

Abstract
A simple and effective high-performance liquid chromatography with diode-array detection method coupled with a liquid-liquid extraction pretreatment has been developed for determining the pharmacokinetics and tissue distribution of a novel structurally modified derivative (8-acetamino-isocorydine) of isocorydine. According to the in vivo experiments data calculations by DAS 2.0 software, a two-compartment metabolic model was suitable for describing the pharmacokinetic of 8-acetamino-isocorydine in rats. 8-Acetamino-isocorydine was absorbed well after oral administration, and the absolute bioavailability was 76.5%. The half-life of 8-acetamino-isocorydine after intravenous and oral administration was 2.2 h and 2.0 h, respectively. In vivo, 8-acetamino-isocorydine was highly distributed in the lungs, kidney and liver; however, relatively little entered the brain, suggesting that 8-acetamino-isocorydine could not easily pass through the blood brain barrier. Our work describes the first characterization of the pharmacokinetic parameters and tissue distribution of 8-acetamino-isocorydine. The acquired data will provide useful information for the in vivo pharmacology of 8-acetamino-isocorydine, and can be applied to new drug research.
Abbreviations: AICD, 8-acetamino-isocorydine; AUC, area under concentration-time curve; F, absolute bioavailability; HPLC-DAD, high-performance liquid chromatography with diode-array detection; HPLC-UV, high-performance liquid chromatography coupled with ultraviolet detection; ICD, isocorydine; IS, internal standard; LC-ESI-MS/MS, high-performance liquid chromatography–electrospray ionization–tandem mass spectrometry; LLE, liquid-liquid extraction; LLOQ, lower limit of quantification; LOD, limit of detection; QC, quality control; RE, relative error; RP, reverse phase; RSD, relative standard deviation; SD, standard deviation.
KEY WORDS: Alkaloids, Pharmacokinetics, Tissue distribution, High-performance liquid chromatography with diode-array detection, 8-Acetamino-isocorydine
Graphical abstract
Pharmacokinetics and tissue distribution of isocorydine derivative 8-acetamino-isocorydine were investigated by HPLC-DAD coupling with a liquid-liquid extracted pretreatment method.

1. Introduction
Aporphine alkaloids belong to benzylisoquinoline alkaloids, existing in 20 families and more than 100 genera of plants, including Magnoliaceae, Menispermaceae, Euphorbiaceae, Lauraceae, Loganiaceae, Annonaceae, Aristolochiaceae, Berberidaceae, Papaveraceae, Ranunculaceae, Rutaceae, etc. These alkaloids share the common characteristic of a tetracyclic skeleton, albeit with different substituents. The aporphine template is associated with a wide range of biological activities, such as antioxidant, anti-platelet aggregation, anticonvulsant, anticancer antimalarial, antiprotozoal, antipoliovirus, cytotoxic and anti-Parkinson׳s disease. Because of their attractive biological activities, many studies have focused on the potential of aporphinoid alkaloids in drug development, and the anticancer activity of these compounds has become a hot pharmaceutical research area in recent years1, 2, 3, 4, 5, 6.
Isocorydine (ICD), an aporphine alkaloid, is widely present in many plants, including Dicranostigma leptopodum (Maxim) Fedde, which mainly distributed in the northwest of China7, 8. ICD was reported many biological activities, for example, the spasmolytic, antiplasmodial, antiarrhythmic, analgesic activity and vasodilative activity9, 10. In 2003, ICD hydrochloride was authorized by China State Food and Drug Administration (SFDA) to be commercialized (Chinese Drug Approval No. H53021977). It was listed as both a prescription drug and a medicare drug to cure diverse endogenous pain. Previous studies demonstrated that ICD prohibited the proliferation of hepatocellular carcinoma cell lines both in vitro and in vivo by inducing G2/M cell cycle arrest. In addition, ICD treatment led to a decrease of the percentage of CD133+ PLC/PRF/5 cells. Furthermore, ICD treatment dramatically decreased the tumorigenicity of SMMC-7721 and Huh7 cells. These findings indicate that ICD might be a potential therapeutic drug for the chemotherapeutic treatment of hepatocellular carcinoma11, 12. However, the short half-life, rapid elimination, and low tolerated dose (LD50=32.2 mg/kg in mice13) present limitations to its development as a novel chemotherapy14, 15. In order to improve the anti-cancer activity, increase the tolerated dose, and extend half-life of ICD, we modified the structure of ICD to obtain a series of derivatives. After screening research, we found that 8-amino-isocorydine possesses the better anti-cancer activity than isocorydine. 8-Amino-isocorydine is synthesized by introducing amino in C-8 site of isocorydine parent structure. But 8-amino-isocorydine is not stable in aqueous solution at room temperature. Even the weak oxidant sodium nitrite could oxidize 8-amino-isocorydine to 6a,7-dihydrogen-isocorydione, because the p-aminophenol segment in the structure of 8-amino-isocorydine could be easily oxidized in aqueous solution under the presence of hydrogen ion. To achieve the stabilized active compounds, based on the pro-drug theory, 8-acetamino-isocorydine (AICD, structure shown in Fig. 1) was synthesized through acetylating 8-amino-isocorydine with acetyl chlorine. The pharmacological experiments showed AICD has a good inhibitory effect on murine hepatoma H22-induced tumors. The inhibitory rate of H22 tumor was 52.71% after a dose 200 mg/kg/d of oral administration16. Thus, we selected AICD as a candidate for new drug development. The results suggested that the structural modifications of isocorydine at C-8 could significantly improve the biological activity of this alkaloid, indicating its suitability as a lead compound in the development of an effective anticancer agent16, 17.
Figure 1.
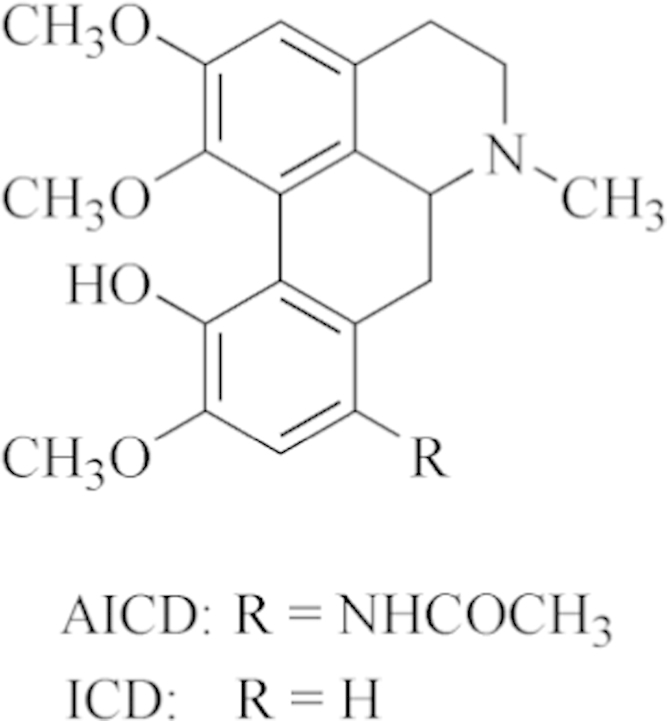
Chemical structures of 8-acetamino-isocorydine (AICD) and isocorydine (ICD).
In order to obtain more information on AICD in vivo, it is necessary to determine its pharmacokinetic properties and tissue distribution. A variety of methods have been used to study the pharmacokinetics of ICD, including high-performance liquid chromatography coupled with ultraviolet detection (HPLC-UV)18, 19 and high-performance liquid chromatography–electrospray ionization–tandem mass spectrometry (LC-ESI-MS/MS)20, 21. However, the use of high-performance liquid chromatography with diode-array detection (HPLC-DAD) is ideal for analyzing ICD in a biological matrix because it is simple, economical, applicable, and provides an acceptable sensitivity and stability. Thus, we used HPLC-DAD to analyze the plasma and tissue homogenates after oral and intravenous administration of AICD in rats. The results showed that AICD enhanced the tolerated dosage and moderately extended the half-life as compared with ICD, which implied that the dosage and the drug delivery time should be adjusted during the new drug research and the pharmacological investigation in vivo about AICD.
2. Materials and methods
2.1. Chemicals and reagents
D. leptopodum (Maxim) was picked from Pingliang, Gansu, China. It was identified by Professor Zhigang Ma (College of Basic Medical Science, Lanzhou University). The reference compound of ICD, used as the internal standard (IS), was extracted, isolated, and purified from D. leptopodum by repeated column chromatography and recrystallization. ICD was identified by Nuclear Magnetic Resonance spectrum and compared with the literature. AICD was synthesized in our laboratory. The purity of AICD and ICD was both more than 98%, as determined by HPLC-DAD analysis, based on the peak area normalization method. Methanol purchased from J&K Chemicals (USA) was of chromatographic grade. Analytical grade dichloromethane, diethylether, ethyl acetate, and ammonia were purchased from Rionlon Tianjin Chemical Co., Ltd. (Tianjin, China). Distilled and deionized water was used for the preparation of samples and solutions.
2.2. Instrumentation
An Agilent 1200 series liquid chromatography system (Agilent Technologies, Palo Alto, USA), equipped with a G1312A binary pump, a G1315B diode array detector performing wavelength scanning from 190 nm to 950 nm, a G1328B manual injector, and Agilent Chemstation software (version A.10.02) was used. The chromatographic separation of the analytes was performed on a SinoChrom ODS-BP C18 analytical column (250 mm×4.6 mm, 5 μm, Dalian Elite Analytical Instruments Co., Dalian, China) at 30 °C. Data analysis was performed on DAS 2.0 software (Drug and Statistics, Mathematical Pharmacology Professional Committee of China). An XK96-A Vortex Mixer (Xinkang Instrument Co., Ltd., Jiangsu, China) and TGL-16G Desk Centrifuge (Anting Instrument Co., Ltd., Shanghai, China) were used.
2.3. Animals
Wistar rats (male, 200–220 g) were obtained from the Animal Center of Gansu University of Traditional Chinese Medicine, Gansu, China (License No. SCXK (Gan) 2004-0006). Animals were bred in a breeding room with a temperature of 25 °C, relative humidity of 50%±10%, and a 12 h dark-light cycle. All of the animals had free access to water and rodent chow at all times, and all of the experimental animals were fed under the above conditions for one week. The night before experiments, the access to food was limited. Research was conducted in accordance with all institutional guidelines and ethics, and was approved by the Laboratories Institutional Animal Care and Use committee of the Gansu University of Traditional Chinese Medicine.
2.4. Preparation of standard solutions and quality control (QC) samples
A stock solution of AICD was prepared in methanol with a final concentration of 1.0 mg/mL. A series of standard solutions with concentration of 0.1, 0.5, 5.0, 15.0, 40.0, 120.0, 360.0 and 1100.0 μg/mL were obtained by further dilution of the stock solution with methanol. The standard solution of the IS was diluted with methanol to a concentration of 100.0 μg/mL. All the solutions were kept at −20 °C, and brought to room temperature before use.
To prepare a series of standard calibration samples, 10 μL of the standard solution at different concentrations were evaporated to dryness by a gentle stream of nitrogen, and 100 μL of the drug-free rat plasma or tissues homogenates was added. The mixture was then processed by a sample extraction procedure, which is described in Section 2.5. The final standard plasma and tissues concentrations were 0.01, 0.05, 0.5, 1.5, 4.0, 12.0, 36.0, and 110.0 μg/mL. QC samples with no IS were prepared as same as standard calibration solutions. QC samples containing AICD in plasma had concentrations of 0.5, 5.0, 50.0 and 100.0 μg/mL (very low, low, middle and high, respectively). All of these solutions were prepared fresh before use.
2.5. Sample pretreatment
In this study, a conventional liquid-liquid extraction (LLE) method was used to extract the AICD from biological samples (plasma, tissue homogenates). Biological samples were thawed at room temperature, and a 100 μL plasma sample was spiked with 10 μL IS standard solution (100 μg/mL). After briefly mixing, 400 μL of dichloromethane was added and mixed by vortex for 1 min. The sample was centrifuged at 10,000 rpm for 10 min at room temperature. The subnatant was collected and evaporated to dryness under a gentle stream of nitrogen at room temperature. The residue was reconstituted with 100 μL of the mobile phase, and 20 μL was injected to the HPLC system.
2.6. Chromatographic conditions
The mobile phase consisted of methanol with 0.02% aqueous ammonia (pH 7.3, 70:30, v/v) with a flow rate of 1 mL/min. Isocratic elution was used for all processes. The analytic column was a SinoChrom ODS-BP C18 (250 mm×4.6 mm, 5 μm). Chromatograms were monitored at 270 nm and the temperature of column was kept at 30 °C.
2.7. Method validation
All methods used on biological samples were validated following the guidelines of the US Food and Drug Administration (FDA)22. The validation parameters included selectivity, linearity, accuracy, precision, recovery and stability.
Selectivity was evaluated by comparing 6 individual blank plasma and spiked plasma samples. The purpose of this assay was to detect the level of interference in blank plasma at the retention time of the AICD and IS.
Calibration curves were obtained by linear regression of the peak area ratio of AICD to IS (Y-axis) and the standard concentration (0.01, 0.05, 0.5, 1.5, 4.0, 12.0, 36.0 and 110.0 μg/mL), which constituted the range of the calibration curve (X-axis). The calibration line was corrected by a weighted factor (1/X2), described as Y=a+bX. Each QC sample was recalculated using the calibration curve. Acceptable values were permitted to differ from the nominal value by ±15%, with the exception of the lower limit of quantification (LLOQ), which was permitted to differ by ±20%. The limit of detection (LOD) was determined as the lowest concentration of AICD in the biological samples that corresponds to 3 times of the baseline noise (S/N=3). The LLOQ was defined as the lowest concentration of AICD in plasma which caused a signal to noise ratio of 10 (S/N=10). The analyte concentration in the calibration curve of the LLOQ should be quantitatively determined with an acceptable precision of <20%.
Accuracy and precision were calculated using the QC samples. Five replicates of each sample were injected on three consecutive days, and the concentration was back calculated from the values on the calibration curve. The accuracy was expressed with a relative error (RE) and the precision was expressed as the relative standard deviation (RSD). The acceptable range of both the RE and RSD was 15%, while LLOQ did not exceed 20%.
The recovery of AICD was performed by comparing the peak area of the added AICD in plasma or tissue homogenates with the same amount of AICD added to the mobile phase. Four concentrations of QC samples (0.5, 5.0, 50.0 and 100.0 μg/mL) were evaluated in the recovery experiments (n=5 for each QC).
The stability of the sample was evaluated in different storage conditions. The amount of AICD in rat plasma was determined using the QC samples under the following 4 storage conditions: (1) after three freeze-thaw cycles (−20 °C/room temperature) with a freezing time of at least 12 h, followed by measurement of the change in the sample; (2) after the sample was maintained at room temperature for 4 h, followed by measurement of the change in the sample; (3) after the sample was maintained at −20 °C (storage conditions) for 40 d, followed by measurement of the change in the sample; and (4) after the sample was maintained in centrifuge tubes at room temperature for 24 h, followed by measurement of the change in the sample. All of these QC samples were prepared fresh, and the acceptable range of stability was less than 15% (n=5 for each QC).
2.8. Pharmacokinetical assay of AICD
Wistar rats (n=12, male) were randomly divided into two equal groups. One group was administrated AICD (100 mg/kg, i.v.) through the tail vein, whereas the other group was administered oral AICD (200 mg/kg) (the doses data were studied by the activity experiment16). The dose formulation of AICD was prepared in 0.9% sterile saline to contain 20 mg/mL or 40 mg/mL of AICD for i.v. or oral administration, respectively. All rats were fasted 12 h before administration. Rats were bled from the retro-orbital sinus by capillary tubes at the following time points: 0 (pre-dose), 0.033, 0.083, 0.25, 0.5, 0.83, 1.33, 2, 3, 4, 6 and 8 h for i.v. administration and 0 (pre-dose), 0.033, 0.083, 0.25, 0.5, 0.83, 1.33, 2, 3, 4, 6, 8, 10 and 12 h for oral administration. Each blood sample (300 μL) was collected in heparinized tubes and centrifuged at 4000 rpm for 15 min to obtain the plasma. The separated plasma was frozen at −20 °C before the assay.
2.9. Tissue distributed assay of AICD
Rats (n=24) were randomly divided into four equal groups, and each rat was administrated AICD (100 mg/kg) through the tail vein. The four groups of rats were euthanized 10 min, 1 h, 3 h or 6 h after administration. Dissected organs and tissues included the heart, liver, brain, lungs and kidneys from each rat. The organs were cleaned with physiological saline, which was absorbed by filter paper. The tissues were then weighed and homogenized in normal saline solution (250 mg/mL), and the obtained tissue homogenates were stored at −20 °C until analysis.
2.10. Calculations
All data was calculated using Microsoft Excel 2007 (Microsoft Co., USA) and OriginPro 8.0 (OriginLab Co., USA) software. The concentration-time curves and pharmacokinetic parameters of AICD were obtained through the DAS 2.0 software (Shanghai, China).
3. Results
3.1. Optimization of HPLC conditions
Methanol with 0.02% aqueous ammonia (pH 7.3, 70:30, v/v) was used as mobile phase and the analytic column was a SinoChrom ODS-BP C18 column (250 mm×4.6 mm, 5 μm); chromatograms were monitored at 270 nm and isocratic elution was used for all processes. Reverse phase (RP)–HPLC is the main tool used for the analysis of alkaloids. However, alkaloids can interact with free silanol groups on the surface of the silica particles which cause the chromatogram tailing in RP chromatography. For this reason, two new generation columns Waters XTerra MS C18 (250 mm×4.6 mm, 5 μm) and SinoChrom ODS-BP C18 (250 mm×4.6 mm, 5 μm) were selected for our experiments. These columns can be endcapped to decrease the number of surface silanols and ensure low silanol activity, thereby improving the ability to separate alkaloids. The pH value of the mobile phase was the most important factor in the RP-HPLC separation of alkaloid compounds, because the retention behavior of alkaloids can be greatly affected by the pH of the mobile phase. The alkaloids tend to easily gain a proton and become ionized in acidic conditions, because they are generally basic and nucleophilic compounds that have one or two nitrogen atoms. Thus, the retention behavior of alkaloids in an RP-column is strongly dependent on the composition of mobile phase. The retention time of aporphine alkaloids was increased due to decreases in their polarity as pH values increased. In contrast, some aporphine alkaloids were converted into their protonated form under acidic conditions, resulting in decreased retention time on C18 column.
Generally, the pKa values of aporphine alkaloids are 7.10–7.16 (ICD, 7.16)23, and introducing a methyl group at nitrogen atom often results in the formation of a strong base. Thus, adjusting the pH value of mobile phase above 7.16 may be useful to resolve the tailing observed in AICD and ICD. Triethylamine or ammonia aqueous solution was therefore added to the mobile phase in order to compete with those free silanol groups on the surface of the silica and overcome the peak broadening and tailing. The XTerra MS C18 column is more suitable when applied under the acidic mobile phase, unlike the SinoChrom ODS-BP C18 column. Thus, we selected the SinoChrom ODS-BP C18 column for our experiment. AICD was prepared by chemical modification of ICD and two compounds that possess the same basic chemical skeleton. As a result, ICD was selected as the IS in order to reduce error.
To improve the peak shape, acetonitrile and methanol were selected as the organic phase of mobile phase, and triethylamine and aqueous ammonia were selected as the additive for the aqueous phase. AICD and ICD have better solubility in methanol than acetonitrile, and the best chromatographic peak shape of the analyte was observed using a methanol and aqueous ammonia solution as the mobile phase. Overall, methanol with 0.02% aqueous ammonia (pH=7.3, 70:30, v/v) and a flow rate at 1 mL/min obtained the best peak shape, appropriate retention time (AICD at 4.0 min and ICD at 6.8 min), and good resolution. Thus, this mixture was selected as the mobile phase for the next pharmacokinetic and tissue distributional analysis.
3.2. Optimization of the sample preparation
Protein precipitation and LLE were used for the pretreatment of biological samples, but the results showed that protein precipitation was not suitable to extract AICD and ICD (IS) from biological samples, due to their low recovery and the interference of biological samples. Several extraction solvents were explored for LLE, such as dichloromethane, ethyl ether, ethyl acetate, and dichloromethane, and were shown to be superior to others in their high recovery of analytes. Further, the extracted samples were relatively purity and had few impurities in the chromatographic peaks or interference peaks. Based on the low number of impurity peaks, low background noise and rapid evaporation, we chose dichloromethane as the best extraction solvent for AICD and ICD (IS). Thus, the LLE method and dichloromethane were finally chosen for the biological sample preparation.
3.3. Validation of the HPLC assay
Selectivity of the method was evaluated by comparing the blank plasma and spiked plasma. Representative chromatograms of the blank plasma, blank plasma spiked with AICD and IS, and the real plasma are shown in Fig. 2. Tissue homogenates results are shown in Fig. S1A–E in Supporting information. There was no peak at the retention time of AICD and IS in the blank plasma and tissue homogenates. This suggested that there was no endogenous interference of the AICD and IS in plasma and tissues.
Figure 2.

Representative chromatograms for the pharmacokinetic investigation of AICD: (a) blank plasma sample; (b) standard solution in blank plasma including AICD and IS (ICD); (c) real plasma sample processed 50 min after oral administration of AICD (200 mg/kg); (d) real plasma sample processed 50 min after i.v. administration of AICD (100 mg/kg). Mobile phase: methanol with 0.02% aqueous ammonia adjusted to pH 7.3 (70:30, v/v) with a flow rate of 1 mL/min; column: SinoChrom ODS-BP C18 (250 mm×4.6 mm, i.d. 5 μm); detection wavelength: 270 nm; retention time: AICD at 4.0 min and ICD at 6.8 min.
The calibration curves for AICD were obtained by extraction of the blank plasma or five tissue homogenates (heart, liver, brain, lungs and kidneys). AICD in all matrices exhibited excellent linearity (all correlation coefficients exceeded 0.9900) over the linear ranges. Thus, all quantitative calculations used the results obtained from the blank plasma as the standard equation for the next investigation. Between 0.05 μg/mL and 110 μg/mL, the calibration curves exhibited a very good linear relationship. The linear equation is Y=0.101X −0.091 and the correlation coefficient was r=0.9985. The LLOQ (n=5, S/N=10) and LOD (n=5, S/N=3) of AICD in rat plasma were 0.10 μg/mL and 0.05 μg/mL, whereas in liver homogenates they were 0.18 μg/ml and 0.10 μg/mL. The linearity and selectivity were suitable for the study of pharmacokinetic and tissue distribution.
The accuracy was represented by the RE of QC samples, RE (%)=(Cdet−Cnom)/Cnom×100, where Cnom was calculated from the nominal concentration and Cdet was the mean value of the detected concentration. Precision was demonstrated as the RSD, using the formula, RSD (%)=(standard deviation (SD))/Cdet×100. The data of intra-day and inter-day accuracy and precision of AICD are shown in Table 1. As the data shows, the intra-accuracy ranged from −1.5% to 8.9%, and the inter-accuracy ranged from −2.8% to 6.9%. Additionally, the intra-precision ranged from −8.6% to 10.7% and the inter-precision ranged from −4.3% to 10.9%. The experimental results for intra-day and inter-day accuracy and precision were within the acceptable limits.
Table 1.
The intra-day and inter-day precision, accuracy and recovery of AICD in rat plasma and biological matrix (n=5).
| Sample matrix | QC concentration (μg/mL) | Intra-day |
Inter-day |
Recovery (mean±SD, %) | ||
|---|---|---|---|---|---|---|
| Accuracy (mean %) | Precision (mean %) | Accuracy (mean %) | Precision (mean %) | |||
| Plasma | 0.5 | 2.5 | 2.6 | 1.6 | 4.3 | 101.2±3.1 |
| 5 | 8.9 | 2.9 | 6.1 | 1.8 | 88.0±3.9 | |
| 50 | 7.4 | 1.2 | 3.2 | 3.3 | 86.4±1.1 | |
| 100 | 5.3 | 1.7 | 3.5 | 3.6 | 84.3±2.5 | |
| Heart | 0.5 | 2.8 | −0.3 | 3.9 | 1.4 | 88.9±2.1 |
| 5 | 1.7 | 4.6 | 3.8 | 7.6 | 81.2±1.4 | |
| 50 | 2.6 | 3.0 | 6.4 | 10.9 | 80.5±1.0 | |
| 100 | 2.1 | −2.3 | 3.0 | 7.1 | 79.8±3.1 | |
| Liver | 0.5 | 2.8 | −8.6 | 5.3 | −3.0 | 86.1±5.8 |
| 5 | 0.7 | 4.0 | 4.9 | 6.5 | 84.9±1.6 | |
| 50 | 0.2 | 1.7 | 5.9 | 0.6 | 82.5±1.3 | |
| 100 | 1.2 | −3.5 | 4.4 | 3.2 | 80.7±2.8 | |
| Brain | 0.5 | 2.5 | −7.6 | 4.4 | −2.0 | 96.2±2.5 |
| 5 | 3.3 | 3.6 | 3.6 | 4.5 | 86.4±1.9 | |
| 50 | 2.6 | −1.6 | 2.0 | 3.2 | 84.1±1.3 | |
| 100 | −1.5 | 3.7 | −2.8 | 5.1 | 82.6±3.2 | |
| Lung | 0.5 | 2.5 | 9.1 | 6.9 | 6.0 | 88.9±3.1 |
| 5 | 1.5 | 10.7 | 2.6 | 9.0 | 82.3±1.7 | |
| 50 | 5.0 | 9.8 | 2.7 | 2.9 | 80.4±1.5 | |
| 100 | 4.7 | 8.9 | 3.0 | 3.6 | 78.9±1.4 | |
| Kidney | 0.5 | 1.7 | −8.2 | 6.5 | −4.3 | 88.3±1.2 |
| 5 | 0.1 | 4.8 | 2.4 | −2.3 | 82.9±1.8 | |
| 50 | 2.1 | 2.9 | 0.8 | 2.0 | 80.6±1.2 | |
| 100 | 2.3 | −2.5 | 1.6 | 3.2 | 78.8±1.7 | |
The extraction recovery of AICD was determined by four QC (0.5, 5.0, 50.0 and 100.0 μg/mL) concentration samples. In both rat plasma and biological samples, the extraction recovery was greater than 78.8%. All the data are shown in Table 1. The data suggest that dichloromethane suitably recovers AICD in a biological matrix.
The stability data are shown in Table 2. The samples were stable when kept at room temperature for 4 h, after three freeze-thaw cycles, and after long-term placement for 40 d and 24 h in a room temperature environment. The results suggest that AICD undergoes negligible degradation under these circumstances. Thus, the method of sample storage at −20 °C was feasible and suitable for further pharmacokinetic and tissue distribution studies.
Table 2.
Stability of AICD in rat plasma (n=5).
| Sample matrix | QC concentration (μg/mL) | Freeze and thaw stability (mean±SD, %) | Short-term stability (mean±SD, %) | Long-term stability (mean±SD, %) | Post-preparative stability (mean±SD, %) |
|---|---|---|---|---|---|
| Plasma | 0.5 | 104.1±3.5 | 100.7±3.2 | 95.4±3.2 | 121.4±3.2 |
| 5 | 99.5±1.6 | 102.2±3.4 | 92.0±10.4 | 103.8±3.1 | |
| 50 | 104.8±6.6 | 101.8±4.1 | 92.9±9.5 | 91.9±1.1 | |
| 100 | 102.5±4.2 | 100.9±3.7 | 96.8±5.6 | 93.6±3.1 | |
| Heart | 0.5 | 104.1±2.9 | 95.8±2.6 | 87.5±2.4 | 100.1±2.8 |
| 5 | 110.5±1.9 | 89.0±1.5 | 99.4±1.7 | 110.6±2.0 | |
| 50 | 108.2±2.9 | 89.4±2.3 | 94.0±2.5 | 91.5±2.4 | |
| 100 | 105.4±3.8 | 93.7±2.8 | 97.3±3.6 | 95.4±3.2 | |
| Liver | 0.5 | 102.2±4.7 | 89.7±3.2 | 108.2±3.8 | 110.7±4.2 |
| 5 | 101.1±0.7 | 103.7±0.7 | 88.2±0.6 | 99.4±0.7 | |
| 50 | 99.6±0.2 | 92.4±0.2 | 87.5±0.2 | 92.7±0.2 | |
| 100 | 102.6±1.5 | 93.7±1.9 | 90.3±3.2 | 95.7±1.4 | |
| Brain | 0.5 | 101.6±2.9 | 115.0±2.8 | 110.0±2.7 | 100.0±5.0 |
| 5 | 101.2±4.0 | 96.3±3.3 | 88.5±3.0 | 92.1±3.1 | |
| 50 | 100.7±2.6 | 92.8±2.4 | 86.6±2.2 | 96.3±2.0 | |
| 100 | 99.3±3.0 | 93.4±2.7 | 90.1±3.5 | 95.3±2.3 | |
| Lung | 0.5 | 103.8±2.6 | 106.7±2.7 | 89.5±2.2 | 92.4±2.3 |
| 5 | 99.9±1.6 | 92.4±1.4 | 90.9±1.4 | 86.6±1.3 | |
| 50 | 103.5±5.3 | 101.1±5.2 | 89.1±4.5 | 92.3±4.7 | |
| 100 | 98.5±4.2 | 94.6±3.9 | 90.2±4.1 | 93.5±3.8 | |
| Kidney | 0.5 | 101.2±1.9 | 98.5±1.7 | 100.2±1.7 | 90.6±1.6 |
| 5 | 95.3±1.1 | 100.1±1.2 | 86.7±1.0 | 87.1±1.0 | |
| 50 | 93.0±2.0 | 91.7±2.0 | 88.4±1.9 | 91.5±1.9 | |
| 100 | 97.1±2.2 | 93.9±2.7 | 90.6±2.1 | 92.3±2.4 |
3.4. Pharmacokinetics
The pharmacokinetic profiles were determined from a rat plasma concentration-time course after oral or i.v. administration of AICD. The mean concentration-time curves in rats receiving i.v. AICD (100 mg/kg) and oral AICD (200 mg/kg) are shown in Fig. 3. The plasma concentration of AICD decreased rapidly within the initial 2 h after i.v. administration, and then gradually decreased to the LLOQ during the next 6 h. In contrast, following oral administration of AICD, the concentration increased quickly, and reached the maximum plasma concentration (Cmax) at 0.8 h after administration. The plasma concentration then decreased to the LLOQ by 12 h.
Figure 3.

Plasma concentration-time profiles of AICD in rats by oral administration (200 mg/kg) and i.v. administration (100 mg/kg).
According to the pharmacokinetic calculations by DAS 2.0 software, which is the authoritative software for the pharmacokinetic calculations, a two-compartment model of in vivo metabolism best fit the data on AICD in rats. The pharmacokinetic parameters after i.v. and oral administration are shown in Table 3. The absolute bioavailability (F) was calculated as 76.5% using the formula: F (%)=(AUC0–∞,oral×Dosei.v.)/(AUC0–∞,i.v.×Doseoral)×10024, 25, 26, based on the AUC0–∞ obtained after i.v. and oral administration. The half-life of AICD after i.v. and oral administration was 2.2 h and 2.0 h, respectively, both of which were longer than the half-life of ICD reported in the literature (24.910 for i.v. and 28.915 min for oral, or 1.21 h for i.v. and 0.906 h for oral)14, 15.
Table 3.
Main pharmacokinetic parameters of AICD after oral administration (200 mg/kg) and i.v. administrations (100 mg/kg) (n=6). MRT: mean retention time, CLz/F and Vz/F are statistical moment parameters; CL/F and V/F are clearance and volume of distribution through bioavailability calibration; Zeta means the tail section slope of the concentration-time curve.
| Oral |
i.v. |
||
| Parameter | Mean±SD | Parameter | Mean±SD |
| A | 76.932±53.879 | A | 20.122±1.414 |
| α | 2.974±1.984 | α | 11.227±6.780 |
| B | 13.057±3.805 | B | 22.088±3.674 |
| β | 0.201±0.042 | β | 0.736±0.210 |
| AUC0–t (mg/L·h) | 57.443±6.912 | AUC0–t (mg/L·h) | 36.195±4.986 |
| AUC0–∞ (mg/L·h) | 59.342±8.377 | AUC0–∞ (mg/L·h) | 38.770±7.954 |
| MRT0–t (h) | 3.697±0.195 | MRT0–t (h) | 1.627±0.399 |
| MRT0–∞ (h) | 4.045±0.458 | MRT0–∞ (h) | 2.224±1.095 |
| t1/2z (h) | 2.003±0.615 | t1/2z (h) | 2.246±1.005 |
| Tmax (h) | 0.830±0.000 | CLz (L/h/kg) | 2.660±0.532 |
| CLz/F (L/h/kg) | 3.420±0.467 | Vz (L/kg) | 8.058±1.974 |
| Vz/F (L/kg) | 9.597±1.582 | Zeta | 0.359±0.158 |
| Zeta | 0.367±0.092 | ||
| Cmax (μg/L) | 14.093±2.067 | ||
| F (%) | 76.500±0.500 | ||
3.5. Tissue distribution
Tissue distribution was performed at 10 min, 1 h, 3 h, and 6 h after i.v. administration at a single dosage at 100 mg/kg. Representative chromatograms for organs and tissues in rats are shown in Fig. S1A–E, and the statistics results are shown in Fig. 4. The lungs, kidneys and liver homogenate had high concentration of AICD at the four time points, suggesting that AICD easily enters these organs. In contrast, low levels of AICD were observed in the brain, suggesting that it may not cross the blood brain barrier (BBB). These data suggest that AICD may be effective in the treatment of liver, lung and kidney cancers.
Figure 4.

Statistical results of the tissue distribution of AICD in rats after i.v. administration (100 mg/kg).
4. Discussion
Modifications at the C-8 position in the D ring of isocorydine were the focus of our screening antitumor agents16. The hydrogen atom located at C-8 possesses high chemical reactivity and is easily lost. So 8-amino-isocorydine has been obtained which was proved to have a good antitumor effect in vitro but was not stable in aqueous solution. So, under the pro-drug theory, AICD was synthesized through acetylating 8-amino-isocorydine with acetyl chlorine, which could not only protect the p-aminophenol segment, but could also be hydrolyzed by enzymolysis and transform to the original compound in vivo. These investigations about their anticancer activities in vitro and in vivo suggested that structural modifications could significantly improve the anticancer activity of ICD alkaloid and have lower side effects on body weight.
The result of the pharmacokinetics and tissue distribution investigation about the AICD supported that t1/2 of AICD in rats after i.v. and oral administration were 2.2 h and 2.0 h, respectively. We suppose the main reason of this phenomenon was that AICD could eliminate fast in intestinal, thus AICD in rats after oral administration can eliminate faster than after i.v. administration. However, the chemical structure modification indeed changed the metabolic behavior of ICD, and the result of pharmacokinetics and tissue distribution study about the AICD indicated that AICD remained in rats for a relatively long time. This implied that AICD could have fast and long-lasting pharmacological effects in vivo, which would be more effective than ICD. According to the tissue distribution data, we found that AICD did not pass through the BBB, unlike its parent compound ICD15, mainly because introducing an acetamino in the D-ring C-8 site of ICD enhanced its polarity and hydrophilicity. The tissue distribution results suggested that the adverse reactions in the central nervous system (such as nausea, vomiting, drowsiness and dizziness) might be reduced when using AICD. Compared with ICD, we found that AICD had a relatively longer effect period, a higher bioavailability and a higher tolerated concentration, suggesting it may be an ideal anti-cancer drug.
During the compartment model simulation of AICD, we found that two-compartment metabolic model was suitable for describing the pharmacokinetic of AICD in rats. In two-compartment model, AICD entered blood-rich tissues and organs (such as heart, liver, kidney, etc.) first, and then AICD distributed into the peripheral compartment (such as skin, muscles, nerves, etc.). This metabolic approach indicated that a large amount of AICD can reach the target organs quickly. Therefore, AICD could play a long-lasting and effective role in therapeutic hepatocellular carcinoma or lung and kidney cancer. Of course, the investigation about metabolic pathways, metabolites and other pharmacokinetic parameters should be carried out for full understanding the disposition of AICD in vivo. Meanwhile, the protective group that has more space volume such as benzoyl group should be applied to extend the half-life of AICD.
The pharmacological activity and pharmacokinetic studies of AICD by our group supported that the structural modification of natural products is an essential method for the new drugs development. The chemical modification would provide the basis to explore the structure-activity relationship about the targeted compound.
5. Conclusions
A simple and effective HPLC-DAD method coupled with an LLE method was developed for determining the metabolic behavior and tissue distribution of AICD. AICD can be quickly absorbed and eliminated, and its Tmax and t1/2 were 0.8 h and 2.0 h, respectively, after oral administration. AICD was absorbed well after oral administration, and its absolute bioavailability was 76.5%. For the first time, the pharmacokinetic and tissue distributional parameters of this isocorydine derivative were reported, which will provide more useful information for AICD in vivo pharmacological investigation and the new drug research.
Acknowledgments
This work was supported by the “West Light Program (No. Y30447YXL1)”, “Build Coalitions of the National Academy of Sciences” of the Chinese Academy of Sciences (No. Y20475YLJ1), and the Science and Technology Program of Gansu (No. 1304FKCA062).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.apsb.2015.03.012.
Contributor Information
Quanyi Zhao, Email: zhaoqy@lzu.edu.cn.
Junxi Liu, Email: liujx@licp.cas.cn.
Appendix A. Supplementary materials
Supplementary data
References
- 1.Wang X., Dong H.J., Yang B., Liu D.H., Duan W.J. Preparative isolation of alkaloids from Dactylicapnos scandens using pH-zone-refining counter-current chromatography by changing the length of the separation column. J Chromatogr B: Biomed Appl. 2011;879:3767–3770. doi: 10.1016/j.jchromb.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 2.Stévigny C., Bailly C., Quetin L.J. Cytotoxic and antitumor potentialities of aporphinoid alkaloids. Curr Med Chem Anticancer Agents. 2005;5:173–182. doi: 10.2174/1568011053174864. [DOI] [PubMed] [Google Scholar]
- 3.Gerhardt D., Horn A.P., Gaelzer M.M. Boldine: a potential new antiproliferative drug against glioma cell lines. Investig New Drugs. 2009;27:517–525. doi: 10.1007/s10637-008-9203-7. [DOI] [PubMed] [Google Scholar]
- 4.Zhang A., Zhang Y., Branfman A.R. Advances in development of dopaminergic aporphinoids. J Med Chem. 2007;50:171–181. doi: 10.1021/jm060959i. [DOI] [PubMed] [Google Scholar]
- 5.Ponnala S., Chaudhary S., Gonzalez S.A. Cytotoxicity of aporphines in human colon cancer cell lines HCT-116 and Caco-2: an SAR study. Bioorg Med Chem Lett. 2011;21:4462–4464. doi: 10.1016/j.bmcl.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Y.J., Liu J.X., Di D.L., Li M., Feng Y. Structural and mechanistic bases of the anticancer activity of natural aporphinoid alkaloids. Curr Top Med Chem. 2013;13:2116–2126. doi: 10.2174/15680266113139990147. [DOI] [PubMed] [Google Scholar]
- 7.Zhang T.C., Ye H.L., Liu J.X. Study on semi-synthetic transforming technology for the natural product of isocorydione. Acta Pharm Sin. 2011;46:1471–1475. [PubMed] [Google Scholar]
- 8.Liu D.H., Liu D.H., Zhang T.C., Liu J.X. Chemical constituents of alkaloids from Dicranostigma leptopodum. Chin Tradit Herb Drugs. 2011;42:1505–1508. [Google Scholar]
- 9.Chen Z.H., Zhang Z.X., Wang M.D. Spasmolytic effects of isocorydine on isolated gallbladder and Oddi׳s sphincter in vitro. Acta Pharmacol Sin. 1985;6:45–48. [PubMed] [Google Scholar]
- 10.Jiang Q.S., Huang X.N., Sun A.S., Wu Q., Xie X.L. Relation of vasodilative action of isocorydine to cyclic nucleotides. Chin J Pharmacol Toxicol. 2001;15:251–255. [Google Scholar]
- 11.Sun H.F., Hou H.L., Lu P., Zhang L.X., Zhao F.Y., Ge C. Isocorydine inhibits cell proliferation in hepatocellular carcinoma cell lines by inducing G2/M cell cycle arrest and apoptosis. PLoS One. 2012;7:e36808. doi: 10.1371/journal.pone.0036808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu P., Sun H.F., Zhang L.X. Isocorydine targets the drug-resistant cellular side population through PDCD4-related apoptosis in hepatocellular carcinoma. Mol Med. 2012;18:1136–1146. doi: 10.2119/molmed.2012.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang B Yun SM. Preliminary study on chemical compositions of Stephania yunnanensis. L. and structrure-antiarrhythmic activity relationship of aporphine type alkaloids. [Dissertation] Yunnan, China: Institute of Traditional Chinese Medicine; 2013
- 14.Guo C.C., Yu C.H., Li L., Wang Y.Q., Wang S.J., Wang W.H. Rapid determination of isocorydine in rat plasma and tissues using liquid chromatography–tandem mass spectrometry and its applications to pharmacokinetics and tissue distribution. Xenobiotica. 2012;42:466–476. doi: 10.3109/00498254.2011.640965. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y.Q., Li H.L., He J.C., Feng E.F., Rao G.X., Xu G.L. Development and validation of a high-performance liquid chromatography coupled with ultraviolet detection method for the determination of isocorydine in rat plasma and its application in pharmacokinetics. Drug Res. 2013;63:558–563. doi: 10.1055/s-0033-1347256. [DOI] [PubMed] [Google Scholar]
- 16.Zhong M., Liu Y.J., Liu J.X., Di D.L., Xu M.R., Yang Y.Y. Isocorydine derivatives and their anticancer activities. Molecules. 2014;19:12099–12115. doi: 10.3390/molecules190812099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bu L.N., Zhao J.X., Li W.G. Antitumor activity of isocorydione in vitro and vivo. Chin Pharm Bull. 2013;29:832–836. [Google Scholar]
- 18.Singh S.K., Mehrotra N., Sabarinath S., Gupta R.C. HPLC-UV method development and validation for 16-dehydropregnenolone, a novel oral hypolipidaemic agent, in rat biological matrices for application to pharmacokinetic studies. J Pharm Biomed. 2003;33:755–764. doi: 10.1016/s0731-7085(03)00308-x. [DOI] [PubMed] [Google Scholar]
- 19.An G.H., Morris M.E. HPLC analysis of mitoxantrone in mouse plasma and tissues: application in a pharmacokinetic study. J Pharm Biomed. 2010;51:750–753. doi: 10.1016/j.jpba.2009.09.027. [DOI] [PubMed] [Google Scholar]
- 20.Li H.L., Peng X.J., He J.C., Feng E.F., Xu G.L., Rao G.X. Development and validation of a LC-ESI-MS/MS method for the determination of swertiamarin in rat plasma and its application in pharmacokinetics. J Chromatogr B. 2011;879:1653–1658. doi: 10.1016/j.jchromb.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Guo C.H., Yan J.G., Li L., Hong L., Wang Y.Q., Shen Q. Application of a liquid chromatography–tandem mass spectrometry method to the pharmacokinetics, tissue distribution and excretion studies of Dactylicapnos scandens in rats. J Pharm Biomed. 2013;74:92–100. doi: 10.1016/j.jpba.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 22.USFDA. Guidance for industry: bioanalytical method validation. 2001. Available from: 〈http://www.fda.gov/downloads/drugs/guidance compliance regulatory information/guidances/uc m070107.pdf〉.
- 23.Kim E.K., Jeong E.K., Han S.B., Jung J.H., Hong J. HPLC separation of isoquinoline alkaloids for quality control of Corydalis species. Bull Korean Chem Soc. 2011;32:3597–3602. [Google Scholar]
- 24.Bhamidipati R., Mujeeb S., Dravid P.V., Khan A.A., Singh S.K., Rao Y.K. Pre-clinical assessment of DRF 4367, a novel COX-2 inhibitor: evaluation of pharmacokinetics, absolute oral bioavailability and metabolism in mice and comparative inter-species in vitro metabolism. Xenobiotica. 2005;35:253–271. doi: 10.1080/00498250500066303. [DOI] [PubMed] [Google Scholar]
- 25.Li X.G., Choi J.S. Effect of genistein on the pharmacokinetics of paclitaxel administered orally or intravenously in rats. Int J Pharm. 2007;337:188–193. doi: 10.1016/j.ijpharm.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 26.Kwon S.H., Kang M.J., Huh J.S., Ha K.W., Lee J.R., Lee S.K. Comparison of oral bioavailability of genistein and genistin in rats. Int J Pharm. 2007;337:148–154. doi: 10.1016/j.ijpharm.2006.12.046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data