determination. Nature. 2002;416:854–60. doi: 10.1038/416854a. [DOI] [PubMed] [Google Scholar]
- PINGAULT V, ENTE D, DASTOT-LE MOAL F, GOOSSENS M, MARLIN S, BONDURAND N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31:391–406. doi: 10.1002/humu.21211. [DOI] [PubMed] [Google Scholar]
- RAPHAEL Y, ALTSCHULER RA. Structure and innervation of the cochlea. Brain Res Bull. 2003;60:397–422. doi: 10.1016/s0361-9230(03)00047-9. [DOI] [PubMed] [Google Scholar]
- READ AP, NEWTON VE. Waardenburg syndrome. J Med Genet. 1997;34:656–65. doi: 10.1136/jmg.34.8.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH SD, KELLEY PM, KENYON JB, HOOVER D. Tietz syndrome (hypopigmentation/deafness) caused by mutation of MITF. J Med Genet. 2000;37:446–448. doi: 10.1136/jmg.37.6.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEEL KP, BARKWAY C. Another role for melanocytes: their importance for normal stria vascularis development in the mammalian inner ear. Development. 1989;107:453–463. doi: 10.1242/dev.107.3.453. [DOI] [PubMed] [Google Scholar]
- STEEL KP, BARKWAY C, BOCK GR. Strial dysfunction in mice with cochleo-saccular abnormalities. Hearing Research. 1987;27:11–26. doi: 10.1016/0378-5955(87)90022-0. [DOI] [PubMed] [Google Scholar]
- STEINGRÍMSSON E. Interpretation of complex phenotypes: lessons from the Mitf gene. Pigment Cell & Melanoma Research. 2010;23:736–740. doi: 10.1111/j.1755-148x.2010.00769.x. [DOI] [PubMed] [Google Scholar]
- STEINGRÍMSSON E, MOORE KJ, LAMOREUX ML, FERRÉ-D’AMARÉ AR, BURLEY SK, SANDERS ZIMRING DC, SKOW LC, HODGKINSON CA, ARNHEITER H, COPELAND NG, JENKINS NA. Molecular basis of mouse microphthalmia (mi) mutations helps explain their developmental and phenotypic consequences. Nat Genet. 1994;8:256–263. doi: 10.1038/ng1194-256. [DOI] [PubMed] [Google Scholar]
- TACHIBANA M, PEREZ-JURADO LA, NAKAYAMA A, HODGKINSON CA, LI X, SCHNEIDER M, MIKI T, FEX J, FRANCKE U, ARNHEITER H. Cloning of MITF, the human homolog of the mouse microphthalmia gene and assignment to chromosome 3p14.1-p12.3. Hum Molec Genet. 1994;3:553–557. doi: 10.1093/hmg/3.4.553. [DOI] [PubMed] [Google Scholar]
- TASSABEHJI M, NEWTON VE, READ AP. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet. 1994;8:251–255. doi: 10.1038/ng1194-251. [DOI] [PubMed] [Google Scholar]
- TIETZ W. A syndrome of deaf-mutism associated with albinism showing dominant autosomal inheritance. Am J Hum Genet. 1963;15:259–64. [PMC free article] [PubMed] [Google Scholar]
- WAARDENBURG PJ. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am J Hum Genet. 1951;3:195–253. [PMC free article] [PubMed] [Google Scholar]
- WU M, HEMESATH TJ, TAKEMOTO CM, HORSTMANN MA, WELLS AG, PRICE ER, FISHER DZ, FISHER DE. c-Kit triggers dual phosphorylations, which couple activation and degradation of the essential melanocyte factor Mi. Genes & Dev. 2000;14:301–312. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Coat color phenotypes of MitfMi‐wh mutant mice. MitfMi‐wh/Mi-wh homozygotes (B) have a white coat due to absence of melanocytes from hair follicles. MitfMi-wh/+ heterozygotes have an intermediate phenotype characterized by a diluted, gray-brown coat (B) on a C57BL/6 background (B) along with an occasionally dorsal white spot (B) and, invariably, a ventral white depigmented spot or patch (A).
Supplementary Figure 2. Representative EP measurement of +/+ and MitfMi-wh/+ mice. Repeated advancing and withdrawal of the electrode revealed the expected EP of ~130 mV in a wild-type mouse at P36 (top panel). Similar measurement attempts with an age-matched MitfMi-wh/+ mouse showed absence of an endocochlear potential (bottom panel).
Supplementary Figure 3. Cochlear and hair cell morphology in +/+ and MitfMi-wh/+ mice. (A) Stria vascularis of P37 wild-type mice. Structure of P37 normal stria vascularis is similar to P28 wild-type stria vascularis in Figure 3, with condensed marginal cell (mc) nuclei and strial interdigitations. Grouped arrowheads designate the boundary between the stria vascularis (sv) and the spiral ligament (sl). (B) Stria vascularis of P37 MitfMi‐wh/+ mice. P37 MitfMi-wh/+ stria vascularis is thinner than wild-type P37 stria vascularis. A sharp, rather than interdigitated, boundary remains between the marginal and basal cell (bc) layers of the MitfMi-wh/+ stria vascularis (grouped arrowheads). Marginal cell (mc) nuclei are open, rather than condensed, in the MitfMi-wh/+ stria vascularis. (C) Detail of P28 MitfMi-wh/+ apical IHC morphology. (D) Detail of P28 MitfMi-wh/+ apical OHC morphology.
Supplementary Figure 4. Representative cytocochleograms of a MitfMi-wh/+ mouse at P28. The percentage of missing inner hair cells (IHC) or outer hair cells (OHC1-3 for separate layers, OHC combined) as a function of distance from the apex is indicated. (A) The percentage of missing cells in all hair cell categories as a function of distance from the apex is indicated. (B) The percentage of missing inner hair cells and outer hair cells (average) as a function of distance from the apex is indicated. (C) The percentage of missing cells in all hair cell categories as a function of fractional distance from the apex is indicated. (D) The percentage of missing inner hair cells and outer hair cells (average) as a function of fractional distance from the apex is indicated.
Supplementary Figure 5. Additional examples of X-gal-stained, Dct-lacZ wild-type and MitfMi-wh/+ cochleas at P1, P7, P14, and P28, depicting progressive and persistent loss of cochlear melanocytes in MitfMi-wh/+ post-natal mice.
Supplementary Figure 6. Cryosections of X-gal-stained, Dct-lacZ wild-type and MitfMi-wh/+ cochleas at P1, P7, P14, and P28, demonstrating the localization of Dct-lacZ-expressing melanocytes in the stria vascularis and their progressive loss from MitfMi-wh/+ striae.