

Abstract
A series of novel N-phenylbenzamide and N-phenylacetophenone compounds were synthesized and evaluated for their antiviral activity against HCV and EV71 (strain SZ-98). The biological results showed that three compounds (23, 25 and 41) exhibited considerable anti-HCV activity (IC50=0.57–7.12 μmol/L) and several compounds (23, 28, 29, 30, 31 and 42) displayed potent activity against EV71 with the IC50 values lower than 5.00 μmol/L. The potency of compound 23 (IC50=0.57 μmol/L) was superior to that of reported compounds IMB-1f (IC50=1.90 μmol/L) and IMB-1g (IC50=1.00 μmol/L) as anti-HCV agents, and compound 29 possessed the highest anti-EV71 activity, comparable to the comparator drug pirodavir. The efficacy in vivo and antiviral mechanism of these compounds warrant further investigations.
KEY WORDS: Synthesis, N-phenylbenzamide, N-phenylacetophenone, Anti-HCV, Anti-EV71, Agents
Graphical abstract
A series of novel N-phenylbenzamide and N-phenylacetophenone compounds were synthesized and evaluated for their antiviral activity against HCV and EV71, and several compounds exhibited considerable anti-HCV and anti-EV71 activity.

1. Introduction
Viral infectious diseases have caused constant threat to human health and the prevention and treatment of viral diseases are a global public health challenge. Hepatitis C virus (HCV) is a single-stranded, positive-sense RNA virus. There was an estimated 170 million people worldwide infected with HCV, and many of them died from HCV-related liver diseases1. In the early years, the combination of PEG-interferon and ribavirin was the only treatment for HCV-infected patients with 40%–50% effectiveness2. Direct-acting antiviral agents (DAA) such as HCV NS3/4A protease inhibitors telaprevir and boceprevir have significantly increased the efficacy in combination with PEG-interferon and ribavirin3. Recently, a new generation of DAAs has become available and currently the interferon-free combination therapy of sofosbuvir (NS5B polymerase inhibitor)4, ledipasvir (NS5A replication complex inhibitor)5 and ribavirin has resulted in a cure rate up to 100% among patients infected with HCV. All of the four marketing drugs, telaprevir, boceprevir, sofosbuvir and ledipasvir were targeting on HCV proteins. However, resistant strains may eventually arise6.
The single-stranded RNA virus EV71 belongs to the Picornaviridae family and usually causes a mild syndrome such as hand-foot-mouth disease (HFMD) and herpangina7, 8. However, some young children infected by EV71 have developed fatal neurological complications such as aseptic meningitis, encephalitis, poliomyelitis-like paralysis and even death9. EV71 infection has been reported worldwide, especially in the Asia-Pacific region10. Currently, there was no effective approved antiviral available for the prevention or treatment of EV71 infection. Supportive therapy is the primary treatment option for severe cases11, 12, 13.
Given the limited efficiency, side effects and drug resistance of these antiviral treatments, continued research is required to develop antiviral drugs with new mechanisms of action. Recently, cellular factors have emerged as useful targets for antiviral therapy and strengthening cellular defense mechanism may be an approach to develop broad-spectrum antivirals. APOBEC3G (Human apolipoprotein B mRNA–editing enzyme catalytic polypeptide-like 3G, hA3G) was initially identified as a host cellular restriction factor of HIV-1 by Sheehy et al.14. In addition, Peng15 has demonstrated that the introduction of external hA3G into HCV-infected Huh7.5 cells effectively inhibited HCV replication. The small molecule compound IMB-26 (3, 4, 5-trimethoxyphenyl-3-(2-bromopropionamido)-4-methoxybenzamide) (Fig. 1) was identified as a stabilizer of hA3G and it was effective against HCV replication in vitro (IC50=1.46±0.62 μmol/L, CC50=11.84±2.87 μmol/L). Our previous work has described a series of substituted N-aryl benzamide compounds such as IMB-1f and IMB-1g (Fig. 1) as anti-HCV agents with the IC50 values ranging from 1.00 μmol/L to 2.00 μmol/L16. In addition, hA3G was also found to be active against other viruses, such as HBV and influenza virus17, 18. Although there has been no report about the relationship between hA3G and EV71, in our laboratory, a series of novel N-phenylbenzamide derivatives were evaluated for their anti-EV71 activity (IC50=5.7–12.0 μmol/L)19. The results showed that IMB-1e (Fig. 1) was the most active compound against EV71.
Figure 1.
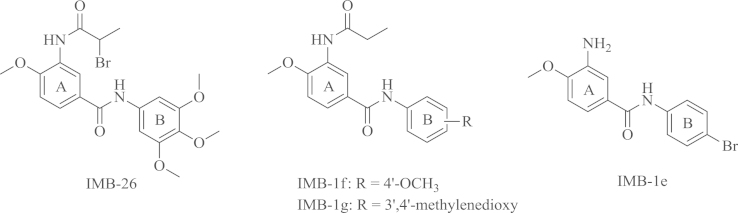
Chemical structures of the antiviral active compounds.
These results prompted us to further optimize the N-phenylbenzamide scaffold to discover more potent antiviral agents. The optimization began with the alkylation of the amino group by a methyl group at the C3-positon on the benzene ring A, and then lipophilic substituents were introduced to the benzene ring B. Meanwhile, we replaced methoxy group at the C4-positon on the benzene ring A with hydroxyl, methyl groups or a hydrogen atom. In addition, to identify the importance of the amide linker between the two aromatic rings A and B, a series of N-phenylacetophenone compounds were also studied. The antiviral activity of the synthesized compounds against HCV and EV71 was evaluated.
2. Results and discussion
2.1. Chemistry
To discover more potent hA3G antiviral agents based on the N-phenylbenzamide scaffold, we designed a series of novel N-phenylbenzamide derivatives. Our previous work has demonstrated that the amino group at the C3-position on the benzene ring A was crucial for antiviral activity. However, due to the cytotoxicity associated with the amino group, the alkylation products of the amino group at the C3-position on the benzene ring A were designed. Meanwhile, our previous results indicated that lipophilic substituents on the benzene ring B would exhibit strong biological activity. Thus, we introduced some lipophilic substituents into the benzene ring B. The amide linker between the two aromatic rings was unstable in vivo, and the hydrolysis of the amide-liker resulted in a loss of antiviral activity. Hence, we inserted a CH2 group between the CO and NH groups to increase the stability of the amide linker. Additionally, we replaced methoxy group with hydroxyl, methyl groups or a hydrogen atom to investigate the influence of the methoxy group at the C4-positon on the benzene ring A for antiviral activity.
A series of N-phenylbenzamide analogs were prepared according to the procedures outlined in Scheme 1, Scheme 2. Compounds 21–34 were synthesized using commercially available 4-methoxy-3-nitrobenzoic acid 1 as the starting material. It was converted into the corresponding benzoyl chloride 2, which was condensed with a variety of anilines using trimethylamine (TEA) as the base to yield the intermediate compounds 3–11. After the nitro groups of the intermediates 3–11 were reduced by hydrogen over 10% dry Pd/C, the amino groups at the C3-positon on the benzene ring A were then acylated by propionyl chloride to afford the 3-propionamido compounds 21–27 or condensed with formaldehyde to afford the 3-methylamido compounds 28–34.
Scheme 1.

Synthetic route for target compounds 22–36. Reagents and conditions: (a) SOCl2, reflux; (b) anilines, TEA, r.t.; (c) H2, 10% dry Pd/C, r.t.; (d) CH3CH2COCl, TEA, r.t.; (e) 40% HCHO, r.t.; (f) BBr3, −78 °C.
Scheme 2.

Synthetic route for target compounds 41–44. Reagents and conditions: (a) anilines, DIC, HOBt, r.t.; (b) CH3CH2COCl, TEA, r.t.; (c) CH3I, K2CO3, 50 °C.
To convert the methoxy group to the hydroxyl group at the C4-position on the benzene ring A, compound 21 or 23 was demethylated using BBr3 at −78 °C to afford the 4-hydroxyl compounds 35 and 36.
To alter the methoxy group to the methyl group or a hydrogen atom at the C4-position on the benzene ring A, compounds 41–44 were prepared using 3-aminobenzoic acid 37 or 3-amino-4-methylbenzoic acid 38 as the starting materials, which was condensed with anilines using diisopropylcarbodiimide (DIC) as a coupling reagent and N-hydroxybenzotriazole (HOBt) as an activating reagent to yield the intermediates 39 or 40. The amino groups were then acylated by propionyl chloride to afford the desired compounds 41 and 43, or alkylated by iodomethane (CH3I) to afford the desired compounds 42 and 44.
To study the importance of the amide linker between the two aromatic rings A and B, we designed a series of N-phenylacetophenone compounds according to the procedures outlined in Scheme 3, Scheme 4. The starting material 1-(4-methoxy-3-nitrophenyl) ethanone 45 was reduced by hydrogen over 10% dry Pd/C and then acylated by propionyl chloride to yield the intermediate 47, which was converted to bromo compound 48 by NBS. The bromo compound was then reacted with anilines to afford the N-phenylacetophenone products 49 and 50. The synthesis of the alkylation products of the amino group was conducted according to the procedure shown in Scheme 4. Firstly, the starting material 45 was converted into the bromo compound 51, which was then coupled with anilines to yield the intermediates 52 and 5320. The desired compounds 54 and 55 were obtained using a one-pot procedure from the corresponding nitro aryls21.
Scheme 3.
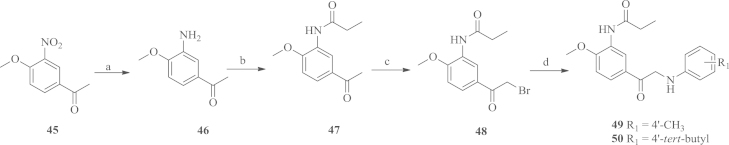
Synthetic route for target compounds 49 and 50. Reagents and conditions: (a) H2, 10% dry Pd/C, r.t.; (b) CH3CH2COCl, TEA, r.t.; (c) NBS, p-TSA, reflux; (d) anilines, NaHCO3, r.t.
Scheme 4.
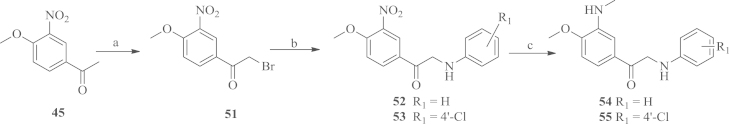
Synthetic route for target compounds 54 and 55. Reagents and conditions: (a) NBS, p-TSA, reflux; (b) anilines, NaHCO3, r.t.; (c) H2, 10% dry Pd/C, 40% HCHO, r.t.
2.2. Evaluation of anti-HCV activity
The antiviral activity of the synthesized compounds against HCV was tested in infected Huh7.5 cells using the RT-PCR method, and VX-950 (telaprevir), a NS3/4A protease inhibitor, was used as a positive control. As shown in Table 1, three compounds 23, 25 and 41 showed relatively potent anti-HCV activity with the IC50 values ranging from 0.57 μmol/L to 7.12 μmol/L. Among these three compounds, the anti-HCV activity of compound 23 (IC50=0.57±0.00 μmol/L) was superior to that of the reported compounds IMB-26 (IC50=1.46±0.62 μmol/L), IMB-1f (IC50=1.90±0.57 μmol/L) and IMB-1f (IC50=1.00±0.05 μmol/L). The cytotoxicity was lower, and especially the selective index (SI) of compound 23 was 53.4. But the potency and selectivity of all the tested compounds against HCV were much lower than that of the control drug telaprevir.
Table 1.
Anti-HCV activity and cytotoxicity of the synthesized compounds.a
| Compd. | IC50 (μmol/L) | CC50 (μmol/L) | SI |
|---|---|---|---|
| 23 | 0.57±0.00 | 30.65±1.25 | 53.4 |
| 25 | 2.36±0.12 | 122.24±2.60 | 51.8 |
| 31 | 32.00±2.14 | 56.63±4.20 | 1.8 |
| 33 | 34.60±3.10 | 39.96±7.25 | 1.20 |
| 41 | 7.12±2.48 | >200 | >28 |
| 42 | 19.51±7.18 | 163.73±9.84 | 8.00 |
| VX-950 | 0.01±0.00 | 21.27±4.70 | 2331 |
All data were average values from three independent assays.
From the results above, we can observe that compounds 23, 25 and 41 possessing the same substituent (propionamido group) at the C3-position on the benzene ring A showed higher anti-HCV activity than those with the methylamino group (31, 33 and 42). Meanwhile, the substituents of these three compounds at the C4′-position on the benzene B were lipophilic (tert-butyl, cyclohexyl, and chloro groups). Thus, these indicated that the propionylamino group at the C3-position on the benzene ring A and lipophilic groups at the C4′-positon on the benzene ring B were the preferred substituents for the activity against HCV. Among these compounds with the propionamido group at the C3-position on the benzene ring A, compound 41 without a substituent has decreased antiviral activity compared with those with a methoxy substituent (23 and 25).
2.3. Evaluation of anti-EV71 activity
The antiviral activity of the synthesized compounds against EV71 (strain SZ-98) were evaluated in Vero cells using the CPE method and a broad-spectrum picornavirus inhibitor, pirodavir, was used as a positive control. The results of antiviral activity are summarized in Table 2. The IC50 values of several N-phenylbenzamide derivatives (23, 28, 29, 30, 31 and 42) were lower than 5.00 μmol/L. Especially, the antiviral activity of 29 (IC50=0.95±0.11 μmol/L) was close to that of the comparator drug (IC50=0.16 μmol/L). Moreover, the SI values of compound 28 and 29 were much larger than 20. Unfortunately, the N-phenylacetophenone compounds 49, 50, 54 and 55 were inactive against EV71 in this experiment. The result might indicate that the amide linker between the two aromatic rings A and B was essential for anti-EV71 activity.
Table 2.
Anti-EV71 (strain SZ-98) activity and cytotoxicity of the synthesized compounds.a
| Compd. | IC50 (μmol/L) | TC50 (μmol/L) | SI |
|---|---|---|---|
| 22 | 8.41±5.94 | >65.35 | >7.77 |
| 23 | 3.53±0.73 | 36.24±0.00 | 10.31 |
| 24 | 14.18±2.94 | 36.24±0.00 | 2.56 |
| 26 | >68.79 | 297.67 | – |
| 27 | >168.36 | 291.60±0.00 | – |
| 28 | 4.93±0.00 | 110.50±0.00 | 22.41 |
| 29 | 0.95±0.11 | >26.09 | >27.44 |
| 30 | 3.56±1.74 | >8.29 | >2.33 |
| 31 | 4.01±0.83 | 34.23±0.00 | 8.58 |
| 32 | 6.89±5.00 | 41.12±0.00 | 5.97 |
| 34 | 16.26±2.88 | >79.07 | >4.86 |
| 35 | 11.73±2.45 | 100.79±0.00 | 8.59 |
| 36 | 9.10±2.06 | 86.08±0.00 | – |
| 41 | 14.17±0.00 | 106.12±0.00 | 7.49 |
| 42 | 4.07±0.00 | >28.5 | >6.99 |
| 43 | 98.74±20.47 | 196.10±0.00 | >2.00 |
| 44 | 19.63±7.42 | 322.65±0.00 | 16.45 |
| 49 | >22.73 | 98.31 | – |
| 50 | >20.14 | >20.14 | – |
| 54 | 82.30±0.00 | 167.11±34.70 | 2.03 |
| 55 | >24.38 | 73.10±0.00 | – |
| Pirodavir | 0.16±0.00 | 49.78±3.96 | 306.17 |
All data were average values from three independent assays.
Based on the results of anti-EV71 activity above, the preliminary structure-activity relationships (SAR) have been established. The replacement of chlorine at the C4′-positon on the benzene ring B with other lipophilic substituents, such as isopropyl, tert-butyl, and n-butyl group (22–24) increased the anti-EV71 activity. However, the N-methyl piperazine derivative (27) did not display antiviral activity against EV71. So we concluded that lipophilic substituents on the benzene ring B was crucial to display anti-EV71 activity. When strong electron-withdrawing groups such as cyano group were introduced at the C4′-positon on the benzene ring B (26), a decreased activity was observed. In addition, altering the propionamido group at the C3-postion on the benzene ring A to the methylamino group (28–34) led to an increase in antiviral activity. In particular, the activity of the 4′-ethyl derivative (29) was close to that of the comparator drug pirodavir. The replacement of the methoxy group at the C4-positon on the benzene ring A with a hydroxy group (35 and 36), or a hydrogen atom (42) increased the antiviral activity. However, when a methyl group was placed at the C4-position on the benzene ring A (43 and 44), the antiviral activity was poor.
Because the amide linker was metabolically unstable in vivo, we inserted a CH2 group between the CO and NH groups of the amide linker and synthesized four N-phenylacetophenone compounds (49, 50, 54, 55). But the antiviral activity against EV71 was much lower than the corresponding N-phenylbenzamide compounds. The result highlighted the importance of the amide linker in anti-EV71 activity.
3. Conclusions
Our previous work has demonstrated that N-phenylbenzamide compounds were a class of potential broad-spectrum antiviral agents. In this paper, a series of novel N-phenylbenzamide and N-phenylacetophenone compounds with a methylamino group on the benzene ring A were synthesized. Lipophilic substituents were introduced at the C4′-position on the benzene ring B, or a CH2 group was inserted between the CO and NH groups of the amide linker, or a methoxy group at the C4-positon on the benzene ring A was replaced with a hydroxy, a methyl group or a hydrogen atom. Totally 23 novel analogs were synthesized and evaluated for their antiviral activity against HCV and EV71 (strain SZ-98). Compounds 23, 25 and 41 exhibited considerable anti-HCV activity with the IC50 values ranging from 0.57 to 7.12 μmol/L, and several compounds (23, 28, 29, 30, 31 and 42) exhibited potent activity against EV71 with IC50 values lower than 5.00 μmol/L. Particularly, the potency of compound 23 was superior to that of the reported compound IMB-26, IMB-1f and IMB-1g for anti-HCV activity, and compound 29 displayed the highest anti-EV71 activity almost comparable to the comparator drug pirodavir. Their efficacy in vivo and antiviral mechanism will be further investigated in the future.
4. Experimental
4.1. Synthesis and characterization
1H NMR and 13C NMR spectra were recorded in CDCl3 or DMSO-d6 on a Bruker BioSpin GmbH 400, 500 or 600 spectrometer. Chemical shifts were reported in parts per million relative to tetramethylsilane as an internal standard. High-resolution mass spectra (HRMS) were obtained on an MDS SCIEX Q-Trap mass spectrometer. Melting points were determined with an X6 microscope melting point apparatus and uncorrected. All reagents and solvents were purchased from J&K and Alfa Aesar Chemicals without purification.
4.1.1. General procedure for the synthesis of compounds 12–20
A solution of 1 (2.5 mmol) in SOCl2 (5 mL) was refluxed for 3 h. The mixture was cooled to ambient temperature and then the solvent was removed under reduced pressure. Aniline (2.5 mmol) and TEA (2.5 mmol) were dissolved in dichloromethane (DCM) (20 mL) and a solution of the acyl chloride above in DCM (5 mL) was added to the reaction. After the mixture was stirred at room temperature for 5 h, the reaction was quenched with saturated aqueous NH4Cl (20 mL), extracted with DCM (20 mL×3), and washed with brine. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (PE/EA=10:1, v/v) to give 3–11, which was then dissolved in CH3OH (20 mL) followed by the addition of 10% dry Pd/C (0.01 mmol). The mixture was stirred under hydrogen for 4 h. The catalyst was filtered off and the combined organic solution was concentrated under reduced pressure to afford the intermediates 12–20.
4.1.2. General procedure for the synthesis of compounds 21–27
To a solution of 12 or 15–20 (2.0 mmol) in DCM (20 mL) was added TEA (2.0 mmol), and then a solution of propionyl chloride (3.0 mmol) in DCM (5 mL) was added slowly. After the mixture was stirred at room temperature for 3 h, the reaction was extracted with DCM (20 mL×3), and washed with brine. The combined organic layers were dried under Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (PE/EA=10:1, v/v) to give 21–27.
N-(4-Iso-propylphenyl)-4-methoxy-3-propionamidobenzamide (22): a white solid; yield: 89%. m.p: 171–173 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.03 (s, 1H), 9.18 (s, 1H), 8.51 (s, 1H), 7.75 (dd, J=8.6, 2.0 Hz, 1H), 7.68–7.64 (m, 2H), 7.21 (d, J=8.5 Hz, 2H), 7.15 (d, J=8.7 Hz, 1H), 3.91 (s, 3H), 2.88–2.85 (m, 1H), 2.42 (q, J=7.5 Hz, 2H), 1.20 (d, J=6.1 Hz, 6H), 1.09 (t, J=7.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 172.72, 165.34, 152.80, 143.92, 137.55, 127.50, 127.36, 126.70, 124.60, 122.65, 120.93, 110.85, 56.45, 33.37, 29.63, 24.45, 10.20. HR-MS (ESI+): 341.1857, Calcd. for C20H24N2O3: 341.1859 [M+H]+.
N-(4-tert-Butylphenyl)-4-methoxy-3-propionamidobenzamide (23): a white solid; yield: 85%. m.p: 165–166 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.03 (s, 1H), 9.18 (s, 1H), 8.51 (s, 1H), 7.74 (dd, J=8.6, 2.2 Hz, 1H), 7.68–7.66 (m, 2H), 7.37–7.34 (m, 2H), 7.15 (d, J=8.7 Hz, 1H), 3.92 (s, 3H), 2.42 (q, J=7.5 Hz, 2H), 1.29 (s, 9H), 1.09 (t, J=7.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 172.73, 165.36, 152.80, 146.16, 137.23, 127.50, 127.36, 125.60, 124.60, 122.66, 120.59, 110.84, 56.45, 34.50, 31.69, 29.63, 10.20. HR-MS (ESI+): 355.2014, Calcd. for C21H26N2O3: 355.2016 [M+H]+.
N-(4-Butylphenyl)-4-methoxy-3-propionamidobenzamide (24): a white solid; yield: 82%. m.p: 165–168 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.02 (s, 1H), 9.18 (s, 1H), 8.50 (s, 1H), 7.74 (dd, J=8.6, 2.2 Hz, 1H), 7.66–7.63 (m, 2H), 7.15 (d, J=8.6 Hz, 3H), 3.91 (s, 3H), 2.55 (t, J=8.0 Hz, 2H), 2.42 (q, J=8.0 Hz, 2H), 1.59−1.51 (m, 2H), 1.31 (m, 2H), 1.09 (t, J=8.0 Hz, 3H), 0.91 (t, J=8.0 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 172.52, 165.35, 151.76, 137.80, 137.46, 128.73, 127.50, 127.39, 124.57, 122.63, 120.85, 110.86, 56.45, 34.73, 33.69, 29.63, 22.16, 14.26, 10.19. HR-MS (ESI+): 355.2014, Calcd. for C21H26N2O3: 355.2016 [M+H]+.
N-(4-Cyclohexylphenyl)-4-methoxy-3-propionamidobenzamide (25): a white solid, yield: 78%. m.p: 167–169 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.01 (s, 1H), 9.18 (s, 1H), 8.50 (s, 1H), 7.73 (dd, J=8.6, 2.2 Hz, 1H), 7.66–7.64 (m, 2H), 7.19–7.14 (m, 3H), 3.91 (s, 3H), 2.42 (m, 3H), 1.79 (d, J=10.5 Hz, 4H), 1.71 (m, 1H), 1.4 –1.37 (m, 4H), 1.28−1.25 (m, 1H), 1.09 (t, J=7.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 172.72, 165.35, 143.18, 137.56, 127.51, 127.39, 127.06, 124.58, 122.63, 120.90, 110.84, 56.45, 43.71, 34.54, 29.63, 26.85, 26.09, 10.19. HR-MS (ESI+): 381.2171, Calcd. for C23H29N2O3: 381.2172 [M+H]+.
N-(4-Cyanophenyl)-4-methoxy-3-propionamidobenzamide (26): a white solid, yield: 75%. m.p: 154–157 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.51 (s, 1H), 9.22 (s, 1H), 8.56 (s, 1H), 8.01−7.98 (m, 2H), 7.83−7.81 (m, 2H), 7.78 (dd, J=8.6, 2.2 Hz, 1H), 7.20 (d, J=8.7 Hz, 1H), 3.94 (s, 3H), 2.44 (q, J=7.5 Hz, 2H), 1.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 172.82, 166.11, 153.20, 144.23, 133.51, 127.68, 126.56, 125.01, 122.64, 120.62, 119.62, 110.95, 105.45, 56.53, 29.64, 10.17. HR-MS (ESI+): 324.1340, Calcd. for C18H17N3O3: 324.1342 [M+H]+.
N-(4-(4-Methylpiperazin-1-yl)phenyl)-4-methoxy-3-propionamidobenzamide (27): a white solid, yield: 65%, m.p: 161–164 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.91 (s, 1H), 9.99 (s, 1H), 9.18 (s, 1H), 8.50 (s, 1H), 7.75 (dd, J=8.6, 2.0 Hz, 1H), 7.65 (d, J=9.0 Hz, 2H), 7.14 (d, J=8.7 Hz, 1H), 6.99 (d, J=9.0 Hz, 2H), 3.91 (s, 3H), 3.34−3.26 (m, 8H), 2.80 (s, 3H), 2.42 (q, J=7.4 Hz, 2H), 1.09 (t, J=7.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 172.73, 165.11, 152.75, 146.20, 132.75, 127.47, 127.38, 124.54, 122.64, 121.98, 116.68, 110.84, 56.46, 52.57, 46.41, 42.43, 29.63, 10.20. HR-MS (ESI+): 397.2231, Calcd. for C22H28N4O3: 397.2234 [M+H] +.
4.1.3. General procedure for the synthesis of compounds 28–34
To the intermediates 13–19 (2.0 mmol) in CH3OH (20 mL) was added 10% dry Pd/C (0.01 mmol) and 40% HCHO solution (3.0 mmol), and the mixture was stirred under hydrogen for 4 h. The catalyst was filtered off and the combined organic solutions were concentrated under reduced pressure. The residue was purified by column chromatography (PE/EA=10:1, v/v) to afford compounds 28–34.
N-(3-Chlorophenyl)-4-methoxy-3-(methylamino)benzamide (28): a white solid, yield: 50%. m.p: 143–144 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.11 (s, 1H), 7.96 (t, J=2.0 Hz, 1H), 7.72 (dd, J=8.3, 1.0 Hz, 1H), 7.37 (t, J=8.1 Hz, 1H), 7.26 (dd, J=8.2, 2.1 Hz, 1H), 7.17–7.07 (m, 1H), 7.02 (d, J=2.1 Hz,1H), 6.91 (d, J=8.3 Hz, 1H), 5.25 (s, 1H), 3.86 (s, 3H), 2.79 (d, J=5.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 166.52, 149.74, 141.51, 139.52, 133.32, 130.66, 127.56, 123.33, 120.08, 119.00, 116.23, 109.01, 107.85, 56.04, 30.19. HR-MS (ESI+): 291.0895, Calcd. for C15H15N2O2Cl: 291.0894 [M+H] +.
N-(4-Ethylphenyl)-4-methoxy-3-(methylamino)benzamide (29): a white solid, yield: 67%. m.p: 156–157 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.88 (s, 1H), 7.66 (d, J=8.5 Hz, 2H), 7.26 (dd, J=8.3, 2.1 Hz, 1H), 7.17 (d, J=8.5 Hz, 2H), 7.04 (d, J=2.1 Hz, 1H), 6.89 (d, J=8.3 Hz, 1H), 5.21 (q, J=5.0 Hz, 1H), 3.85 (s, 3H), 2.79 (d, J=5.0 Hz, 3H), 2.58 (q, J=7.6 Hz, 2H), 1.18 (t, J=7.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 166.09, 149.45, 139.43, 139.07, 137.64, 128.15, 128.10, 120.93, 116.04, 108.96, 107.92, 56.00, 30.22, 28.09, 16.21. HR-MS (ESI+): 285.1597, Calcd. for C17H20N2O2: 285.1597 [M+H] +.
N-(4-iso-propylphenyl)-4-methoxy-3-(methylamino)benzamide (30): a white solid, yield: 60%. m.p: 166–167 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.88 (s, 1H), 7.71–7.61 (m, 2H), 7.26 (dd, J=8.3, 2.1 Hz, 1H), 7.20 (d, J=8.5 Hz, 2H), 7.03 (d, J=2.1 Hz, 1H), 6.89 (d, J=8.4 Hz, 1H), 5.20 (m, 1H), 3.85 (s, 3H), 2.86 (m, 1H), 2.79 (d, J=5.1 Hz, 3H), 1.20 (d, J=4.0 Hz, 6H). 13C NMR (101 MHz, DMSO-d6): δ 166.08, 149.45, 143.74, 139.43, 137.71, 128.09, 126.65, 120.92, 116.03, 108.95, 107.93, 56.00, 33.36, 30.22, 24.46. HR-MS (ESI+): 299.1752, Calcd. for C18H22N2O2: 299.1754 [M+H] +.
N-(4-tert-Butylphenyl)-4-methoxy-3-(methylamino)benzamide (31): a white solid, yield: 69%. m.p: 145–146 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.89 (s, 1H), 7.74−7.60 (m, 2H), 7.42−7.31 (m, 2H), 7.27 (dd, J=8.3, 2.1 Hz, 1H), 7.04 (d, J=2.1 Hz, 1H), 6.89 (d, J=8.4 Hz, 1H), 5.21 (q, J=4.7 Hz, 1H), 3.86 (s, 3H), 2.79 (d, J=5.0 Hz, 3H), 1.29 (s, 9H). 13C NMR (101 MHz, DMSO-d6): δ 166.09, 149.46, 145.98, 139.44, 137.39, 128.08, 125.54, 120.58, 116.04, 108.94, 107.94, 56.00, 34.48, 31.70, 30.23. HR-MS (ESI+): 313.1906, Calcd. for C19H24N2O2: 313.1910 [M+H] +.
N-(4-Butylphenyl)-4-methoxy-3-(methylamino)benzamide (32): a white solid, yield: 60%. m.p: 160–162 °C. 1H NMR (500 MHz, DMSO-d6): δ 9.89 (s, 1H), 7.65 (d, J=8.5 Hz, 2H), 7.25 (dd, J=8.3, 2.1 Hz, 1H), 7.14 (d, J=8.5 Hz, 2H), 7.02 (d, J=2.1 Hz, 1H), 6.89 (d, J=8.4 Hz, 1H), 5.23 (m, 1H), 3.85 (s, 3H), 2.78 (d, J=5.1 Hz, 3H), 2.55 (t, J=7.6 Hz, 2H), 1.58−1.52 (m, 2H), 1.33–1.29 (m, 2H), 0.90 (t, J=7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 166.09, 149.44, 139.43, 137.62, 128.68, 128.11, 120.87, 116.02, 108.97, 107.91, 56.00, 34.73, 33.70, 30.22, 22.17, 14.27. HR-MS (ESI+): 313.1907, Calcd. for C19H24N2O2: 313.1910 [M+H] +.
N-(4-Cyclohexylphenyl)-4-methoxy-3-(methylamino)benzamide (33): a white solid, yield: 65%. m.p: 165–167 °C. 1H NMR (500 MHz, DMSO-d6): δ 9.87 (s, 1H), 7.66−7.64 (m, 2H), 7.25 (dd, J=8.3, 2.1 Hz, 1H), 7.17 (d, J=8.5 Hz, 2H), 7.02 (d, J=2.1 Hz, 1H), 6.89 (d, J=8.4 Hz, 1H), 5.21 (m, 1H), 3.85 (s, 3H), 2.78 (d, J=5.1 Hz, 3H), 2.46 (s, 1H), 1.79 (m, 4H), 1.71 (m, 1H), 1.4−1.31 (m, 4H), 1.22 (m, 1H). 13C NMR (101 MHz, DMSO-d6): δ 166.08, 149.44, 143.01, 139.43, 137.73, 128.11, 127.01, 120.89, 116.03, 108.96, 107.92, 56.00, 43.70, 34.56, 30.22, 26.86, 26.09. HR-MS (ESI+): 339.2065, Calcd. for C21H27N2O2: 339.2067 [M+H]+.
N-(4-Cyanophenyl)-4-methoxy-3-(methylamino)benzamide (34): a white solid, yield: 73%. m.p: 165–167 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.36 (s, 1H), 8.01−7.97 (m, 2H), 7.82−7.79 (m, 2H), 7.28 (dd, J=8.3, 2.2 Hz, 1H), 7.02 (d, J=2.1 Hz, 1H), 6.93 (d, J=8.4 Hz, 1H), 5.28 (m, 1H), 3.87 (s, 3H), 2.78 (t, J=7.8 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 166.87, 149.94, 144.38, 139.57, 133.50, 127.35, 120.52, 119.66, 116.44, 109.01, 107.88, 105.26, 56.07, 30.17. HR-MS (ESI+): 282.1236, Calcd. for C16H15N3O2: 282.1237 [M+H]+.
4.1.4. General procedure for the synthesis of compounds 35–36
To a solution of 21 or 23 (1.0 mmol) in DCM (20 mL) was added BBr3 (1.2 mmol) in DCM (5 mL) over 15 min at −78 °C. The resulting solution was allowed to warm to 25 °C and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl, extracted with DCM (20 mL×3), and washed with brine. The combined organic layers were dried with Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (PE/EA=10:1, v/v) to give 35–36.
N-(4-Chlorophenyl)-4-hydroxy-3-propionamidobenzamide (35): a white solid, yield: 34%. m.p: 179–181 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.55 (s, 1H), 10.15 (s, 1H), 9.31 (s, 1H), 8.36 (d, J=1.2 Hz, 1H), 7.82−7.78 (m, 2H), 7.62 (dd, J=8.4, 2.0 Hz, 1H), 7.41−7.37 (m, 2H), 6.96 (d, J=8.4 Hz, 1H), 2.43 (q, J=7.5 Hz, 2H), 1.10 (t, J=7.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 173.17, 165.73, 138.91, 128.90, 127.31, 126.61, 125.76, 125.00, 123.18, 122.20, 115.48, 40.19, 39.77, 39.35, 29.54, 10.21. HR-MS (ESI+): 319.0842, Calcd. for C16H15N2O3Cl: 319.0844 [M+H]+.
N-(4-tert-Butylphenyl)-4-hydroxy-3-propionamidobenzamide (36): a white solid, yield: 40%. m.p: 182–184 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.46 (s, 1H), 9.95 (s, 1H), 9.32 (s, 1H), 8.33 (s, 1H), 7.65−7.61 (m, 3H), 7.35 (d, J=8.6 Hz, 2H), 6.95 (d, J=8.4 Hz, 1H), 2.43 (m, 2H), 1.28 (s, 9H), 1.10 (t, J=7.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 173.18, 165.44, 151.60, 146.05, 137.30, 126.50, 126.21, 125.58, 124.92, 123.26, 120.53, 115.48, 34.49, 31.69, 29.52, 10.22. HR-MS (ESI+): 341.1859, Calcd. for C20H25N2O3: 341.1859 [M+H]+.
4.1.5. General procedure for the synthesis of 39, 40
To a solution of 37 or 38 (3.6 mmol) in DCM (30 mL) and DMF (5 mL) was added DIC (4.3 mmol), HOBt (4.3 mmol) and aniline (3.8 mmol). After the mixture was stirred at room temperature for 24 h, the reaction was extracted with DCM (20 mL×3), and washed with brine. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (PE/EA=5:1, v/v) to give 39 or 40.
4.1.6. General procedure for the synthesis of 41 or 43
To a solution of 39 or 40 (2.0 mmol) in DCM (30 mL) was added TEA (2.0 mmol), and then the solution of propionyl chloride (3.0 mmol) was added slowly. After the mixture was stirred at room temperature for 3 h, the reaction was extracted with DCM (20 mL×3), and washed with brine. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (PE/EA =10:1, v/v) to give 41 or 43.
N-(4-Chlorophenyl)-3-propionamidobenzamide (41): a white solid, yield: 80%. m.p: 160–163 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.38 (s, 1H), 10.08 (s, 1H), 8.10 (s, 1H), 7.83 (m, 3H), 7.60 (d, J=7.8 Hz, 1H), 7.44 (m, 3H), 2.35 (q, J=7.5 Hz, 2H), 1.10 (t, J=7.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 172.70, 166.17, 140.02, 138.62, 135.88, 129.21, 129.00, 127.69, 122.52, 122.32, 122.25, 118.95, 29.98, 10.07. HR-MS (ESI+): 303.0893, Calcd. for C16H15N2O2Cl: 303.0894 [M+H]+.
N-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-4-methyl-3-propionamidobenzamide (43): a white solid, yield: 77%. m.p: 167–168 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.02 (s, 1H), 9.42 (s, 1H), 7.94 (s, 1H), 7.68 (dd, J=7.9, 1.6 Hz, 1H), 7.38 (d, J=2.4 Hz, 1H), 7.34 (d, J=8.0 Hz, 1H), 7.20 (dd, J=8.8, 2.5 Hz, 1H), 6.82 (d, J=8.7 Hz, 1H), 5.76 (s, 1H), 4.23 (q, J=5.0 Hz, 4H), 2.38 (q, J=7.6 Hz, 2H), 2.26 (s, 3H), 1.12 (t, J=7.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 172.60, 165.14, 143.23, 140.01, 137.04, 136.32, 133.33, 133.17, 130.55, 125.19, 124.60, 117.01, 114.14, 109.98, 64.53, 29.35, 18.47, 10.40. HR-MS (ESI+): 341.1494, Calcd. for C19H20N2O4: 341.1495 [M+H]+.
4.1.7. General procedure for the synthesis of compounds 42 or 44
To a solution of 39 or 40 (2.0 mmol) in acetone (15 mL) was added K2CO3 (3.0 mmol) and CH3I (5.00 mmol), and then the resulting solution was stirred at 50 °C for 10 h. The mixture was cooled to ambient temperature and the solvent was removed under reduced pressure. The residue was dissolved in DCM (35 mL), and washed with brine. The organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (PE/EA=10:1, v/v) to afford 42 or 44.
N-(4-Chlorophenyl)-3-(methylamino)benzamide (42): a white solid, yield: 45%. m.p: 164–167 °C. 1H NMR (400 MHz, CDCl3): δ 7.76 (s, 1H), 7.60 (d, J=8.8 Hz, 2H), 7.33 (d, J=8.8 Hz, 2H), 7.12 (s, 1H), 7.07 (d, J=7.6 Hz, 1H), 6.78 (dd, J=8.1, 2.1 Hz, 1H), 2.90 (s, 3H). 13C NMR (151 MHz, DMSO-d6): δ 166.92, 150.38, 138.79, 136.08, 129.29, 128.92, 127.47, 122.22, 115.31, 115.01, 110.93, 30.17. HR-MS (ESI+): 261.0789, Calcd. for C14H13N2OCl: 261.0789 [M+H]+.
N-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-4-methyl-3-(methylamino)benzamide (44): a white solid, yield: 77%. m.p: 170–173 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.85 (s, 1H), 7.39 (d, J=2.4 Hz, 1H), 7.19 (dd, J=8.8, 2.4 Hz, 1H), 7.10 (m, 2H), 6.98 (d, J=1.0 Hz, 1H), 6.81 (d, J=8.7 Hz, 1H), 5.24 (q, J=4.9 Hz, 1H), 4.23 (m, 4H), 2.80 (d, J=4.9 Hz, 3H), 2.13 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 166.31, 147.99, 143.24, 139.86, 134.11, 133.52, 129.63, 125.86, 116.99, 115.17, 114.05, 109.89, 107.73, 64.64, 64.41, 30.52, 23.76. HR-MS (ESI+): 299.1388, Calcd. for C17H18N2O3: 299.1390 [M+H]+.
N-(5-(2-Bromoacetyl)-2-methoxyphenyl)propionamide (48): to a solution of 45 (1.0 mmol) in CH3OH (15 mL) was added 10% dry Pd/C (0.01 mmol), the mixture was reacted with hydrogen (40 psi) for 4 h. The catalyst was filtered off and the combined organic solutions were concentrated under reduced pressure. The residue was dissolved in DCM (25 mL) followed by the addition of TEA (1.0 mmol), and then a solution of propionyl chloride (1.5 mmol) was added slowly. After the mixture was stirred at room temperature for 3 h, the reaction mixture was extracted with DCM (20 mL×3), and washed with brine. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue 47 (1.5 mmol) was dissolved in CCl4 (25 mL) followed by the addition of NBS (1.2 mmol) and p-TSA (0.01 mmol). After the mixture was refluxed for 5 h, the mixture was cooled to ambient temperature and then the reaction was quenched with saturated aqueous NH4Cl (30 mL), extracted with DCM (20 mL×3), and washed with brine. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (PE/EA=10:1, v/v) to give 48: a white solid, yield: 70%. m.p: 101–103 °C. 1H NMR (400 MHz, CDCl3): δ 9.37 (s, 1H), 8.22 (d, J=2.0 Hz, 1H), 7.68 (dd, J=8.9, 2.0 Hz, 1H), 7.12 (d, J=8.7 Hz, 1H), 4.72 (s, 2H), 3.92 (s, 3H), 2.71 (q, J=7.7 Hz, 2H), 1.21 (t, J=7.9 Hz, 3H). ESI-MS (m/z): 300.1 [M+H]+.
4.1.8. General procedure for the synthesis of 49 or 50
To a solution of 48 (0.5 mmol) in ethanol (15 mL) was added NaHCO3 (1.0 mmol) and aniline (0.55 mmol). After the mixture was stirred at room temperature for 24 h, the solvent was removed under reduced pressure. The residue was extracted with DCM (20 mL×3), and washed with brine. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (PE/EA=10:1, v/v) to give 49 or 50.
N-(2-Methoxy-5-(2-(p-tolylamino)acetyl)phenyl)propionamide (49): a yellow solid, yield: 34%. m.p: 132–134 °C. 1H NMR (500 MHz, DMSO-d6): δ 9.23 (s, 1H), 8.62 (s, 1H), 7.91 (dd, J=8.6, 1.8 Hz, 1H), 7.17 (d, J=8.7 Hz, 1H), 6.90 (d, J=8.1 Hz, 2H), 6.58 (d, J=8.3 Hz, 2H), 5.59 (t, J=5.3 Hz, 1H), 4.55 (d, J=5.4 Hz, 2H), 3.94 (s, 3H), 2.43 (q, J=7.5 Hz, 2H), 2.15 (s, 3H), 1.08 (t, J=7.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 195.62, 172.91, 154.21, 146.33, 129.72, 128.11, 128.04, 125.81, 124.94, 113.04, 111.14, 56.61, 50.17, 29.64, 20.55, 10.13. HR-MS (ESI+): 327.1700, Calcd. for C19H22N2O3: 327.1703 [M+H]+.
N-(5-(2-((4-(tert-Butyl)phenyl)amino)acetyl)-2-methoxyphenyl)propionamide (50): yellow solid. yield: 25%. m.p: 130–132 °C. 1H NMR (500 MHz, DMSO): δ 9.24 (s, 1H), 8.63 (s, 1H), 7.91 (dd, J=8.6, 2.2 Hz, 1H), 7.18 (d, J=8.7 Hz, 1H), 7.12–7.09 (m, 2H), 6.60−6.58 (m, 2 H), 5.62 (t, J=5.4 Hz, 1H), 4.55 (d, J=5.5 Hz, 2H), 3.94 (s, 3H), 2.43 (d, J=7.5 Hz, 2H), 1.22 (s, 9H), 1.08 (t, J=7.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 195.76, 172.91, 154.20, 146.23, 138.80, 128.12, 128.06, 125.87, 125.79, 121.68, 112.67, 111.15, 56.61, 50.12, 33.91, 31.93, 29.65, 10.13. HR-MS (ESI+): 369.2169, Calcd. for C22H28N2O3: 369.2172 [M+H]+.
4.1.9. General procedure for the synthesis of 52 or 53
To a solution of 45 (0.5 mmol) in CCl4 (15 mL) was added NBS (0.6 mmol) and p-TSA (0.01 mmol). After the mixture was refluxed for 5 h, the mixture was cooled to ambient temperature and then the reaction was quenched with saturated aqueous NH4Cl (30 mL), extracted with DCM (20 mL×3), and washed with brine. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (PE/EA =10:1, v/v) to give 51, which was then dissolved in ethanol (25 mL) followed by the addition of NaHCO3 (1.0 mmol) and anilines (0.55 mmol). After the mixture was stirred at room temperature for 24 h, the solvent was removed under reduced pressure. The residue was extracted with DCM (20 mL×3), and washed with brine. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (PE/EA=10:1, v/v) to give 52 or 53.
1-(4-Methoxy-3-nitrophenyl)-2-(phenylamino)ethanone (52): a yellow solid, yield: 79%. ESI-MS (m/z): 287.1 [M+H]+.
2-((4-Chlorophenyl)amino)-1-(4-methoxy-3-nitrophenyl)ethanone (53): a yellow solid, yield: 60%. ESI-MS (m/z): 321.0 [M+H]+.
4.1.10. General procedure for the synthesis of 54 or 55
To a solution of 52 or 53 (1.0 mmol) in CH3OH (15 mL) was added 10% dry Pd/C (0.01 mmol) and 40% HCHO (2.5 mmol). The mixture was stirred under hydrogen for 4 h. The catalyst was filtered off and the combined organic solutions were concentrated under reduced pressure. The residue was purified by column chromatography (PE/EA =10:1, v/v) to afford 54 or 55.
1-(4-Methoxy-3-(methylamino)phenyl)-2-(phenylamino)ethanone (54): a yellow solid, yield: 32%. m.p: 135–137 °C. 1H NMR (500 MHz, DMSO-d6): δ 7.45 (dd, J=8.3, 1.7 Hz, 1H), 7.08 (t, J=7.7 Hz, 2H), 7.04 (d, J=1.5 Hz, 1H), 6.92 (d, J=8.3 Hz, 1H), 6.68 (d, J=8.2 Hz, 2H), 6.56 (t, J=7.2 Hz, 1H), 5.76 (t, J=5.3 Hz, 1H), 5.32 (q, J=4.8 Hz, 1H), 4.58 (d, J=5.4 Hz, 2H), 3.88 (s, 3H), 2.77 (d, J=5.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 195.94, 151.27, 148.67, 139.79, 129.26, 129.01, 117.81, 116.50, 112.96, 109.09, 106.85, 56.14, 49.72, 30.10. HR-MS (ESI+): 271.1440, Calcd. for C16H18N2O2: 271.1441 [M+H]+.
2-((4-Chlorophenyl)amino)-1-(4-methoxy-3-(methylamino)phenyl)ethanone (55): a yellow solid, yield: 25%. m.p: 140–141 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.44 (dd, J=8.3, 2.1 Hz, 1H), 7.12−7.08 (m, 2H), 7.03 (d, J=2.0 Hz, 1H), 6.92 (d, J=8.4 Hz, 1H), 6.71−6.68 (m, 2H), 6.01 (t, J=5.4 Hz, 1H), 5.30 (q, J=5.1 Hz, 1H), 4.58 (d, J=5.4 Hz, 2H), 3.88 (s, 3H), 2.77 (d, J=5.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 195.61, 151.30, 147.70, 139.79, 128.93, 128.90, 119.68, 117.83, 114.34, 109.08, 106.84, 56.14, 49.73, 30.10. HR-MS (ESI+): 305.1051, Calcd. for C16H17N2O2Cl: 305.1051 [M+H]+.
4.2. Cells and virus
Huh7.5 human liver cells were kindly provided by Vertex Pharmaceuticals (Boston, USA), and were cultured in Dulbecco׳s Modified Eagle׳s Medium, which was supplemented with 10% inactivated fetal bovine serum and 1% penicillin-streptomycin. The cells were cultured at 37 °C in 5% CO2, released with 0.05% trypsin-EDTA and split twice a week. The plasmid pFL-J6/JFH/JC1, which contains the full-length chimeric HCV cDNA was kindly provided by Vertex Pharmaceutical (Boston, USA). Vero cells were purchased from the American Type Culture Collection and were cultured in Minimum Essential Medium (MEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 U/mL penicillin G and 100 mg/mol streptomycin). EV71 strain SZ-98 was kindly provided by Dr Qi Jin, Institute of Pathogen Biology, Chinese Academy of Medical Sciences and Peking Union Medical School.
4.3. Cytotoxicity assay
The Huh7.5 cells were used in the test; Huh7.5 cells (1×104 cells/well) were planted into 96-microwell plates. Six hours later the culture media was replaced with fresh medium containing the tested compounds at various concentrations. Cytotoxicity was evaluated by the MTT assay at 96 h. The 50% cytotoxic concentration (CC50) was calculated with the Reed & Muench method.
The cytotoxic effect of the target compounds on Vero cells was assayed by the CPE method. Briefly, cells were seeded into 96-well culture plates (3×104 cells/well) and were incubated overnight. Then, different concentrations of the test compounds were applied in triplicate. After incubation for 3 d, Median toxic concentration (TC50) was defined as the concentration that inhibits 50% cellular growth in comparison with untreated controls and calculated by the Reed and Muench method.
4.4. Anti-HCV assay
The Huh7.5 cells were seeded into 6-well plates (Costar) at a density of 3×104 cells/cm2. After 6 h incubation, cells were infected with HCV viral stock (45 u/cell) and treated simultaneously with the test compounds or the control. The culture medium was removed after 96 h inoculation and the intracellular total RNA was extracted with RNeasy Mini Kit (Qiagen, Hilden, Germany). The HCV RNA was quantified directly with a one-step RTPCR kit (Invitrogen). The 50% inhibition concentration (IC50) was calculated with the Reed & Muench method. SI value was calculated as the ratio of CC50/IC50.
4.5. Anti-EV71 assay
The anti-EV71 activity of the target compounds was also assayed by the CPE method. Briefly, cells (3×104 cells/well) were plated into 96-well culture plates. The cells were infected with EV71 of 100TCID50. Then, various concentrations of the test compounds were supplemented immediately for incubation of another 48 h. The IC50 was determined by the Reed and Muench method. The SI value was calculated as the ratio of TC50/IC50.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 81273439 and 81202414) and Student Fund of Innovation Project of Peking Union Medical College (No. 2013-1007-10).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Contributor Information
Yuhuan Li, Email: yuhuanlibj@126.com.
Zhuorong Li, Email: l-z-r@263.net.
References
- 1.Davila JA, Morgan RO, Shaib Y, McGlynn KA, EI-Serag HB. HCV and increasing incidence of hepato cellular carcinoma: apopulation-based study. Gastroenterology. 2004;127:1372–1380. doi: 10.1053/j.gastro.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 2.McHutchison JG, Lawitz EJ, Shiffman ML, Muir AJ, Galler GW, McCone J. Peginterferon α-2b or α-2a with ribavirinfor treatment of hepatitis infection. N Engl J Med. 2009;361:580–593. doi: 10.1056/NEJMoa0808010. [DOI] [PubMed] [Google Scholar]
- 3.Stankiewicz-Drogoń A, Dörner B, Erker T, Boguszewska-Chachulska AM. Synthesis of new acridone derivatives, inhibitors of NS3 helicase, which efficiently and specifically inhibit subgenomic HCV replication. J Med Chem. 2010;53:3117–3126. doi: 10.1021/jm901741p. [DOI] [PubMed] [Google Scholar]
- 4.Kayali Z, Schmidt WN. Finally sofosbuvir: an oral anti-HCV drug with wide performance capability. Pharmgenomice Pers Med. 2014;7:387–398. doi: 10.2147/PGPM.S52629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gentile I, Buonomo AR, Borgia F, Castaldo G, Borgia G. Ledipasvir: a novel synthetic antiviral for the treatment of HCV infection. Expert Opin Investig Drugs. 2014;23:561–571. doi: 10.1517/13543784.2014.892581. [DOI] [PubMed] [Google Scholar]
- 6.Lin C, Lin K, Luong YP, Rao BG, Wei YY, Brennan DL. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN2061: structural analysis indicates different resistance mechanisms. J Biol Chem. 2004;279:17508–17514. doi: 10.1074/jbc.M313020200. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt NJ, Lennette EH, Ho HH. An apparently new enterovirus isolated from patients with disease of the central nervous system. J Infect Dis. 1974;129:304–309. doi: 10.1093/infdis/129.3.304. [DOI] [PubMed] [Google Scholar]
- 8.Chang LY, Lin TY, Hsu KH, Huang YC, Lin KL, Hsueh C. Clinical features and risk factors of pulmonary oedema after enterovirus-71-related hand, foot, and mouth disease. Lancet. 1999;354:1682–1686. doi: 10.1016/S0140-6736(99)04434-7. [DOI] [PubMed] [Google Scholar]
- 9.Shimizu H, Utama A, Yoshii K, Yoshida H, Yoneyama T, Sinniah M. Enterovirus 71 from fatal and nonfatal cases of hand, foot and mouth disease epidemics in Malaysia, Japan and Taiwan in 1997–1998. Jpn J Infect Dis. 1999;52:12–15. [PubMed] [Google Scholar]
- 10.Yang F, Ren L, Xiong Z, Li J, Xiao Y, Zhao R. Enterovirus 71 outbreak in the People׳s Republic of China in 2008. J Clin Microbiol. 2009;47:2351–2352. doi: 10.1128/JCM.00563-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu KX, Ng MM, Chu JJ. Developments towards antiviral therapies against enterovirus 71. Drug Discov Today. 2010;15:1041–1051. doi: 10.1016/j.drudis.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shang LQ, Xu MY, Yin Z. Antiviral drug discovery for the treatment of enterovirus 71 infections. Antivir Res. 2013;97:183–194. doi: 10.1016/j.antiviral.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Kuo RL, Shih SR. Strategies to develop antivirals against enterovirus 71. Virol J. 2013;10:28–35. doi: 10.1186/1743-422X-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 15.Peng ZG, Zhao ZY, Li YP, Wang YP, Hao LH, Fan B. Host apolipoprotein B messenger RNA–editing enzyme catalytic polypeptide-like 3G is an innate defensive factor and drug target against hepatitis C virus. Hepatology. 2011;53:1980–1989. doi: 10.1002/hep.24160. [DOI] [PubMed] [Google Scholar]
- 16.Li YP, Peng ZG, Hao LH, Wu ZY, Zhu YP, Hu LX. Synthesis of novel substituted N-aryl benzamides as hA3G stabilizers and their inhibitory activity against hepatitis C virus replication. Acta Pharm Sin B. 2013;3:312–321. [Google Scholar]
- 17.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 18.Hao LH, Li YP, He WY, Wang HQ, Shan GZ, Jiang JD. Synthesis and antiviral activity of substituted bisarylamide compounds as novel influenza virus inhibitors. Eur J Med Chem. 2012;55:117–124. doi: 10.1016/j.ejmech.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Ji XY, Wang HQ, Hao LH, He WY, Gao RM, Li YP. Synthesis and antiviral activity of N-phenylbenzamide derivatives, a novel class of enterovirus 71 inhibitors. Molecules. 2013;18:3630–3640. doi: 10.3390/molecules18033630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pal MJ, Swamy NK, Hameed SH, Padakanti S, Yeleswarapu KR. A rapid and direct access to symmetrical/unsymmetrical 3, 4-diarylmaleimides and pyrrolin-2-ones. Tetrahedron. 2004;60:3987–3997. [Google Scholar]
- 21.Lunde SA, Sydnes MO. One-pot procedures for the formation of secondary aryl amines from nitro aryls. Synlett. 2013;24:2340–2344. [Google Scholar]