

Significance
Type 1 diabetes is an autoimmune disease in which the insulin-producing beta cells within the islets of Langerhans of the pancreas are destroyed by T cell-mediated immune attack. The peptide epitopes derived from islet proteins that are targeted by CD4+ T cells have been difficult to determine. We show in the nonobese diabetic (NOD) mouse model of the disease that a peptide (WE14) derived from chromogranin A is likely posttranslationally modified to create a target epitope. We hypothesize that the modification is caused by transpeptidation in which other peptides are fused to the N terminus of WE14. We propose that, in autoimmunity, new epitopes created in the target organs can be attacked by T cells that are normally nonreactive to natural self-antigens.
Keywords: autoimmunity, antigen processing, posttranslational modification, crystallography, transpeptidation
Abstract
Chromogranin A (ChgA) is an autoantigen for CD4+ T cells in the nonobese diabetic (NOD) mouse model of type 1 diabetes (T1D). The natural ChgA-processed peptide, WE14, is a weak agonist for the prototypical T cell, BDC-2.5, and other ChgA-specific T-cell clones. Mimotope peptides with much higher activity share a C-terminal motif, WXRM(D/E), that is predicted to lie in the p5 to p9 position in the mouse MHC class II, IAg7 binding groove. This motif is also present in WE14 (WSRMD), but at its N terminus. Therefore, to place the WE14 motif into the same position as seen in the mimotopes, we added the amino acids RLGL to its N terminus. Like the other mimotopes, RLGL-WE14, is much more potent than WE14 in T-cell stimulation and activates a diverse population of CD4+ T cells, which also respond to WE14 as well as islets from WT, but not ChgA−/− mice. The crystal structure of the IAg7–RLGL–WE14 complex confirmed the predicted placement of the peptide within the IAg7 groove. Fluorescent IAg7–RLGL–WE14 tetramers bind to ChgA-specific T-cell clones and easily detect ChgA-specific T cells in the pancreas and pancreatic lymph nodes of NOD mice. The prediction that many different N-terminal amino acid extensions to the WXRM(D/E) motif are sufficient to greatly improve T-cell stimulation leads us to propose that such a posttranslational modification may occur uniquely in the pancreas or pancreatic lymph nodes, perhaps via the mechanism of transpeptidation. This modification could account for the escape of these T cells from thymic negative selection.
Type 1 diabetes (T1D) is an autoimmune disease characterized by infiltration of T cells into the pancreatic islets of Langerhans, resulting in destruction of insulin-secreting beta cells (reviewed in ref. 1). Numerous autoantigens, including insulin/proinsulin itself and chromogranin A (ChgA), have been reported to be T-cell targets in the disease in mice and/or humans (reviewed in refs. 2 and 3). In the nonobese diabetic (NOD) mouse model of T1D, epitopes in the insulin B chain have been shown to be essential for T1D development (4), and CD4+ T-cell clones specific for insulin or ChgA have been shown to be particularly potent in induction of T1D in immune-deficient NOD-scid mice (5). ChgA (gene name CHGA) is a member of the granin family of neuroendocrine secretory proteins. Full-length ChgA is proteolytically processed to yield several functional peptides including a small peptide, WE14 (6). Our group participated in the study that first identified ChgA as the autoantigen and the WE14 peptide as the key epitope for the prototypical BDC-2.5 and other NOD-derived CD4+ T cells (7).
WE14 is a weak agonist for these ChgA-specific T cells, but we (7) and others (8, 9) have identified a series of mimotope peptides that are very strong agonists for these T cells. Comparison of the sequence of WE14 with these mimotope peptides reveals the presence of a common amino acid motif, WXRM(D/E), at the C terminus where it is predicted to occupy positions p5 to p9 in the IAg7 binding groove. WE14 shares the motif with the mimotopes, WSRMD, but it lies at the N terminus of the peptide. Therefore, we have suggested that, to bind similarly to IAg7 as the mimotopes, the N-terminal motif would fill p5 to p9, leaving p1 to p4 empty, resulting in a very unstable complex (3, 7, 10).
In this article, we show that extending the N terminus of WE14 with four amino acids optimal for the p1-to-p4 positions of the IAg7 groove creates a “super agonist” that stimulates a variety of ChgA-specific T cells much better than WE14 itself. The crystal structure of this peptide bound to IAg7 confirmed the validity of the strategy, showing the WSRMD motif in the p5-to-p9 positions of the IAg7 binding groove. We suggest that, in vivo, a unique pancreatic N-terminal posttranslational modification of WE14, perhaps via transpeptidation, might be the mechanism for greatly improving WE14 presentation in the pancreas. Poor presentation of unmodified WE14 in the thymus could explain how ChgA-specific T cells escape negative selection in the thymus.
Results
Improvement of WE14 Presentation by IAg7 After N-Terminal Modification.
Our previous experiments showed that adding the four natural ChgA amino acids (EDKR) upstream of WE14 in unprocessed ChgA (Fig. 1A) in fact destroyed WE14 presentation (7). Therefore, we replaced these amino acids with RLGL from the N-terminal end of one of our previously reported mimotopes, S3 (SRLGLWVRME), to yield RLGL-WE14 (Fig. 1A). To characterize the activity of this fused peptide, we prepared a soluble version and compared it with WE14 in its ability to stimulate two prototypical ChgA-reactive T-cell hybridomas, BDC-2.5 and BDC-10.1 (11, 12), when presented by an IAg7-expressing antigen-presenting cell (Fig. 1B). As we previously reported for the S3 mimotope (7), the RLGL-WE14 peptide was many orders of magnitude more potent than WE14 in activating the hybridomas.
Fig. 1.

ChgA tetramer can detect diabetogenic T cells. (A) The aligned sequences of four peptides are shown: WE14, WE14 with the natural EDRK N-terminal extension, the S3 mimotope, and modified WE14 containing the RLGL N-terminal extension from S3. (B) RLGL-WE14 can stimulate the BDC-2.5 and BDC-10.1 T-cell hybridomas in a much lower concentration than WE14. (C) RLGL-WE14/IAg7 tetramer stains the BDC-2.5 and BDC-10.1 T-cell hybridomas but not a control hybridoma, B3K-506, specific for IAb plus the p3K peptide. (D) A representative experiment showing that the RLGL-WE14/IAg7 tetramer detects CD4+, but not CD8+, pancreatic and pancreatic lymph node T cells from two 19-wk-old NOD female mice from the same cage, one with diabetes and one without diabetes.
We also prepared a fluorescent tetramer of IAg7 covalently occupied with the RLGL-WE14 peptide (13, 14). This tetramer stained with very high avidity both the BCD-2.5 and BDC-10.1 cells, but not a control T-cell hybridoma (Fig. 1C). We also tested the tetramer with CD4+ T cells from the pancreas and pancreatic lymph nodes of prediabetic and new onset diabetic NOD mice (Fig. 1D). In all cases, high avidity tetramer-positive cells were easily detected in the CD44 high CD4+, but not CD8+, T cells. In the pancreas, the frequency of tetramer-positive T cells was higher in the prediabetic than in the diabetic mice, consistent with the hypothesis that ChgA-reactive T cells leave the pancreas as the source of antigen decreases via the destruction of beta cells. The frequency of tetramer-positive T cells in the pancreatic lymph nodes was much lower than in the pancreas, and, in this case, there was no reduction in the frequency with the onset of diabetes, suggesting that these T cells may be longer-lived central memory CD4+ T cells persisting after the destruction of pancreatic beta cells.
Crystal Structure of IAg7 in Complex with RLGL-WE14.
Our data strongly suggested that RLGL-WE14 represents a version of WE14 whose activity in detecting and stimulating ChgA-specific T cells is greatly improved because the N-terminal modification places the critical WSRMD motif of WE14 in the correct position in the IAg7 binding groove. To confirm this conclusion, we solved the crystal structure of the RLGL–WE14–IAg7 complex to a resolution of 2.4 Å (Table S1). Electron density clearly showed, as predicted, that the WSRMD motif of WE14 lay in the p5-to-p9 positions of the IAg7 binding groove with the RLGL N-terminal extension in positions p1 to p4 (Fig. 2A). A surface view of the complex (Fig. 2B) shows that the side chains of the anchor residues (colored blue), two from RLGL (p1R and p4L) and two from WE14 (p6S and p9D), are buried in the binding groove and not exposed on the surface. Fig. 2 C–F shows that the side chains of these anchor residues fit particularly well in the usual four pockets of the groove. The side chains of peptide amino acids that are well exposed on the surface for potential T-cell receptor (TCR) contact (colored red in Fig. 2B) include those of p5W, p7R, and p8M from WE14, which are also common to nearly all previously identified mimotopes, as well as that of p2L from the RLGL extension.
Table S1.
Table of crystallographic data and refinement statistics
| Data Collection | |
| Space group | P 43 21 2 |
| Cell dimensions | |
| a, b, c, Å | 161.77, 161.77, 204.16 |
| α, β, γ, ° | 90, 90, 90 |
| Resolution, Å | 2.45–50 (2.45–2.48)* |
| Rsym or Rmerge† | 0.05 (0.26)* |
| I/σI | 37 (1.35) |
| Completeness, % | 94 (93.58) |
| Redundancy | 2.8 (1.6) |
| Refinement | |
| Resolution, Å | 2.45–44.9 |
| No. of reflections | 93,184 |
| Rwork/Rfree‡ | 18.9/24.2 (34/41) |
| No. of atoms | 12,201 |
| Protein | 11,143 |
| Ligand/ion | 26 |
| Water | 1,032 |
| B-factors | |
| Protein | 28.86 |
| Ligand/ion | 28.81 |
| Water | 23.66 |
| Rms deviations | |
| Bond lengths, Å | 0.008 |
| Bond angles, ° | 1.120 |
| Ramachandran plot | |
| Most favored region, % | 95.4 |
| Additional allowed region, % | 4.17 |
| Generously allowed region, % | 0.0 |
| Disallowed region, % | 0.43 |
All data (outer shell).
Rmerge = Σ(|I −〈I〉|)/Σ(I).
Rfree = ΣTest||Fobs| − |Fcalc||/ΣTest |Fobs|, where “Test” is a set of ∼5% of the total reflections randomly chosen and set aside.
Fig. 2.

The crystal structure of RLGL-WE14 binding to IAg7. (A) Overview of the crystal structure of the α1/β1 portion of IAg7 in complex with RLGL-WE14 peptide solved at a resolution of 2.4 Å. IAg7 α1 (cyan) and β1 (magenta) are shown as ribbon structures. The RLGL-WE14 peptide is represented as a wireframe with CPK coloring (carbon white, oxygen red, nitrogen blue). The electron density (2Fo − Fc, peptide omit, contoured at 1σ) is shown around the peptide. (B) The solvent accessible surface of the IAg7 complex with RLGL-WE14 (p1 to p9) is shown: IAg7 α1 (cyan), IAg7 β1 (magenta), peptide backbone (white), side chains of anchors p1, p4, p6, and p9 (blue) and p2, p5, p7, and p8 (red). (C) Interactions between amino acids in the p1 pocket and the p1R of RLGL-WE14. (D) Same for p4L. (E) Same for p6S. (F) Same for p9D. (C–F) Wireframe representations O (red), N (blue), S (yellow), C-αIAg7 (cyan), C-βIAg7 (magenta), C-RLGL-WE14 (white), and H-bonds/salt bridges (green).
CD4+ T Cells with a Variety of TCRs Cross-React Among RLGL-WE14, WE14, and ChgA.
The RLGL-WE14 tetramer detected two previously identified ChgA-specific CD4+ T cells, BDC-2.5 and BDC-10.1, that had very different T-cell receptors (TCRs) (Table S2) and also identified a substantial population of activated CD4+ T cells present in NOD mice (Fig. 1D). To test the generality of this relationship among ChgA, WE14, and the RLGL-WE14 peptide, we produced additional RLGL-WE14 reactive T-cell hybridomas directly from NOD splenic T cells after activation in vitro with the peptide, but without previous priming in vivo. We selected three hybridomas for extensive characterization, whose TCRs were not identical to each other or to those of BDC-2.5 or BDC-10.1 (Table S2).
Table S2.
TCR sequences of ChgA-specific T cells
| T cell | Vα/Jα | αCDR3 | Vβ/Jβ | CDR3 |
| BDC-2.5 | 1.5/22 | CASS- -LA-GSWQLIF | 4/1.2 | CASSQGGTTNSDYTF |
| BDC-10.1 | 9.2/32 | CALEGHYGGSGNKLIF | 15/2.6 | CGAR-GSGTD- -QYF |
| G7W-68 | 14/47 | CVVG- -DA- --NKMIF | 12/2.6 | CAS- -RLGGY-EQYF |
| G7W-120 | 9.2/32 | CALEGHYGGSGNKLIF | 15/2.6 | CGAR-EQGG- -EQYF |
| G7W-149 | 10/49 | CAME- -RR-GYQNFYF | 12/2.1 | CAS-RAEGADYAEGF |
Bold and underlined, N region.
The new T-cell hybridomas, as well as BDC-2.5 and BDC-10.1, were tested in vitro for their production of IL-2 in response to various concentrations of the RLGL-WE14 peptide presented by M12.C3.G7, an IAg7-expressing variant of the B-cell lymphoma, M12.C3 (15). All five hybridomas responded well to the RLGL-WE14 peptide (Fig. 3A). They also all responded to high concentrations of the weakly stimulatory natural WE14 peptide, but not to another ChgA peptide (amino acids 11–24, DTKVMKCVLEVISD) that has been suggested by others (16, 17) to be the natural ChgA epitope for BDC-2.5 (Fig. 3B). Most importantly, as previously demonstrated with BDC-2.5 and BDC-10.1 (7), the three new hybridomas responded to antigen supplied by pancreatic islets from WT, but not ChgA−/− mice (18) (Fig. 3C). A control insulin-specific hybridoma, PCR-1.10 (19), responded well to islets from both mice.
Fig. 3.

Diabetogenic T cells with a variety of TCRs cross-react with modified WE14, WE14, and ChgA. Three new T-cell hybridomas were generated from NOD splenic CD4+ T cells activated in vitro with RLGL-WE14 peptide. The sequences of their TCR alpha and beta V-domains are listed in Table S2 along with those of BDC-2.5 and BDC-10.1 The IL-2 response of the five hybridomas to (A) various doses of RLGL-WE14 or (B) no peptide (white), 400 μg/mL ChgA Vasostatin-1 (11-24) peptide (green), 400 μg/mL (red) or 800 μg/mL (blue) WE14 presented by M12.C3.G7. (C) The IL-2 response of the five hybridomas and an insulin reactive control hybridoma (PCR1-10) to pancreatic islets cells from ChgA+/+ (red) or ChgA−/− (blue) mice using spleen cells from IAg7+/+/ChgA−/− mice as antigen-presenting cells . Results were averaged from three or more experiments. Error bars are SEMs.
Confirming the Position of Natural WE14 Binding in the IAg7 Peptide Groove.
As a further test of the location of WE14 in the IAg7 binding groove, we used an approach with which we previously determined the location of the insulin B:9–23 peptide in the IAg7 groove (14, 20). Our structure of the RLGL-WE14 bound to IAg7 and previous IAg7 structures (21, 22) showed the proximity of the side chain of the peptide p6 amino acid to that of α62N of the IAg7 alpha-chain (Fig. 4A). Therefore, we prepared soluble and membrane-bound versions of IAg7-WE14 with the S in the WSRMD motif and α62N of IAg7 mutated to cysteines. These cysteines should form a disulfide bond between the IAg7 β-chain–linked peptide and IAg7 α-chain, but only if the WCRMD were to bind in the p5-to-p9 positions of the IAg7 groove (modeled in Fig. 4B). The soluble form was purified from the supernatant of infected High Five insect cells. SDS/PAGE analysis with and without 2-mercaptoethanol reduction confirmed the complete formation of this disulfide (Fig. S1A).
Fig. 4.

Disulfide link between IAg7 and WE14 can stabilize the binding of the peptide to MHCII groove. (A) Top view of a portion (p5 to p8) of RLGL-WE14 bound to IAg7 showing the proximity of the side chains of 62N of the IAg7 alpha-chain and RLGL-WE14 p6S. (B) The same view but with a model of a predicted disulfide between α62N and p6S mutated to C (WE14-SS). (C) IL-2 production by the five ChgA-specific and one control insulin-specific (PCR1-10) hybridomas to linked versions of HEL control (white), WE14 (green), RLGL-WE14 (red), and WE14-SS (blue) peptides. For presentation, each of the IAg7-linked peptides was expressed via baculovirus on the surface of ICAM+/B7+ SF9 insect cells (36). Results are the average and SEM of three or more experiments.
Fig. S1.

Expression and properties of the IAg7-WE14-SS construct. As described in SI Materials and Methods and Fig. S3, a sequence encoding the WE14 peptide with an S-to-C mutation at the second position was cloned into a baculovirus linked to either a secreted or membrane-bound form of IAg7 that carried an N-to-C mutation at position 62 of the IAg7 α1 domain. (A) Virus containing the soluble form of IAg7-WE14-SS was used to infect High Five cells, and the protein was purified from the culture supernatant. The figure shows a Coomassie blue-stained SDS page gel analysis of the protein before and after treatment with 2-mercaptoethanol. (B) ICAM+/B7+ SF9 cells were infected with virus encoding the membrane-anchored version of the IAg7-WE14-SS construct or with membrane-anchored versions of IAg7-WE14 or IAg7-RLGL-WE14. After 3 d, the cells were stained with the anti-IAg7–specific mAb, OX-6, and the anti-baculovirus GP64-specific mAb, ACV1. Infected cells were gated via the ACV1 mAb, and the overlaid histograms show the level of staining with OX-6. Uninfected SF9 cells were used as a negative control.
The membrane-anchored form was expressed in intercellular adhesion molecule (ICAM)+/B7+ SF9 insect cells (13) and IAg7 surface expression compared by flow cytometry to that of WE14 or RLGL-WE14 in the same construct but without the mutations (Fig. S1B). IAg7-WE14-SS expressed as well as the IAg7-WE14 but somewhat less than the IAg7–RLGL–WE14 complex. These cells were tested for their ability to stimulate the ChgA-specific hybridomas and a control insulin-specific hybridoma (Fig. 4C). As expected, the ChgA-specific T cells responded very strongly to the RLGL-WE14. The ChgA-specific T cells responded weakly or not at all to the tethered unmodified WE14 peptide. However, all but the weakest responding ChgA hybridoma, G7W-149, also responded to the disulfide-linked WE14. As expected, the insulin-specific control hybridoma failed to respond to any of these constructs. The responses to the disulfide-linked WE14 peptide were not as strong as those to the RLGL-WE14 peptide. This result might have been because the surface expression level was lower but more likely was due to the contribution of the upwardly pointing p2L from the RLGL extension to TCR recognition.
In summary, all five T-cell hybridomas bearing various TCRs and identified independently were demonstrated to be both ChgA-dependent and both RLGL-WE14– and WE14-reactive. We conclude that the natural epitope for a variety of ChgA-reactive T cells is some version of WE14 that places the WSRMD motif in positions p5 to p9.
Analysis of the Contribution of Individual Amino Acids of the Extension to WE14.
The extremely large improvement in presentation by the RLGL N-terminal extension to WE14 led us to examine the contributions of individual amino acids of the extension to its activity. We tested the activity of WE14 to which the four amino acids were added, one at a time, creating intermediate synthetic peptides of L-WE14, GL-WE14, and LGL-WE14. These peptides were compared with RLGL-WE14 and WE14 in their ability to activate two of the strongest responders in our group of ChgA-specific T-cell hybridomas, BDC-10.1 (Fig. 5A) and G7W-68 (Fig. 5B).
Fig. 5.

Successive N-terminal modification of WE14 greatly increases the response of ChgA-specific hybridomas. The response of the BDC-10.1 (A) and G7W-68 (B) hybridomas to various doses of WE14 (black), or WE14 successively extended by L (orange), GL (green), LGL (blue), and RLGL (red). The structural contributions of the L, GL, and LGL extensions are shown in C, D, and E, respectively. (F) Based on the structural features of IAg7, peptide binding, and elution studies with IAg7 and the sequences of mimotopes for BDC-2.5 and other ChgA-specific T cells, potential amino acids at p1, p2, p3, and p4 of an N-terminally extended WE14 peptide predicted to be compatible with creating a strong agonist peptide are shown. The possibility that a p1 amino acid may not be essential for a strong agonist is indicated.
The simple addition of an L to WE14 increased the activity of the peptide 10-fold for BDC-10.1 (Fig. 5A) or 100-fold for G7W-68 (Fig. 5B). This increase in activity was undoubtedly linked in part to its providing a third anchor residue at p4, whose side chain is well captured in the p4 pocket (Fig. 2D). However, crystallographic studies (21, 22), as well as those involving screening peptide libraries (8, 9) or examining peptides naturally bound to IAg7 (23), show that other amino acids such as I, V, A, G, and T, should suffice as p4 anchors. As shown in Fig. 5C, perhaps more important than the nature of the p4 side chain, the backbone O of p4 completes a pair of H-bonds with the side chain of IAg7 α62N, part of the conserved interactions of MHCII with the peptide backbone seen in nearly all MHCII–peptide complexes. Furthermore, the backbone N of the 4L peptide bond makes an H-bond to the backbone O of Y9 in the first beta-strand of the IAg7 alpha chain.
The addition of a p3G to L-WE14 did not further improve the activity and, for BDC-10.1, even lessened the activity of the previous L addition (Fig. 5A). Because it lacks a side chain, the only contribution this p3G could make to improve peptide binding is via IAg7 interactions with its backbone. However, the backbone of the p3 does not participate in any of the conserved MHCII–peptide interactions (Fig. 5D). On the other hand, side chains of amino acids at p3 position of MHCII bound peptides are often important in TCR interaction. It is noteworthy that, in our previous characterization of the S3 mimotope, substitutions for G of A, S or T, but not K, W, D, or I, were accepted at the p3 position for stimulation of BDC-2.5 or BDC-10.1. Likewise in other studies, in which libraries were used to find mimotopes for BDC-2.5 and other ChgA-specific T cells, A and P were favored at the p3 position (8, 9). These results suggest that the p3 side chain primarily needs to be small and needs to keep out of the way rather than contribute positively to TCR recognition.
The further addition of the p2L to the peptide resulted in another large increase in activity, creating a peptide 10,000-fold better than the WE14 peptide (Fig. 5 A and B). Our structure suggested two ways that this p2L improves presentation (Fig. 5E). First, whereas in MHCII structures p2 is not an anchor residue, the N and O of its backbone form H-bonds to the IAg7 helix β80N side chain, another one of the highly conserved backbone interactions mentioned above that is important for binding and positioning the peptide within the binding groove. Secondly, as seen in Fig. 2B, the side chain p2L points straight up from the binding groove and, as mentioned above, is likely to contribute to TCR interaction as well. Furthermore, whereas in our experiments L is sufficient for all of the WE14 T cells we have studied, others have suggested that, in other mimotopes, I, A, or R is also compatible at least for some ChgA-specific T cells (8, 9).
The LGL-WE14 peptide was still about 10-fold less active than the full-length RLGL-WE14 peptide (Fig. 5 A and B), presumably due to the effect of adding the fourth p1 anchor. We chose R at this position based on the previous crystal structure of HEL peptide bound to IAg7, which showed extensive interaction of the p1R side chain with the IAg7 p1 pocket (22), a rationale borne out by our structure (Fig. 2C). However, as in the case of the IAg7 p4 anchor, other studies (8, 9, 21, 23) have shown that the p1 pocket of IAg7 is very forgiving, accepting a variety of other amino acids, including Q, E, V, A, S, I, K, G, H, M, and T.
Therefore, based on our structure, the properties of the p1 and p4 IAg7 pockets, and the variations in p1-to-p4 amino acids in the mimotopes reported by ourselves and others, we conclude that many different peptide extensions to WE14 should create highly active agonists for WE14-specifc CD4+ T cells (Fig. 5F). In the discussion below, we consider the implications of these findings for the possible posttranslational modifications of WE14 in vivo that might generate the natural ligand driving CD4+ ChgA-specific T cells involved in T1D.
Discussion
CD4+ T cells specific for ChgA were among the first identified from the NOD mouse model of T1D (11, 12), yet ChgA as the source of the antigen and WE14 as the relevant epitope were only recently reported (7). Our functional and structural data presented here led us to conclude that WE14 is an essential part of the epitope for a diverse set of ChgA-specific CD4+ T cells in vivo. Another laboratory has reported that a different unrelated ChgA peptide is the natural epitope for transgenic BDC-2.5 T cells (16, 17), but we were not able to reproduce this finding here with the BDC-2.5 hybridoma, perhaps due to sensitivity differences between hybridomas and the transgenic T cells. However, we show here that a set of T-cell hybridomas, including BDC-2.5, whose TCRs contain a variety of Vα, Jα, Vβ, and Jβ segments and CDR3 loops, all respond to the RLGL-WE14 peptide at low concentrations, to the WE14 peptide at high concentrations, and to pancreatic islets from WT ChgA mice, but not ChgA knockout mice. It is highly unlikely that all of these T cells could fortuitously cross-react as well with another unrelated ChgA peptide.
Although we conclude that WE14 is part of the functional epitope for ChgA-specific T cells, our collaborators in a previous study showed that a partially purified antigen preparation from an insulinoma tumor had a much higher specific activity than synthetic WE14 in stimulating T cells (7). We suggested that in vivo there was likely a posttranslational modification to WE14 to improve its presentation. One possibility considered has been modification by transglutaminase because there is a potential glutamine target for this enzyme in WE14 and in vitro treatment of WE14 with transglutaminase increased its stimulatory activity (24). However, the active principle in the reaction product has not yet been determined.
Based on our results reported here, we suggest alternatively that the posttranslational modification of WE14 in vivo to increase its activity is an addition of amino acids to its N terminus after this end of the peptide has been freed from ChgA by prohormone convertase processing. We propose that the most likely mechanism for this modification is reverse proteolysis, also known as transpeptidation (reviewed in ref. 25). Shown schematically in Fig. S2, it involves the reversal of the proteolytic mechanism before completion, in which the N-terminal cleavage product is still bound via its C terminus to the protease, but the C-terminal cleavage fragment is left covalently bound in the active site. Although a molecule of water usually finishes the reaction by releasing the bound peptide from the enzyme, this cleavage can also be accomplished by the N-terminal NH2 of another peptide, thus reestablishing a peptide bond to form a new chimeric peptide.
Fig. S2.

Schematic representation of the mechanism of transpeptidation by serine, threonine, or cysteine proteases.
In vitro, the use of transpeptidation in peptide synthesis began many years ago (26), but this mechanism plays a role in the natural processing of proteins and peptides in plants (27), insects (28), and microorganisms (29). More relevantly, a number of novel CD8+ T-cell tumor epitopes have been shown to arise in the proteasome of mammalian cells by transpeptidation via intra- or interprotein peptide fusions (30, 31). Certain conditions favor the transpeptidation reaction: first, a high degree of proteolysis in a confined space; second, proximity of the enzyme-recipient peptide intermediate to the N terminus of the attacking donor peptide. This condition can be established by a very high concentration of the free donor peptide or more efficiently by the tethering of the donor peptide to the enzyme-peptide intermediate, such as would occur during natural internal transpeptidation involving two parts of the same protein.
Pancreatic beta cells would seem a perfect environment for transpeptidation. They contain granules with an extremely high concentration of insulin, ChgA, and other beta-cell proteins. Granules contain the products of natural processing via prohormone convertases and carboxypeptidase E, but islets also contain many catabolic breakdown products apparently generated by lysosomal proteases during granule turnover (32). Our work and those of others suggest that many different N-terminal modifications of WE14 could create a greatly improved WE14 epitope. Mass spectrometry analysis of the partially purified preparation of antigen for ChgA-specific T cells found a high concentration of at least four granule proteins: insulin, ChgA, secretogranin-1, and secretogranin-2 (7). A scan of the sequences of these proteins (without signal peptide) for four amino acid peptides that match the possible amino acids listed in Fig. 5F yields 24 potential donor peptides for WE14 transpeptidation (Table S3). Although the required C termini of these donors would not be generated during normal convertase/carboxypeptidase processing, they could be generated by lysosomal cathepsins.
Table S3.
Potential donor peptides derived from pancreatic proteins for N-terminal WE14 modification to create strong agonists
| Protein source* | aa nos.† | IAg7 groove position (p) 1‡ 2 3 4 |
| Chromogranin A | 48–51 | I L S I |
| 147–150 | E A T T | |
| 197–200 | Q A T G | |
| 203–206 | E R G L | |
| 205–208 | G L S A | |
| 349–352 | E L T A | |
| 424–427 | S L S A | |
| Insulin-1/2 | 43–46 | E L G G |
| 53–56 | T L A L | |
| Secretogranin-1 | 85–88 | G A P V |
| 183–186 | E A S A | |
| 273–276 | G R S G | |
| 303–306 | V A T V | |
| 495–498 | R L G A | |
| 627–630 | V L T A | |
| Secretogranin-2 | 26–29 | I R A L |
| 166–169 | S L A T | |
| 179–182 | K L T G | |
| 255–258 | K R S G | |
| 259–262 | Q L G L | |
| 358–361 | E L A V | |
| 389–392 | K A P G | |
| 402–405 | G L S V | |
| 538–541 | R I P V |
Islet-specific proteins highly enriched in a partially purified antigen preparation (7).
After cleavage of the signal peptide.
May not be required.
The organ-specific posttranslational modification of self-peptides by enzymes such as transglutaminase (33) and peptidylarginine deiminases (34) has become an attractive hypothesis to explain how the T cells driving autoimmunity escape negative selection in the thymus but find their antigen in the target tissue. In the examples reported thus far, the modification can either improve MHCII binding or introduce a new site for T-cell recognition. Several reviews of transpeptidation have pointed out its potential for a role in the posttranslational generation of the targets of autoimmunity (25, 35). We propose here that transpeptidation modification of WE14 may be such a case. Finally, there is a striking parallel between how modifications of WE14 and the insulin B:9–23 peptide improve peptide presentation. In the case of B:9–23, C-terminal rather than N-terminal modifications are required to generate the optimal peptides (14, 20). These ideas spur us, and we hope others, to delve more deeply into the catabolic breakdown products of self-proteins looking for direct evidence of autoantigenic fused peptides created by transpeptidation.
Materials and Methods
Mice.
Nonobese diabetic (NOD) mice were purchased from The Jackson Laboratory. ChgA−/− mice were originally obtained from Dr. Sushil Mahata (University of California, San Diego) (18). They were partially backcrossed to NOD mice by Dr. Katherine Haskins (National Jewish Health), who provided us with H-2g7 +/+/ChgA+/− mice from this cross. We intercrossed these mice to obtain ChgA−/− and ChgA+/+ mice as sources of pancreatic islets for our T-cell stimulation studies. All mice were housed at the National Jewish Health Biological Resource Center under the Institutional Animal Care and Use Committee-approved protocols.
Reagents.
Peptides at >98% purity were synthesized and purified by CHI Scientific. The sequence of the ChgA vasostatin-1 peptide (amino acids 11–24, DTKVMKCVLEVISD) (16) was reconfirmed by mass spectrometry at the time of its use in our experiments. Phycoerythrin-streptavidin (PE-SA) was obtained from Prozyme. Fluorescently labeled mAbs for flow cytometry were as follows: FITC-B220, FITC-F4/80, APCeFluor780-CD8, PE-Cy7-CD4, and PerCP-Cy5.5-CD44 (eBioScience).
Baculovirus Constructions and Expression.
All recombinant proteins used here were produced in baculovirus as previously described (13, 14, 36). Details are in SI Materials and Methods, and the sequences of constructs containing versions of WE14 are shown in Fig. S3.
Fig. S3.
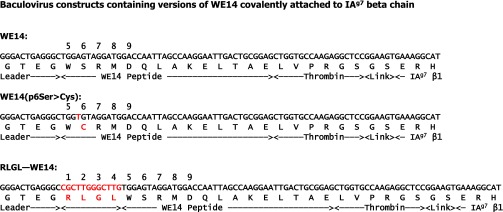
Construction of baculovirus with IAg7 containing covalently tethered versions of WE14. As described in SI Materials and Methods the DNA sequences shown were incorporated into a baculovirus vector bound to versions of IAg7. The sequences were placed between sequences encoding the IAg7 beta leader and the β1 domain of IAg7 such that the encoded peptide would be expressed covalently attached to the N terminus of the β1 domain via a flexible linker. For the WE14 and RLGL-WE14 constructs, the virus encoded wild type IAg7 alpha-chain and for the WE14-SS construct, the virus encoded the mutation in the peptide, p6S > C, such that, when expressed connected to IAg7 with the codon for α1 62N mutated to encode cysteine, a p6-to-IAg7 α64 disulfide is predicted.
T-cell/B-cell Lines.
The BDC-2.5 and BDC-10.1 T-cell hybridomas were provided by Dr. Haskins (National Jewish Health Center, Denver). The insulin B:9–23 reactive PCR-1.10 T-cell hybridoma was provided by Dr. Nakayama (Barbara Davis Center for Childhood Diabetes, Aurora, CO). The M12.C3.G7 B-cell lymphoma line was provided by Dr. Emil Unanue (Washington University, St. Louis, MO).
Production of T-Cell Hybridomas.
T-cell hybridomas specific for RLGL-WE14 were prepared as previously described (37). Details are in SI Materials and Methods.
Preparation of NOD Pancreatic and Pancreatic Lymph Node Cells.
Fresh isolated NOD pancreases were cut into small pieces and digested in 50 mL of balanced salt solution (BSS) containing 5% (vol/vol) FBS plus 5 µM CaCl2 and 100 µg/mL collagenase (C9407; Sigma) at 37° C for 15 min. The digested mixture was then washed with BSS, crushed, and passed through a 100-μm nylon mesh screen to remove residual tissue, and the cells were resuspended in culture medium. Pancreatic lymph nodes were crushed, and the cells were resuspended in culture medium.
Flow Cytometric Analysis of IAg7 Tetramer Binding.
A total of 2–10 × 105 hybridoma or 1–10 × 106 pancreatic or pancreatic lymph node cells were incubated in 25 µL of PE-SA tetramer (15–20 µg/mL) in culture medium containing excess 24G2 FcR-specific mAbs for 2 h at 37 °C in a humidified 10% CO2 incubator, with gentle agitation every 30 min. The surface-staining antibodies (Fluorescent anti-B220, -F4/80, -CD8, -CD44, and -CD8 mAbs) were added if necessary, and cells were incubated at 4° C for another 20 min. The cells were washed and analyzed on a FACScan (Becton Dickinson) flow cytometer for hybridomas or the Cyan flow cytometer (Dako) for pancreatic and pancreatic lymph node cells.
IL-2 Assay.
The responses of T-cell hybridomas to various stimuli were assessed by IL-2 production as previously described (37). Details are in SI Materials and Methods.
Protein Crystallization.
Soluble IAg7-RLGL-WE14 protein was expressed in baculovirus-infected High Five insect cells. After removal of the C-terminal zippers and the BirA tag from the protein with papain, it was concentrated to 7 mg/mL. Crystals were grown by the hanging-drop vapor-diffusion method at room temperature using a mother liquor of 1.6 M ammonium citrate, pH 6.5. The crystals were cryoprotected by the well solution plus 25% glycerol.
Structure Data Collection and Processing.
Diffraction data were collected at beamline 24-ID-C at the Advanced Photon Source, Argonne National Laboratory using the Pilatus detector. The data were processed with HKL2000 (38). The structures were solved by Phaser (39) using the molecular replacement method and refined by REFMAC5 (40). Manual adjustments to the structure were performed with Coot (41). Data collection and refinement statistics are shown in Table S1.
SI Materials and Methods
Baculovirus Constructions and Expression.
A baculovirus encoding in a single virus the extracellular domains of the α- and β-chains of IAg7 with a covalently attached peptide were constructed as previously described (13, 14, 36). Briefly, common features in all of the constructs were a flexible linker attaching the peptide via its C terminus to the N terminus of the IAg7 β-chain plus a stabilizing acid-base leucine zipper attached to the C termini of the IAg7, the basic half of the zipper to the IAg7 α-chain and the acidic half to the β-chain. For insect cell surface expression, a sequence encoding the transmembrane-cytoplasmic tail of the baculovirus gp64 protein was attached to the C-terminal end of the acidic half of the zipper. ICAM+/B7+ SF9 insect cells infected 3 d previously with baculovirus expressing these constructs were used for antigen presentation. For soluble protein and fluorescent tetramers, the transmembrane region was replaced with a sequence encoding a biotinylation peptide tag. Soluble IAg7 was purified by immunoaffinity chromatography from the culture supernatants of High Five insect cells 6 d after infection with virus encoding these constructs. Purified IAg7 was biotinylated with the BirA enzyme, and an excess of bio-IAg7 was incorporated into PE-SA tetramers. Tetramers were separated from monomeric IAg7 by FPLC size exclusion chromatography on a Superdex200 10/300GL (GE Healthcare). For crystallization, the biotinylation tag and zippers were removed enzymatically with papain (42). The sequences of the portions of the constructs containing various versions of WE14 are shown in Fig. S2.
Production of T-Cell Hybridomas.
T-cell hybridomas were prepared as previously described (37). Briefly, a single cell suspension from the spleens of 10-wk-old unimmunized NOD female mice was cultured at 10 × 106 cells per mL with 10 µg/mL RLGL-WE14 peptide for 2 d. The cell blasts were purified with Lymphocyte Separation Medium (ICN Biomedicals) and then cultured in medium containing IL-2 for another 2 d. The cells were then fused to BW α-β- cells and cultured in hypoxanthine–aminopterin–thymidine for a week at limiting dilution, and clones were picked and tested for IL-2 production after stimulation in RLGL-WE14. Positive cells were cloned, retested, and stored frozen. cDNA isolated from each hybridoma was used as template in PCRs with a set of Vα, Vβ, Cα, and Cβ oligos. These PCR fragments were sequenced to determine the TCR sequences of the hybridomas.
IL-2 Assay.
Hybridoma cells (105) and 105 antigen-presenting cells (M12C3.G7) were plated in 96-well plates with various concentration of peptides for a total volume per well of 250 μL. For the islet stimulation assay, 105 hybridoma cells and 105 ChgA−/− splenic cells as antigen-presenting cells were cultured with 50 hand-picked ChgA+/+ or ChgA−/− pancreatic islets in a final volume of 250 μL. For presentation of IAg7 tethered peptides, 7.5 × 104 ICAM+/B7+ SF9 insect cells (36) expressing transmembrane-anchored versions of the constructs were used as antigen-presenting cells with 105 hybridoma T cells. Cells were incubated at 37° C in a humidified 10% CO2 incubator overnight. Supernatants were collected the next day and assayed as previously described (37) for the presence of IL-2 using the IL-2–dependent HT-2 cell line.
Acknowledgments
We thank Dr. David Neau for assistance at the Advanced Photon Source, Argonne National Laboratory, Beamline 24 ID-C. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (Grant P41 GM103403). The Pilatus 6M detector on the 24-ID-C beamline is funded by NIH Office of Research Infrastructure Programs High-End Instrumentation Grant S10 RR029205. This work was also supported in part by NIH Grants AI-18785 (to J.W.K.), CCTSI KL2 TR001080 (to S.D.), and ES025797 (to S.D.), as well as by funding from the Boettcher Foundation (S.D).
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates and structure factors have been deposited in the Research Collaboratory for Structural Bioinformatics Protein Data Bank, www.pdb.org (PDB ID code 5DMK).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1517862112/-/DCSupplemental.
References
- 1.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464(7293):1293–1300. doi: 10.1038/nature08933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DiLorenzo TP. Multiple antigens versus single major antigen in type 1 diabetes: Arguing for multiple antigens. Diabetes Metab Res Rev. 2011;27(8):778–783. doi: 10.1002/dmrr.1251. [DOI] [PubMed] [Google Scholar]
- 3.Stadinski B, Kappler J, Eisenbarth GS. Molecular targeting of islet autoantigens. Immunity. 2010;32(4):446–456. doi: 10.1016/j.immuni.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Nakayama M. Insulin as a key autoantigen in the development of type 1 diabetes. Diabetes Metab Res Rev. 2011;27(8):773–777. doi: 10.1002/dmrr.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burton AR, et al. On the pathogenicity of autoantigen-specific T-cell receptors. Diabetes. 2008;57(5):1321–1330. doi: 10.2337/db07-1129. [DOI] [PubMed] [Google Scholar]
- 6.Barkatullah SC, Curry WJ, Johnston CF, Hutton JC, Buchanan KD. Ontogenetic expression of chromogranin A and its derived peptides, WE-14 and pancreastatin, in the rat neuroendocrine system. Histochem Cell Biol. 1997;107(3):251–257. doi: 10.1007/s004180050110. [DOI] [PubMed] [Google Scholar]
- 7.Stadinski BD, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. 2010;11(3):225–231. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Judkowski V, et al. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J Immunol. 2001;166(2):908–917. doi: 10.4049/jimmunol.166.2.908. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida K, et al. Evidence for shared recognition of a peptide ligand by a diverse panel of non-obese diabetic mice-derived, islet-specific, diabetogenic T cell clones. Int Immunol. 2002;14(12):1439–1447. doi: 10.1093/intimm/dxf106. [DOI] [PubMed] [Google Scholar]
- 10.Marrack P, Kappler JW. Do MHCII-presented neoantigens drive type 1 diabetes and other autoimmune diseases? Cold Spring Harb Perspect Med. 2012;2(9):a007765. doi: 10.1101/cshperspect.a007765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haskins K, Portas M, Bergman B, Lafferty K, Bradley B. Pancreatic islet-specific T-cell clones from nonobese diabetic mice. Proc Natl Acad Sci USA. 1989;86(20):8000–8004. doi: 10.1073/pnas.86.20.8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haskins K, Wegmann D. Diabetogenic T-cell clones. Diabetes. 1996;45(10):1299–1305. doi: 10.2337/diab.45.10.1299. [DOI] [PubMed] [Google Scholar]
- 13.Crawford F, Kozono H, White J, Marrack P, Kappler J. Detection of antigen-specific T cells with multivalent soluble class II MHC covalent peptide complexes. Immunity. 1998;8(6):675–682. doi: 10.1016/s1074-7613(00)80572-5. [DOI] [PubMed] [Google Scholar]
- 14.Crawford F, et al. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci USA. 2011;108(40):16729–16734. doi: 10.1073/pnas.1113954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carrasco-Marin E, Shimizu J, Kanagawa O, Unanue ER. The class II MHC I-Ag7 molecules from non-obese diabetic mice are poor peptide binders. J Immunol. 1996;156(2):450–458. [PubMed] [Google Scholar]
- 16.Nikoopour E, et al. Cutting edge: Vasostatin-1-derived peptide ChgA29-42 is an antigenic epitope of diabetogenic BDC2.5 T cells in nonobese diabetic mice. J Immunol. 2011;186(7):3831–3835. doi: 10.4049/jimmunol.1003617. [DOI] [PubMed] [Google Scholar]
- 17.Nikoopour E, et al. Vasostatin-1 antigenic epitope mapping for induction of cellular and humoral immune responses to chromogranin A autoantigen in NOD mice. Eur J Immunol. 2014;44(4):1170–1180. doi: 10.1002/eji.201343986. [DOI] [PubMed] [Google Scholar]
- 18.Montesinos MS, et al. The crucial role of chromogranins in storage and exocytosis revealed using chromaffin cells from chromogranin A null mouse. J Neurosci. 2008;28(13):3350–3358. doi: 10.1523/JNEUROSCI.5292-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakayama M, et al. Germline TRAV5D-4 T-cell receptor sequence targets a primary insulin peptide of NOD mice. Diabetes. 2012;61(4):857–865. doi: 10.2337/db11-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stadinski BD, et al. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci USA. 2010;107(24):10978–10983. doi: 10.1073/pnas.1006545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corper AL, et al. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science. 2000;288(5465):505–511. doi: 10.1126/science.288.5465.505. [DOI] [PubMed] [Google Scholar]
- 22.Latek RR, et al. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity. 2000;12(6):699–710. doi: 10.1016/s1074-7613(00)80220-4. [DOI] [PubMed] [Google Scholar]
- 23.Suri A, Walters JJ, Gross ML, Unanue ER. Natural peptides selected by diabetogenic DQ8 and murine I-A(g7) molecules show common sequence specificity. J Clin Invest. 2005;115(8):2268–2276. doi: 10.1172/JCI25350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delong T, et al. Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes. 2012;61(12):3239–3246. doi: 10.2337/db12-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berkers CR, de Jong A, Ovaa H, Rodenko B. Transpeptidation and reverse proteolysis and their consequences for immunity. Int J Biochem Cell Biol. 2009;41(1):66–71. doi: 10.1016/j.biocel.2008.08.036. [DOI] [PubMed] [Google Scholar]
- 26.Bergmann M, Fraenkel-Conrat H. The enzymatic synthesis of peptide bonds. J Biol Chem. 1938;124:1–6. [Google Scholar]
- 27.Bowles DJ, et al. Posttranslational processing of concanavalin A precursors in jackbean cotyledons. J Cell Biol. 1986;102(4):1284–1297. doi: 10.1083/jcb.102.4.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu M, Min T, Eliezer D, Wu H. Native chemical ligation in covalent caspase inhibition by p35. Chem Biol. 2006;13(2):117–122. doi: 10.1016/j.chembiol.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 29.Mazmanian SK, Liu G, Ton-That H, Schneewind O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science. 1999;285(5428):760–763. doi: 10.1126/science.285.5428.760. [DOI] [PubMed] [Google Scholar]
- 30.Vigneron N, et al. An antigenic peptide produced by peptide splicing in the proteasome. Science. 2004;304(5670):587–590. doi: 10.1126/science.1095522. [DOI] [PubMed] [Google Scholar]
- 31.Hanada K, Yewdell JW, Yang JC. Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature. 2004;427(6971):252–256. doi: 10.1038/nature02240. [DOI] [PubMed] [Google Scholar]
- 32.Boonen K, et al. Neuropeptides of the islets of Langerhans: A peptidomics study. Gen Comp Endocrinol. 2007;152(2-3):231–241. doi: 10.1016/j.ygcen.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 33.Sollid LM, Jabri B. Celiac disease and transglutaminase 2: A model for posttranslational modification of antigens and HLA association in the pathogenesis of autoimmune disorders. Curr Opin Immunol. 2011;23(6):732–738. doi: 10.1016/j.coi.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wegner N, et al. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol Rev. 2010;233(1):34–54. doi: 10.1111/j.0105-2896.2009.00850.x. [DOI] [PubMed] [Google Scholar]
- 35.Cresswell P. Cell biology. Cutting and pasting antigenic peptides. Science. 2004;304(5670):525–527. doi: 10.1126/science.1097508. [DOI] [PubMed] [Google Scholar]
- 36.Crawford F, Huseby E, White J, Marrack P, Kappler JW. Mimotopes for alloreactive and conventional T cells in a peptide-MHC display library. PLoS Biol. 2004;2(4):E90. doi: 10.1371/journal.pbio.0020090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White J, Kappler J, Marrack P. Production and characterization of T cell hybridomas. Methods Mol Biol. 2000;134:185–193. doi: 10.1385/1-59259-682-7:185. [DOI] [PubMed] [Google Scholar]
- 38.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 39.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dai S, et al. Crystal structure of HLA-DP2 and implications for chronic beryllium disease. Proc Natl Acad Sci USA. 2010;107(16):7425–7430. doi: 10.1073/pnas.1001772107. [DOI] [PMC free article] [PubMed] [Google Scholar]