

Abstract
The Keap1–Nrf2–ARE pathway is an important antioxidant defense mechanism that protects cells from oxidative stress and the Keap1–Nrf2 protein–protein interaction (PPI) has become an important drug target to upregulate the expression of ARE-controlled cytoprotective oxidative stress response enzymes in the development of therapeutic and preventive agents for a number of diseases and conditions. However, most known Nrf2 activators/ARE inducers are indirect inhibitors of Keap1–Nrf2 PPI and they are electrophilic species that act by modifying the sulfhydryl groups of Keap1׳s cysteine residues. The electrophilicity of these indirect inhibitors may cause "off-target" side effects by reacting with cysteine residues of other important cellular proteins. Efforts have recently been focused on the development of direct inhibitors of Keap1–Nrf2 PPI. This article reviews these recent research efforts including the development of high throughput screening assays, the discovery of peptide and small molecule direct inhibitors, and the biophysical characterization of the binding of these inhibitors to the target Keap1 Kelch domain protein. These non-covalent direct inhibitors of Keap1–Nrf2 PPI could potentially be developed into effective therapeutic or preventive agents for a variety of diseases and conditions.
Abbreviations: 1O2, singlet oxygen; AD, Alzheimer׳s disease; ARE, antioxidant response element; Bach1, BTB and CNC homology 1; BTB, broad complex, tramtrack and bric-a-brac; CBP, cAMP response element binding (CREB) protein; CDDO-Me, bardoxolone methyl; COPD, chronic obstructive pulmonary disease; CTR, C-terminal region; CVD, cardiovascular disease; DGR, double glycine repeats; FITC, flurescein isothiocyanate; FP, fluorescence polarization; GCL, glutamate-cysteine ligase; GPx, glutathione peroxidase; GST, glutathione S-transferase; H2O2, hydrogen peroxide; HO-1, heme-oxygenase-1; HTS, high-throughput screening; IBS, inflammatory bowel disease; IVR, intervening region; Keap1, Kelch-like ECH-associated protein 1; MD, molecular dynamics; NMR, .; NO, nitric oxide; NQO1, NAD(P)H quinone oxidoreductase I; Nrf2, nuclear factor erythroid 2–related factor 2; NTR, N-terminal region; , superoxide, OH·, hydroxyl radical; , peroxynitrate; PD, Parkinson׳s disease; PPI, protein–protein interaction; RNS, reactive nitrogen species; ROS, reactive oxygen species; SOD, superoxide dismutase; SPR, surface plasmon resonance; STZ, streptozotocin; THIQ, tetrahydroisoquinoline; TRX, thioredoxin; vitamin C, ascorbate; vitamin E, tocopherols
KEY WORDS: Oxidative stress, Keap1, Nrf2, Direct inhibitors of protein–protein interaction, High throughput screening assays, Structure–activity relationships, X-ray crystallography
Graphical abstract
The Keap1–Nrf2–ARE pathway is an important antioxidant defense mechanism that protects cells from oxidative stress. Non-covalent direct inhibition of the Keap1–Nrf2 protein–protein interaction (PPI) has become an important approach to upregulate the expression of ARE-controlled cytoprotective oxidative stress response enzymes in the development of therapeutic and preventive agents for a number of diseases and conditions.
1. Introduction
Redox reactions are a vital component of many natural physiological processes, and as a result, the human body is constantly exposed to numerous oxidative and electrophilic chemicals. The imbalance between biochemical processes leading to the production of oxidative and electrophilic species and those responsible for the removal of these chemicals is referred to as oxidative stress1. Oxidative stress can be caused by excess reactive oxygen species (ROS) and reactive nitrogen species (RNS) generated from both exogenous and endogenous sources. Exogenous oxidative sources include carcinogenic chemicals, environmental carcinogens, and radiation. Endogenous oxidative sources include chemicals involved in intracellular processes such as cellular signaling, metabolic processes, and inflammation that produce oxidative conditions within the body1, 2. ROS include superoxide , hydrogen peroxide (H2O2), hydroxyl radical (OH·), and singlet oxygen (1O2) and they can oxidize DNA, leading to DNA damage. RNS include peroxynitrate and nitric oxide (NO), which are also DNA oxidants1, 2. ROS and RNS are generated in the body as the result of natural physiological processes such as aerobic respiration in mitochondria and during inflammatory responses that protect our body from foreign pathogens and, in some cases, serve as signaling molecules. Sustained oxidative damage is associated with inflammation, aging and a number of diseases including cancer, diabetes, atherosclerosis, hypertension, cystic fibrosis, Parkinson׳s and Alzheimer׳s diseases2, 3. Since sustained oxidative stress conditions can cause damage to DNA and vital cellular structures, the human body has developed antioxidative and cytoprotective mechanisms against various kinds of oxidative stress4, 5.
The antioxidant defense system is the major protective mechanism used by cells to defend against and neutralize the damaging effects of oxidants and electrophiles4, 5. As shown in Fig. 1, the antioxidant defense system can involve the direct reduction of the reactive oxygen or nitrogen species by low molecular weight compounds from endogenous sources or our diet. These antioxidants are redox-active, short-lived, and consumed or modified during the process and therefore they need to be replenished or regenerated to offer further protection. Examples of these antioxidants include glutathione, ascorbate (vitamin C), tocopherols (vitamin E), lipoid acid, vitamin K, and ubiquinol, and other polyphenolic compounds4. In addition, there are various antioxidant enzymes that are involved in the more effective, catalytic detoxification of reactive oxygen or nitrogen species. These enzymes include NAD(P)H, NAD(P)H quinone oxidoreductase I (NQO1), superoxide dismutase (SOD), glutathione S-transferase (GST), glutathione peroxidase (GPx), heme-oxygenase-1 (HO-1), glutamate-cysteine ligase (GCL), catalase, and thioredoxin (TRX)4, 6. These cytoprotective proteins have relatively long half-lives, are not consumed in their antioxidant actions, and can catalyze a wide variety of chemical detoxification reactions; some of them are involved in regeneration of the small molecule antioxidants4. Many of these antioxidant cytoprotective enzymes are controlled by the same three-component transcription pathway: the antioxidant response element (ARE), the nuclear factor erythroid 2–related factor 2 (Nrf2), and the Kelch-like ECH-associated protein 1 (Keap1)4, 7.
Figure 1.

The antioxidant defense system employed by our body to defend against and neutralize the damaging effects of oxidative stress. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are constantly produced by normal cellular processes and environmental sources. Their damaging effects are mitigated through direction reduction by dietary or endogenous antioxidants or through the more efficient catalytic detoxification by various antioxidant enzymes under the control of transcription factor Nrf2. Keap1 serves as an important redox sensor involved in the feedback regulation of oxidative stress response.
2. Components of the Keap1–Nrf2–ARE pathway
2.1. Antioxidant response element (ARE)
ARE, also known as the electrophile response element (EpRE), is a cis-regulatory element or enhancer sequence, which is found in the promoter region of numerous genes encoding detoxification enzymes and cytoprotective proteins8. The nucleotide sequence of ARE has been investigated in numerous mutagenic analysis studies9, 10, 11. The exact ARE sequence varies between genes; however, the typical functionally active ARE is a 16 nucleotide sequence of 5′-TA/CAnnA/GTGAC/GTGAC/GnnnGCA/G-3′, where n is any nucleotide7, 11. Under conditions of oxidative stress, stabilized Nrf2 translocates to the nucleus, where it forms a heterodimer with Maf, and binds to the ARE sites, leading to the activation of downstream target genes8, 12, 13. Bach1 (BTB and CNC homology 1) is a transcriptional repressor of ARE. Under normal physiological conditions, Bach1 forms a dimer with Maf protein, preventing Nrf2 from binding to DNA. In response to ARE inducers, Bach1 undergoes rapid nuclear export and proteasomal degradation.
2.2. Nuclear factor erythroid 2–related factor 2 (Nrf2)
Nrf2 is a transcription factor which is essential for maintaining cellular homeostasis14. It is a 66-kDa cap ‘n’ collar (CNC) protein with a basic leucine zipper (bZip) DNA binding motif that is characteristic of NF-E215. Nrf2 contains 6 highly conserved domains named Nrf2-ECH homology domains (Neh1-6, Fig. 2)16. The first domain, Neh1 domain, corresponds to the bZip motif necessary for dimerization with Maf and binding to DNA17. Additionally, the DNA binding domain within Neh1 was found to have a nuclear localization sequence (NLS, residues 494–511), which is necessary for the nuclear localization of Nrf218. The highly conserved Neh2 domain lies at the N-terminal region of the protein. It serves as a negative regulatory domain in Nrf2 transcriptional activity. Neh2 contains DLG and ETGE motifs which correspond to the two binding sites for the Keap1 Kelch domain that facilitate the formation of a complex composed of one molecule of Nrf2 and two molecules of Keap119, 20. The presence of seven lysine residues within Neh2 allows for negative regulation of Nrf2 transcriptional activity via proteasome-mediated Nrf2 degradation21. The presence of a serine residue (Ser40) in the Neh2 domain is essential for release of Nrf2 from Keap1. Phosphorylation at Ser40 is required for Nrf2 to dissociate from Keap1 and thus avoid Keap1-mediated ubiquitination. However, Ser40 is not needed for Nrf2 stabilization and accumulation in the nucleus14. The Neh3 domain of Nrf2 is among members of the CNC bZIP transcription factors. It is located at the C-terminus of the protein and is essential for the transactivation of ARE gene by Nrf222. The Neh4 and Neh5 domains are considered transactivation domains that cooperatively bind to cAMP response element binding (CREB) protein (CBP), which has been shown to be essential co-activator for many transcription factors. Finally, Neh6 domain which is located in the middle of Nrf2 and has been reported to be associated with redox-insensitive degradation of the Nrf222, 23.
Figure 2.

The organization and domain structure of Nrf2.
2.3. Kelch-like ECH-associated protein 1 (Keap1)
Keap1 is a 69.7-kD actin-binding protein composed of 625 amino acid residues, 27 of which are cysteine residues24. As shown in Fig. 3, Keap1 consists of five distinct domains: (i) the N-terminal region (NTR), (ii) the broad complex, tramtrack and bric-a-brac (BTB) domain, (iii) the intervening region (IVR), (iv) the double glycine repeats (DGR) or Kelch domain, (v) and the C-terminal region (CTR)25. The BTB domain is an evolutionary conserved domain also found in actin-binding proteins and zinc finger transcription factors25. Keap1 forms a homodimer through the BTB domain and dimerization is required for binding to Nrf226. In addition, the BTB domain is also responsible for the interaction between Keap1 and Cullin3-Rbx1 E3 ubiquitin ligase (Cul3-E3-ligase)24, 27. The cysteine rich IVR is sensitive to oxidation and the nuclear export signal (NES) motif, and is necessary for Keap1 activity26, 28. In the IVR domain of Keap1, four especially reactive cysteine residues have been identified: Cys257, Cys273, Cys288 and Cys297. Cys273 and Cys288 are essential for Keap1-dependent ubiquitination of Nrf2 and Keap1-mediated repression of Nrf2 activity28, 29. Both the BTB and (IVR) domains were shown to be essential for Nrf2 degradation30. The Kelch domain consists of six repeating Kelch motifs (KR1–KR6) that form a six-bladed β-propeller structure31. The Kelch domain is where Keap1 binds to the Neh2 domain of Nrf232.
Figure 3.
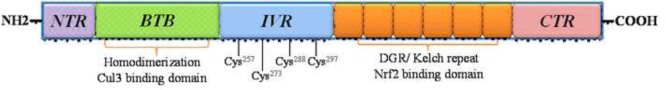
The organization and domain structure of Keap1.
3. Mechanism and regulation of the Keap1–Nrf2–ARE pathway
Keap1 functions as a master regulator of the Keap1–Nrf2–ARE pathway by controlling the steady state level of Nrf2 based on cellular redox conditions33. Under basal conditions, Nrf2 is bound as shown in Fig. 4 to Keap1 and targeted for ubiquitination and proteasomal degradation by Cul3-E3-ligase, with a t1/2 of less than 20 min. The rapid turnover of Nrf2 prevents the unnecessary expression of Nrf2 target genes34, 35, 36, 37. Keap1 forms a homodimer via its BTB domain. The Neh2 domain of Nrf2 contains two binding motifs: the high affinity ETGE and the low affinity DLG motifs36, 38. The ETGE and DLG motifs each bind to a separate Kelch domain in the Keap1 dimer. The binding of each motif to a Kelch domain (“two-site substrate recognition”) is required for the ubiquitination of Nrf2 that leads to its rapid degradation by 26S proteasome under basal conditions. The ubiquitination of Nrf2 occurs at an α-helix with seven lysine residues located between the binding motifs39. Under induced conditions, the Keap1–Nrf2–Cul3 complex is disturbed. As a consequence, Nrf2 is stabilized (t1/2 of up to 200 min) and can translocate to the nucleus. Two mechanistic models have been proposed for Nrf2 stabilization: the “Keap1-Cul3 dissociation model” and the “hinge and latch model”37, 40.
Figure 4.

The Keap1-Nrf-ARE pathway. In the "hinge" and "latch" mechanism of Nrf2 regulation, the high affinity ETGE motif of Nrf2 initially binds to Kelch domain of Keap1 and the lower affinity affinity DLG motif binds to the second Keap1 to close the conformation. Nrf2 is polyubiquitinated at its Lys rich (7K) region and targeted for subsequent degradation by the 26S proteasome.
In the Keap1-Cul3 dissociation model, it is proposed that inducers stabilize Nrf2 by dissociating the Keap1-Cul3 complex, resulting in the inhibition of ubiquitination and stabilization of Nrf237. Under induced conditions, covalent modification of cysteine residues in the BTB domain of Keap1 leads to a “steric clash” between Keap1 and Cul339. This results in the dissociation of the Keap1–Cul3 interaction and, therefore, disruption of Keap1-Cul3-E3-ligase activity39, 41. Cys151 has been found to be necessary to achieve this effect21, 41. Substitution of serine for Cys151 in the BTB domain renders Keap1 unable to dissociate from Nrf2 even in the presence of oxidative stress. This suggests that Cys151 functions as a sensor for oxidants and electrophiles and plays a crucial role in Nrf2 activation29, 34, 39, 42, 43, 44, 45. Other mechanisms for Nrf2 stabilization in response to inducers have been proposed, but will not be discussed here. These alternative mechanisms include nucleocytoplasm shuttling of Keap1, ubiquitination of Keap1, and Nrf2 as a direct sensor37.
In the “hinge and latch model” shown in Fig. 4, Nrf2-Keap1 contact is mediated by a strong binding interaction between the ETGE motif and one Kelch domain of Keap1 (the “hinge”), and a weaker binding interaction between the DLG motif and the other Kelch domain of Keap1 (the “latch”)33, 36. The high-affinity ETGE motif functions as a “hinge” by fixing Nrf2 to Keap1. The low-affinity DLG motif functions as a “latch” by locking or unlocking the position of Nrf2 depending on the redox state of the cell. Under basal conditions, the DLG motif locks the Neh2 domain in the correct position to enable the ubiquitination and degradation of Nrf2 in proteasomes36, 40, 46. When under oxidative stress, cysteine residues in Keap1 become oxidized and this modification unlocks the “latch”. Under these conditions, the orientation of Nrf2 prevents ubiquitination by the Keap1-Cul3 complex and this process leads to Nrf2 stabilization33, 40, 46, 47. As a result, Nrf2 bypasses proteasomal degradation and accumulates in the cell, translocates to the nucleus, forms a heterodimer with Maf, binds to ARE and therefore promotes the transcription of ARE-dependent genes30, 32, 34, 48.
4. The Keap1–Nrf2–ARE pathway as a therapeutic target
Inflammation and oxidative stress play an essential role in the pathogenesis of many human diseases and conditions49. Inflammation in the body produces large amounts of ROS and RNS that can induce oxidative damage to DNA and other cellular molecules including membrane lipids and proteins25. The Keap1–Nrf2–ARE pathway is a major defense mechanism used to counteract oxidative stress. This pathway protects many organs and cells and the pathway׳s protective role has been implicated in many human disorders50, including cancer, neurological diseases, airway disorders, cardiovascular diseases, diabetes, inflammatory bowel disease (IBS), and autoimmune diseases. Regulation of the Nrf2–ARE signaling has also been implicated in basic health, lifespan, and aging50. The role of the Keap1–Nrf2–ARE pathway in oxidative stress and age related diseases offers novel therapeutic and pharmacologic opportunities as we reviewed previously7. This section discusses briefly the major diseases and conditions that involve oxidative stress and the Keap1–Nrf2–ARE pathway and that could potentially be treated by modulators of this pathway.
4.1. Cancer
ROS and oxidative stress are a hallmark of human cancer49. The initiation of the formation of many tumors results from damage to DNA by electrophilic carcinogen metabolites or by ROS. The hypothesis that oxidative-stress induced lesions contribute to carcinogenesis is supported by the increased susceptibility to cancer observed in patients with a variety of chronic inflammatory diseases including ulcerative colitis, viral hepatitis, prostatitis, Helicobacter pylori infection, parasitic diseases, and many others. In patients with inflammatory diseases such as these, cancer induction may be a pathological consequence of elevated ROS levels which lead to increased levels of oxidative DNA damage which increases the risk of mutations that may lead to the development of cancer2. Given the ubiquitous involvement of oxidative damage in carcinogenesis, the Keap1–Nrf2–ARE pathway has been widely regarded as a potential therapeutic target for chemoprevention.
Various studies have revealed that Nrf2 plays a central role in cancer chemoprevention by promoting the expression of detoxification enzymes and cytoprotective proteins. Inducers of Nrf2 function as chemopreventive agents by preventing carcinogens from reaching their target, inhibiting parent molecules from undergoing metabolic activation, or preventing carcinogens from interacting with vital biomolecules such as DNA, RNA, and proteins. Disruption of the Nrf2 gene leads to increased susceptibility to environmental carcinogenesis by altering the expression of detoxifying enzymes and leads to the loss of chemopreventive efficacy by inducers. Therefore, induction of the Nrf2–ARE has been recognized as an important molecular and therapeutic target for chemoprevention.
Despite its promising potential for chemoprevention in normal and premalignant tissues, Nrf2 has also been shown to have a role in tumor cell growth and survival in malignant cells51. High levels of Nrf2 have been found in several types of human cancer cells, resulting from mutations in Keap1 or Nrf2 that result in constitutive expression of up-regulated genes52, 53. Nrf2 overexpression appears to exert its protective role in both normal and cancer cells. Studies show that elevated levels of Nrf2 can lead to increased expression of detoxification enzymes, cytoprotective proteins, and transporters. This gives cancer cells an advantage by enhancing cell proliferation and can cause resistance to chemotherapy51, 52, 53, 54, 55. Recent studies have shown that inhibition of Nrf2 in malignant cells suppresses tumor growth and enhances the efficacy of chemotherapy56, 57. Therefore, Nrf2 could be targeted for the treatment of cancer by either inducing activity for chemoprevention or inhibiting activity in existing tumors.
4.2. Neurodegenerative diseases
The brain is highly susceptible to oxidative damage due to its high lipid content, high oxygen consumption, and the high presence of redox-active metals, including Cu and Fe capable of catalytic ROS production. Neurodegenerative diseases share several pathological features including the accumulation of aberrant protein aggregates and mitochondrial dysfunction, excitotoxicity, and proteasomal dysfunction58. Enhanced ROS production and oxidative damage play the pivotal role in the onset and advancement of neurodegenerative diseases such as Alzheimer׳s disease (AD), Parkinson׳s disease (PD)58. The protective effect of Nrf2 against oxidative stress and neurotoxicity has been reported and the Keap1–Nrf2–ARE pathway have been proposed and investigated as a potential therapeutic target in AD and PD59, 60. Activation of Nrf2 has been investigated for its potential therapeutic applications in other neurological disorders such as Huntington׳s disease, amyotrophic lateral sclerosis (ALS or Lou Gehrig׳s disease), multiple sclerosis, traumatic brain injury and cerebral hemorrhages3, 58, 61.
4.3. Diabetes and diabetic complications
Experimental evidence has established that oxidative stress is involved in the pathogenesis of diabetes and the development of diabetic complications, including diabetic cardiomyopathy and nephropathy. Hyperglycemia has been shown to induce oxidative stress due to an increase in glucose metabolism and, thus, mitochondrial production of ROS62, 63, 64. The Nrf2–ARE pathway has been shown to play an important role in the regulation of energy metabolism, which has led to interest in the pathway as a potential target for the prevention and treatment of metabolic diseases such as diabetes. Nrf2 levels have also been shown to be lower in pre-diabetic and diabetic patients as compared to patients without diabetes, which suggests that diminished Nrf2 expression is involved in the development of oxidative stress in diabetes65, 66. Induction of Nrf2–ARE regulated genes attenuates insulin resistance and even inhibits the accumulation of fat67, 68. The role of Nrf2 in the regulation of metabolism and blood glucose levels has generated interest in targeting the pathway for the prevention and treatment of diabetes.
In addition to metabolic regulation and the pathogenesis of diabetes, Nrf2 appears to have an important role in diabetic complications. Oxidative stress is known to have a role in diabetic complications, including diabetic cardiomyopathy and nephropathy. Studies have indicated increased production of ROS in diabetic cardiomyocytes. This suggests that high levels of glucose induce ROS production and, thus, oxidative damage to the vasculature that directly contributes to the evolution of diabetic cardiomyopathy. Nrf2 has been demonstrated to be required for protection against glucose-induced oxidative stress and diabetic cardiomyopathy. Multiple studies have shown experimental evidence that demonstrates the involvement of Nrf2 in diabetic nephropathy. Streptozotocin (STZ) treated-Nrf2-null mice were determined to be more susceptible to oxidative damage and renal impairment than wild type mice62, 69, 70. The protective role of Nrf2 in diabetic nephropathy suggests that activation of Nrf2 could be used to prevent or impede the advancement of the disease. For example, bardoxolone methyl (CDDO-Me) is a potent activator of Nrf2 and was clinically evaluated for the treatment of chronic kidney disease in patients with type 2 diabetes71.
4.4. Chronic obstructive pulmonary disease (COPD) and other respiratory diseases
The respiratory system can be particularly susceptible to oxidative stress. Since the airways are the first point of contact for inhaled oxidants, the redox balance in the airway can be continuously and repeatedly disturbed by the increased accumulation of oxidants72, 73. Pulmonary expression of Nrf2 is primarily found in the epithelium and alveolar macrophages. The absence or depletion of Nrf2 expression has been shown to aggravate lung toxicity caused by multiple oxidative sources including cigarette smoke, allergens, viral infections, bacterial endotoxins, hyperoxia, and various environmental pollutants72. Several studies have also revealed that Nrf2 deficiency is associated with a greater susceptibility to COPD, emphysema, asthma, pulmonary fibrosis, acute respiratory distress syndrome and sepsis73, 74, 75, 76, 77. Therefore, activation of Nrf2 in alveolar macrophages appears to be a promising therapeutic target for the treatment of numerous respiratory diseases.
4.5. Cardiovascular disease (CVD)
Oxidative stress has been implicated in a number of cardiovascular diseases (CVDs) including atherosclerosis, hypertension, and cardiomyopathy. Therefore, the role of Nrf2 in CVD and its potential as a therapeutic target for the treatment of CVD has been of recent interest. Nrf2 is ubiquitously expressed in the cardiovascular system and plays a crucial role in maintaining cardiovascular homeostasis via the induction of ARE-dependent genes78. Nrf2 has been investigated as a therapeutic target for the treatment of cardiomyopathy and atherosclerosis; however, the results of these experiments have been inconclusive79. Although Nrf2 has shown vascular protective effects and has been suggested as a potential strategy for the treatment of atherosclerosis, several studies have proposed that Nrf2 promotes the pathogenesis of atherosclerosis through a different mechanism80, 81. Therefore, the potential pro-atherosclerosic effects of Nrf2 activation should be considered when designing Nrf2-targeted therapies for the treatment of CVD and other diseases.
4.6. Other diseases and conditions involving the Nrf2-ARE pathway
In addition to the diseases described above, the role of Nrf2 and its therapeutic potential have been investigated in numerous other diseases and health issues. The role of Nrf2 in gastrointestinal diseases, such as ulcerative colitis and chronic gastritis, has been investigated and appeared to strongly inhibit pro-inflammatory signaling associated with these conditions82, 83, 84, 85. The involvement of Nrf2 in the pathogenesis and treatment of liver disease and hepatotoxicity has been extensively investigated. Nrf2 was found to be crucial in combatting hepatotoxicity and liver injury86, 87, 88, 89, 90. Nrf2 also regulates the innate immune response and modulation of the Nrf2 pathway has been involved in diminishing various immune and inflammatory responses associated with infections91, 92, 93, autoimmune diseases94, 95, 96, and other innate immune responses97, 98, 99. The vast number of diseases and biological mechanisms that involve the Keap1–Nrf2–ARE clearly indicate its importance. The potential applications make the pathway a very interesting and promising target for drug design.
5. Direct inhibition of Keap1–Nrf2 protein–protein interaction (PPI)
The large amount of evidence indicating the importance of Nrf2 activation to human health has prompted interest in the discovery of small molecule and peptide activators of the Keap1–Nrf2–ARE pathway. Numerous natural (e.g., curcumin, sulforaphane, and isothiocyanate) and synthetic (e.g., bardoxolone methyl, oltipraz and Tecfidera™) small molecules that induce ARE-dependent gene expression have been investigated for their medicinal and therapeutic properties. However, most of these Nrf2 activators currently known are indirect inhibitors of the Keap1–Nrf2 interaction. The indirect inhibitors are electrophilic species or are metabolically transformed in vivo to become electrophilic, and subsequently react with the sulfhydryl groups of cysteine residues in Keap1 by oxidation or alkylation100, 101. Indirect inhibitors and their molecular mechanisms of action have been reviewed previously7. The electrophilicity of indirect inhibitors poses a problem. Their lack of specificity and selectivity increases the risk of “off-target” toxic effects due to their ability to react with the cysteine residues of other enzymes and proteins. Therefore, direct inhibition of the Keap1–Nrf2 PPI has recently become an appealing strategy for activation of Nrf2. The discovery of non-reactive direct small molecule inhibitors of the Keap1–Nrf2 pathway appears to be the most promising strategy due to the diminished possibility of toxic effects, as compared with indirect inhibitors, and increased stability and bioavailability, as compared with peptide inhibitors102, 103.
5.1. Screening assays for the discovery of small molecule direct inhibitors of Keap1–Nrf2 PPI
Several different assays have been developed for the screening and identification of small molecule inhibitors of the Keap1–Nrf2 PPI. These include surface plasmon resonance (SPR)-based solution competition assay, fluorescence polarization (FP) assay and the cell-based Neh2-luciferase assay.
5.1.1. SPR-based solution competition assay
The SPR-based solution competition assay selectively screens for Nrf2 activators that directly inhibit Keap1–Nrf2 interaction7, 104. In this assay, the Kelch domain of Keap1 is allowed to flow in solution over an SPR sensor chip with the 16mer Nrf2 peptide immobilized on the sensor chip surface. The optimal immobilization method is the use of a biotin-labeled 16mer Nrf2 peptide immobilized as the ligand on a streptavidin sensor chip. These conditions provided sensitive and stable surfaces for both kinetic analysis of the Keap1–Nrf2 PPI and detection of free Keap1 Kelch domain protein concentration in solution competition assays. This method was used to determine the minimal Nrf2 peptide sequence required to bind Keap1 Kelch domain104. The advantage of this assay is that it allows for the measurement of direct inhibition of Keap1–Nrf2 interaction. However, the limited throughput of the SPR-based assay prevents it from being used as the primary assay in high-throughput applications7, 104.
5.1.2. Fluorescence polarization (FP) assay
FP is a powerful tool used to study the interactions between biomolecules in solution. FP competition assays can be used to screen for small molecules that inhibit ligand-receptor interactions105. We previously reported the development of an FP assay that can be used for high-throughput screening of large chemical libraries in an effort to identify small molecule inhibitors of Keap1–Nrf2 interaction106. Fluorescently-labeled-Nrf2 peptides containing the ETGE motif were designed and synthesized as tracers to detect direct inhibitors of Keap1–Nrf2 interaction. Flurescein isothiocyanate (FITC)-labeled Nrf2 9mer peptide amide was determined to be the optimal tracer and was used in our FP assay. We have successfully used this assay in the high-throughput screening (HTS) of the NIH MLPCN small molecule library to discover small molecule inhibitors of Keap1–Nrf2 interaction7, 102, 106.
5.1.3. Cell-based Neh2-luciferase assay
In the cell-based Neh2-luciferase assay, a Neh2-luciferase reporter system is constructed with the Neh2 domain of Nrf2 fused to a luciferase gene as a tool to monitor Nrf2 activation in real time107. The overexpressed Neh2-luciferase fusion protein competes with endogenous Nrf2 for Keap1 binding and subsequent ubiquitination and degradation. Nrf2 activators disrupt the Neh2-luc-Keap1-Cul3 complex and, thus, the Neh2-lucisferase protein is not degraded. The increase in luciferase activity serves as a direct measure of the ability of a compound to disrupt the Keap1–Nrf2 interaction. The advantage of this assay is that there is an immediate response upon the addition of Nrf2 activators, which allows for differentiation of Nrf2 activators by monitoring their kinetics of reporter activation. This system is suitable for HTS with Z׳-values of >0.77, 107.
5.2. Peptide inhibitors and Nrf2-based peptide probes
The elucidation of the structure of the Keap1–Nrf2 binding interaction provided important insight into the development of peptide inhibitors of the interaction. Specific amino acids that are critical for Keap1–Nrf2 binding were first determined using extensive alanine-scan mutagenesis of the Keap1 protein108. Replacement of the residues Tyr334, Asn382, His436, Tyr525, and Tyr572 with alanine residues considerably disrupted the ability of Keap1 to bind to Nrf2. It was also determined that the Phe478 residue is not required for Keap1–Nrf2 binding, but it is required for suppression of Nrf2-dependent gene expression. When Phe478 was replaced with an alanine residue, the mutant was unable to direct Nrf2 ubiquitination. Additionally, there are three arginine residues that were determined to be critical for Keap1–Nrf2 binding: Arg380, Arg415 and Arg483. By understanding the structure Keap1–Nrf2 binding interface, various peptides have been designed to either simply inhibit the interaction or serve as a probe to gauge the activity of small molecule inhibitors in screening assays108.
5.2.1. Peptide inhibitors of Keap1–Nrf2 PPI
Our laboratory designed a series of truncated peptides based on the ETGE motif of Nrf2 and evaluated them as direct inhibitors of the Keap1–Nrf2 PPI using the SPR assay and the FP assay we developed104, 106. The affinity of the non-acetylated Nrf2 peptides increases with increasing peptide length as shown in Table 1104, 106. The 7mer peptide (entry 1, Table 1) was totally inactive and the 8mer (entry 2, Table 1) was only weakly active, while the 9mer (entry 4, Table 1) was shown to be significantly more active with a Kd of 350 nmol/L in the SPR assay and IC50 of 3.48 μmol/L in FP assay corresponding to an Ki of 865 nmol/L. Longer Nrf2 peptides (10mer to 16mer peptides (entries 8–12, Table 1)) are much more active with Kd ranging from 22 to 31 nmol/L and IC50 values from 0.163 to 0.298 μmol/L. We observed the N-terminal acetylation of 9mer Nrf2 peptide significantly increased the binding affinity to Keap1 Kelch domain to a level that is similar to the binding affinities of the longer Nrf2 peptides while C-terminal amidation of 9mer Nrf2 peptide had little effect104, 106. Based on these studies, we concluded that minimal binding sequence of Nrf2 ETGE motif to Keap1 Kelch domain is the 9mer sequence of LDEETGEFL104, 106.
Table 1.
The inhibition of the Keap1–Nrf2 interaction by Nrf2 peptides as determined using SPR and FP assay.
| Entry | Peptide name | Peptide sequence | (nmol/L)a | IC50 (μmol/L)b | Ki (nmol/L)b |
|---|---|---|---|---|---|
| 1 | 7mer Nrf2 | H-EETGEFL-OH | >>1000 | >>100 | — |
| 2 | 8mer Nrf2 | H-DEETGEFL-OH | >>1000 | 21.7±20.1 | 7010 |
| 3 | 8mer Nrf2-NH2 | H-DEETGEFL-NH2 | — | 30.5±22.7 | 9870 |
| 4 | 9mer Nrf2 | H-LDEETGEFL-OH | 352 | 3.48±0.92 | 865 |
| 5 | 9mer Nrf2-NH2 | H-LDEETGEFL-NH2 | 355 | 3.57±2.20 | 1140 |
| 6 | Ac-9mer Nrf2 | Ac-LDEETGEFL-OH | 23.1 | 0.194±0.049 | 47.4 |
| 7 | Ac-9mer Nrf2-NH2 | Ac-LDEETGEFL-NH2 | 21.4 | 0.196±0.032 | 48.1 |
| 8 | 10mer Nrf2 | H-QLDEETGEFL-OH | 27.3 | 0.272±0.026 | 72.7 |
| 9 | 11mer Nrf2 | H-LQLDEETGEFL-OH | 31.3 | 0.298±0.033 | 81.1 |
| 10 | 12mer Nrf2 | H-QLQLDEETGEFL-OH | 23.8 | 0.249±0.022 | 65.2 |
| 11 | 14mer Nrf2 | H-FAQLQLDEETGEFL-OH | 22.5 | 0.243±0.020 | 63.3 |
| 12 | 16mer Nrf2 | H-AFFAQLQLDEETGEFL-OH | 23.9 | 0.163±0.011 | 37.4 |
Wells and co-workers also obtained a series of synthetic peptide inhibitors of Keap1–Nrf2 PPI based on the ETGE or DLG motif of the Neh2 domain of Nrf2 using peptide phage display library and investigated their ability to bind the Kelch domain of human Keap1 using an FP assay109. They determined that the minimal sequence required for binding between the ETGE motif and the Kelch domain is the seven-amino-acid sequence Ac-DEETGEF-OH and that the optimal sequence was Ac-DPETGEL-OH. They also determined that the minimal sequence required for binding between the DLG motif and the Kelch domain is Ac-WRGDIDL-OH (Fig. 5)109. In a later study, they modified the peptides by replacing the acetyl groups at the N-terminus with benzoyl or stearoyl groups to increase the lipophilicity of the molecules and assess their effect on binding with the Kelch domain using an FP assay. They found that the sequence stearoyl (St)-DPETGEL-OH demonstrated potent activity (IC50=22 nmol/L), and promoted expression of Nrf2-dependent gene expression in a cell-based assay110.
Figure 5.

Summary of structure–activity relationship data for the DLG motif (residues 24–30; blue) and the ETGE motif (residues 76–83; red).
In another study of Keap1–Nrf2 peptide inhibitors, Searcey and co-workers synthesized a series of TAT-conjugated ETGE peptides that target the interaction. They evaluated Nrf2 activation by measuring the expression of the downstream target gene heme oxygenase–1 (HO-1). They found that only the TAT-ETGE-14mer (TAT-14: YGRKKRRQRRRLDEETGEFLPIQ) was able to induce HO-1 expression in a time and dose dependent manner111.
5.2.2. Fluorescently labeled Nrf2 peptide probe optimization
The need for an assay adaptable to HTS of large chemical libraries to aid in the identification and design of small molecule inhibitors of Keap1–Nrf2 PPI lead us to develop the FP assay described above106. Fluorescently-labeled Nrf2-peptides containing the ETGE motif were designed and synthesized as probes to detect the direct inhibition of the Keap1–Nrf2 PPI. In our effort to optimize the fluorescent Nrf2 peptide probe in terms of Keap1 Kelch domain–binding affinity and the dynamic range of the assay, we prepared and evaluated a series of FITC-labeled 8–16mer Nrf2. As shown in Table 2106, the FITC-labeled 9mer Nrf2 peptide demonstrated a binding affinity comparable to the longer FITC-labeled Nrf2 peptides (entry 3 vs. entries 5–9, Table 2), which suggests that most of the bonding interactions between the Kelch domain and Nrf2 reside within the 9mer peptide sequence (LDEETGEFL). Thus, the binding affinity was not significantly affected by peptide length as long as the peptide contains the nine amino acids in the DxETGE motif. In addition, the FITC-labeled 9mer Nrf2 peptide amide (entry 4, Table 2) demonstrated the highest dynamic range among the peptides tested. Therefore, the FITC-labeled 9mer Nrf2 peptide amide is the optimal sequence and has been used as our probe in our FP assay7, 106.
Table 2.
FITC-labeled Nrf2 peptides of different length (8 mer to 16 mer): their binding affinities and dynamic rangea.
| Entry | Peptide name | Peptide sequence | Kd (nmol/L) | Dynamic range (ΔmA) |
|---|---|---|---|---|
| 1 | FITC-8mer Nrf2 | FITC-DEETGEFL-OH | ~750b | –c |
| 2 | FITC-8mer Nrf2-NH2 | FITC-DEETGEFL-NH2 | ~1000b | –c |
| 3 | FITC-9mer Nrf2 | FITC-LDEETGEFL-OH | 65.1±9.7 | 97.3 |
| 4 | FITC-9mer Nrf2-NH2 | FITC-LDEETGEFL-NH2 | 25.6±10.8 | 109.8 |
| 5 | FITC-10mer Nrf2 | FITC-QLDEETGEFL-OH | 30.1±6.1 | 73.5 |
| 6 | FITC-11mer Nrf2 | FITC-LQLDEETGEFL-OH | 47.7±7.4 | 96.3 |
| 7 | FITC-12mer Nrf2 | FITC-QLQLDEETGEFL-OH | 44.5±12.9 | 70.6 |
| 8 | FITC-14mer Nrf2 | FITC-FAQLQLDEETGEFL-OH | 61.9±16.5 | 64.2 |
| 9 | FITC-16mer Nrf2 | FITC-AFFAQLQLDEETGEFL-OH | 28.7±5.7 | 80.1 |
Anisotropy measurements were performed using FP assay106.
The Kd of the FITC-labeled 8mer Nrf2 peptides were assessed using the anisotropy of the fully bound FITC-9mer Nrf2 peptide.
Because of low binding affinity, the higher end of the dynamic range could not be determined.
5.3. Small molecule direct inhibitors of Keap1–Nrf2 PPI
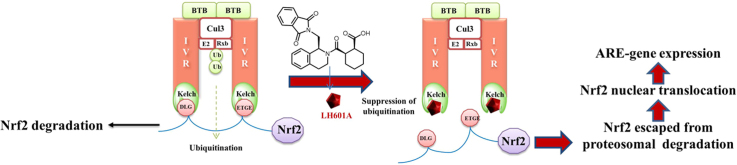
As stated earlier, the vast majority of inhibitors of Keap1–Nrf2 PPI is electrophilic species that work by covalent modification of the cysteine residues in Keap1. Recently, several small molecule direct inhibitors of Keap1–Nrf2 PPI through non-covalent binding to the Keap1 Kelch domain have been reported102, 103, 112, 113, 114. We first reported in 2013 the discovery of small molecule direct inhibitors of the Keap1–Nrf2 PPI102. The FP assay106 that we developed and reported in 2012 was successfully used to screen the MLPCN library of 337,116 compounds (PubChem Assay ID: 504523, 504540). The primary screen generated 489 hits at 10 μmol/L, which was reduced to 460 hits after excluding fluorescent compounds. These 460 hits were subjected to confirmation assays using the eight-point dose-response FP assay and a thermal shift secondary assay, generating 8 confirmed hits. From these eight hits, hit 1 (LH601) was the most promising, with an IC50 of 3 μmol/L in the FP assay and a Kd of 1.6–1.9 μmol/L in our SPR assay102. In addition, there are no chemically reactive functional groups present in LH601, therefore it is not expected to modify the sulfhydryl groups of cysteine residues in Keap1 or other proteins.
LH601 has three chiral centers and four possible stereoisomers. We separated the four isomers (LH601A–D) using a combination of flash silica gel chromatography and chiral HPLC purification; we then compared their Keap1 Kelch domain–binding activity using our SPR and FP assays. It was found that LH601A is the most active stereoisomer, which is about 100 times more potent than its corresponding enantiomer LH601B while their diastereomers LH601C&D are inactive. The stereospecific binding activity of LH601 isomers made us more confident about the true binding of LH601A to Keap1 Kelch domain. X-ray crystallography was then used to assign the absolute stereochemistry of LH601 isomers; the active stereoisomer LH601A was determined to be of (S,R,S)-configuration (1)102. We also synthesized a number of analogs to determine the structure-activity relationships of LH601A. Preliminary SAR studies provided the following conclusions as shown in Fig. 6: (i) Keap1-binding activity resides primarily in one stereoisomer of (S,R,S)-configuration; (ii) acid functionality on the cyclohexane ring is required for optimal activity; (iii) a one-carbon linker between the tetrahydroisoquinoline (THIQ) and phthalimido group is optimal; (iv) one of the carbonyls in the phthalimido group can be removed to give a lactam that retains strong binding affinity to Keap1 Kelch domain.
Figure 6.
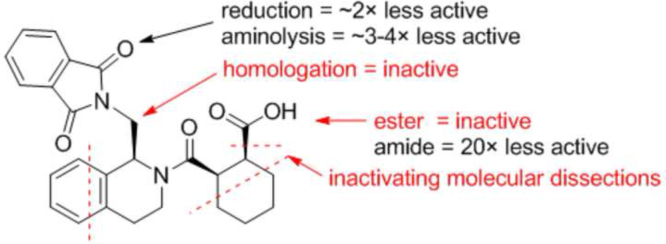
Structure–activity relationships around LH601A (1)102. The activity noted was based on the solution competition SPR assay.
After confirming the Keap1-binding activity of LH601A in our FP and SPR assays, we determined its cellular activity in two cell-based functional assays102. In the CellSensor® ARE-bla HepG2 cell line where ARE controls the expression of β-lactamase, LH601A was found induce ARE-controlled genes with an EC50 of 18 μmol/L as compared to >100 μmol/L for its enantiomer LH601B and its diastereomers LH601C/D. In the PathHunter® U2OS Keap1–Nrf2 functional assay that uses β-galactosidase-based enzyme fragment complementation technology and luminescence for the detection of Nrf2 nuclear translocation, LH601A promoted the nuclear translocation of Nrf2 with a similar EC50 of 12 μmol/L. All these data indicate that LH601A is cell-permeable and is capable of inhibiting the Keap1–Nrf2 interaction, leading to the dissociation of Nrf2 from Keap1 in the cytosol, its subsequent translocation to the nucleus, and the upregulation of ARE-controlled genes.
Jnoff and colleagues113 at UCB Pharma recently confirmed most of our earlier findings and provided X-ray co-crystal structure evidence that LH601A binds to the Nrf2 binding site on the Keap1 Kelch domain. The cocrystal structures of Keap1 Kelch domain with LH601A and its analogs provide further confirmation of the stereochemistry of the active isomer LH601A and the nature of its direct binding interaction to Keap1 Kelch domain. Several analogs of LH601A were synthesized and evaluated by UCB scientists leading to a more potent Keap1 binder, compound 4 (Fig. 7), where one of the phthalimide carbonyl was reduced to form the lactam and an additional methyl group was introduced at the 5 position of THIQ. Compound 4 is 3-fold more potent than 1 as an inhibitor of Keap1–Nrf2 PPI in the FP assay. It was suggested that the methyl group in compound 4 likely acts as a "lipophilic plug" providing a good shape fit toward the pore of the Kelch domain resulting in the increase in potency as compared to 1113.
Figure 7.

Structures of direct inhibitors of Keap1–Nrf2 PPI that have recently been reported.
Silvian and colleagues114 at Biogen used a high throughput homogeneous confocal fluorescence anisotropy assay to screen the lead discovery library of 267,551 compounds from Evotech plus 1911 compounds selected from a virtual screening and identified two compounds as inhibitors of Keap1–Nrf2 PPI: the benzenesulfonyl-pyrimidone 2 and the N-phenyl-benzenesulfonamide 3. Compound 2 was found to have an IC50 value of 118 μmol/L while 3 was found to have an IC50 of 2.7 μmol/L in the confocal FP assay. Using an ARE-driven luciferase reporter assay, 3 was shown to increase the levels of both Nrf2 and one of Nrf2 target genes NQO1 at 100 mol/L. Native mass spectrometry (NMR) and X-ray crystallography were used to confirm that 3 binds specifically in the cavity of the Keap1 Kelch domain114.
You and coworkers112 used virtual screening of the Specs database and identified compound 7 as a small molecule inhibitor of Keap1–Nrf2 PPI. Based on the crystal structure of the interaction between the ETGE and DLG motifs of Nrf2 and the Kelch domain of Keap1, they found that a negative ionizable center should be included in Keap1–Nrf2 PPI inhibitors. Before screening the library, 90% of the compounds in the Specs database were excluded because they possessed a formal charge of more than −1. This reduced the number of compounds from 251,774 to 21,119. The virtual screening of the 21,119 compounds leads to 17 virtual hits that were experimentally evaluated in the FP assay to identify the small molecule inhibitors of the Keap1–Nrf2 PPI. Compound 7 with a symmetrical structure containing two benzoic acids at the ends and a carbodihydrazide in the middle was reported to have an IC50 of 9.80 μmol/L in the FP assay but relatively low ARE-inducing activity in a cell-based assay due to its poor cell permeability112.
In a more recent study by You, Sun and coworkers103, a potent direct inhibitor 6 of the Keap1–Nrf2 PPI was derived from the Biogen inhibitor 3 as shown in Fig. 7. Compound 6 was reported to have an IC50 of 28.6 nmol/L and a Kd of 3.59 nmol/L in the FP assay. Studies on the molecular binding determinants and molecular dynamics (MD) simulations of Keap1–Nrf2 PPI suggested that the incorporation of two acetic acid side chains to 3 would provide favorable binding interactions with the Keap1 Kelch domain. The activity of 6 was also demonstrated through the cell-based ARE-luciferase reporter assay and the qRT-PCR103.
In another study reported by Sham, Xing and coworkers115, rapid structure-based virtual screening and hit-based substructure search were utilized to identify small molecules that disrupt Keap1–Nrf2 PPI. The noncovalent inhibitor 5 was reported to have comparable Keap1 binding affinity to 3 in an FP assay, but is 3-times more active than compound 3 in a cell-based assay115.
Another interesting but simple compound (8) containing a furanyloxadiazole linked to a phenoxyacetic acid, was reported to be a direct inhibitor of Keap1–Nrf2 PPI in a Japanese patent application116. Two co-crystal structures of Keap1 Kelch domain with 8 were deposited to PDB databank (PDB ID: 3VNG, 3VNH) but neither the binding affinity of 8 to Keap1 Kelch domain nor any cellular ARE-inducing activity have been reported.
5.4. X-ray co-crystal structures of Keap1 Kelch domain with small molecule direct inhibitors of Keap1–Nrf2 PPI
The 3-D structures of the human and mouse Keap1 Kelch domain with and without Nrf2-derived peptides were determined by X-ray crystallography31, 108, 117. The crystal structure of the human Keapl Kelch domain was determined at 1.35 Å resolution31 while the complex of human Keap1 Kelch domain with the 16mer Nrf2 peptide bound was reported at 1.5 Å resolution (PDB ID: 2FLU)108, 117. Furthermore, the structures of the mouse Kelch domain of Keap1 with and without Nrf2 peptide were also determined by X-ray crystallography (PDB ID: 1X2J and 1X2R). The human and mouse Keap1 are very similar in sequence with sequence identity of 94% overall and 97% in the Kelch domain. Both cocrystal structures of the human and mouse Keap1 Kelch domain–Nrf2 peptide complexes overlay very well with each other and the apo structure of human Keap1 Kelch domain. After alignment of the three crystal structures, our analysis indicates that the RMSDs for the Cα atoms range between 0.40 and 0.48 Å and for all atoms between 0.87 and 0.95 over the 285 Keap1 Kelch domain residues. They all show that the Kelch domain folds up into a highly symmetric 6-bladed β-propeller structure with each blade consisting of 44–51 amino acids (Fig. 8A). The 16mer Nrf2-derived peptide has two antiparallel β-strands connected by a turn region that has two tight overlapping type-1 β turns (residues 77–80 and 78–81). The Nrf2 peptide binds to the top face of the β-propeller with all six blades contributing to the complex formation (Fig. 8A and B). Side chains from six residues in Keap1 (Ser363, Asn382, Arg380, Arg415, Arg483 and Ser508) participate in H-bond interactions to the carboxylate oxygen atoms from E79 and E82 in the Nrf2 peptide (Fig. 8C) and several Keap1 residues are involved in H-bond interactions to the peptide backbone and in van der Waals interactions between the Kelch domain and the Nrf2 peptide (Fig. 8B). Another interesting feature of the Keap1 Kelch domain with relevance to peptide-binding site is the positively charged region which is primarily due to the highly conserved Arg residues. Only the side chains of peptide glutamate residues E79 and E82 make specific interactions with the binding site. The carboxylate group of peptide E79 interacts with the side chains of Ser508, Arg415, and Arg483 while the carboxylate group atoms of peptide E82 interacts with the side chains of Ser363, Asn382, and Arg380 (Fig. 8C). These structure details revealed in the high resolution (1.5 Å) co-crystal structure of the human Keap1 Kelch domain-Nrf2 peptide complex suggest that inhibitors that interfere with the Keap1–Nrf2 PPI can derive their inhibition by binding Keap1 Kelch domain at the site where Nrf2 peptide is binding.
Figure 8.

Structures of the Kelch domain of human Keap1 bound to an Nrf2 peptide. (A) A top-down view showing the six-bladed β-propeller structure in red ribbon and the peptide as a yellow tube. Each blade of the β-propeller is numbered I–VI. Both the N- and C-termini of the domain are located in blade I and are labeled N and C, respectively. The four β-strands found in each blade are designated A–D as shown in white font on blade VI. (B) A surface representation of the Kelch propeller (gray) and peptide (yellow tube). Selected residues are shown in blue (basic), orange (polar) and green (apolar). (C) Charge–charge and H-bonding interactions between the side chain atoms of the Nrf2 peptide and residues in the Kelch domain. Not shown are 5 H-bond interactions between the peptide backbone atoms and residues in the Kelch domain (reproduced with permission from reference108).
Moreover, several structural and functional evidences substantially support the concept that the DxETGE motif is the principal Keap1 binding site in Nrf2 peptide. The lysine-rich residues in Nrf2 required for ubiquitination are located at a distance of 10–30 amino acids on the DxETGE N-terminal side 21, and these residues would be positioned for ubiquitin transfer upon binding of Nrf2 to Keap1 via the DxETGE motif. The second low-affinity Keap1 binding site in Nrf2 containing the LxxQDxDLG sequence located at a distance of approximately 50 amino acids on the N-terminal side of the DxETGE motif.
Based on the cocrystal structure of Keap1 Kelch domain with the 16mer Nrf2 peptide (PDB ID: 2FLU), we docked LH-601A to the Nrf2 peptide binding site in Keap1 Kelch domain. The strength of binding between LH-601A and Keap1 increases by interactions between THIQ and Arg415 (π-cation), phthalimido and Arg380 (π-cation), and the hydrogen-bonding interactions which were observed between Keap1 and Nrf2 peptide. Jnoff and colleagues also docked LH601A to the Nrf2 peptide binding site in Keap1 Kelch domain based on the cocrystal structure of Keap1 Kelch domain with the 16mer Nrf2 peptide (PDB ID: 2FLU) and the crystal structure of Kelch domain of human Keap1 PDB ID: (1ZGK)113. In their top pose, the LH-601A cyclohexyl group posits in a similar pocket to our docked pose while the remainder of LH-601A forming completely different interaction patterns. In their co-crystal structure of a mutant Keap1 Kelch domain with LH601A (PDB ID: 4L7B)113, the aromatic ring of the THIQ group oriented into the central pore, while the phthalimide and cyclohexane carboxylic acid moieties extending outward. Regarding the phthalimide group, the first carbonyl group is hydrogen-bonded to Ser602, the second carbonyl group is hydrogen-bonded to Ser555 through water molecule, and finally the phenyl ring interacts with Tyr572 through a π-stacking. The cyclohexane carboxylic acid group is hydrogen-bonded to both Arg415 and Asn414.
Silvian and colleagues114 from Biogen cocrystallized compounds 2 and 3 mentioned above with Keap1 Kelch domain. Compound 2 (PDB ID: 4IN4) co-crystallized to 2.6 Å resolution with two molecules of 2 binding side-by-side in each central cavity of Keap1 Kelch domain (i.e. 2 binds to the Keap1 Kelch domain protein in a 2:1 stoichiometry (Fig. 9B)). Both electrostatic and hydrophobic interactions are involved between compound 2 and the Keap1 Kelch domain protein. Three serine residues (Ser508, Ser555 and Ser602) and two arginines (Arg415 and Arg483) form H-bonds with the two molecules of 2 while Tyr525 and Phe477 residues form π-π stacking interactions with two molecules and there exists hydrophobic interaction between the CF3 group in one molecule (A) of 2 and the meta-dimethylphenyl group in the other molecule (B) of 2. There seems to be no cooperative interaction between the two molecules of 2 in each binding site with Hill coefficient close to 1.0. The more potent compound 3 (PDB ID: 4IQK) co-crystallized to 2.0 Å resolution where 3 binds to the Keap1 Kelch domain protein in a 1:1 stoichiometry. Compound 3 interacted through four π-π stacking interactions with Keap1 side chains of residues Tyr334, Tyr525, Tyr572 and Arg415. The electron-rich naphthalene ring stacks with Arg415 and the second naphthalene ring inserted deep into the polar hole of the central cavity. Furthermore, Serine residues, Ser508 and Ser602, form H-bond interactions with compound 3114.
Figure 9.

The interactions observed in X-ray co-crystal structures of Keap1 Kelch domain with small molecule direct inhibitors of Keap1–Nrf2 PPI: (A) 1 or (B) 2 or (C) 3. Ionic interactions are indicated with red dotted lines and hydrophobic interactions are indicated with blue dotted double sided arrows. There are two ligands occupying the binding site in the co-crystal structure of Keap1 Kelch domain with 2. (A was reproduced with permission from reference113).
6. Conclusions
The Keap1–Nrf2–ARE pathway is a critical antioxidant defense mechanism that protects cells from oxidative stress. Since oxidative stress has been implicated in numerous human diseases and conditions, the Keap1–Nrf2–ARE pathway has been become an important cellular target for the development of potential therapeutic and preventive agents for a number of diseases and conditions. Nrf2 is the master transcription factor of ARE-dependent genes and Keap1 is the major negative regulator of Nrf2. Most ARE inducers known are indirect inhibitors of Keap1–Nrf2 PPI that are electrophilic species acting by modifying the sulfhydryl groups of Keap1׳s cysteine residues. However, the electrophilicity of indirect inhibitors is problematic due to the potential for "off-target" reactions with cysteine residues of other important cellular proteins. To circumvent the potential toxic side effects caused by these "off-target" reactions, several direct inhibitors of Keap1–Nrf2 PPI have been developed. These direct inhibitors function by inhibiting the Keap1–Nrf2 PPI via a non-covalent mechanism and could potentially be developed into effective therapeutic or preventive agents, representing a novel therapeutic strategy for the treatment and/or prevention of a variety of diseases and conditions.
Acknowledgments
We gratefully acknowledge the financial support of Grants CA133791, CA125868, and MH093197 from the National Institutes of Health, United States. Dhulfiqar Ali Abed is a recipient of a graduate scholarship from the Higher Committee of Education Development in Iraq. Haifa Albanyan is a recipient of a graduate scholarship from the Cultural Mission of the Royal Embassy of Saudi Arabia.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19:169–185. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- 2.Sedelnikova OA, Redon CE, Dickey JS, Nakamura AJ, Georgakilas AG, Bonner WM. Role of oxidatively induced DNA lesions in human pathogenesis. Mutat Res. 2010;704:152–159. doi: 10.1016/j.mrrev.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li J, Wuliji O, Li W, Jiang ZG, Ghanbari HA. Oxidative stress and neurodegenerative disorders. Int J Mol Sci. 2013;14:24438–24475. doi: 10.3390/ijms141224438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dinkova-Kostova AT, Talalay P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res. 2008;52 Suppl 1:S128–S138. doi: 10.1002/mnfr.200700195. [DOI] [PubMed] [Google Scholar]
- 5.Lakowicz JR. 3rd ed. Springer Science and Business Media; New York: 2006. Principles of fluorescence spectroscopy. [Google Scholar]
- 6.Lyakhovich VV, Vavilin VA, Zenkov NK, Menshchikova EB. Active defense under oxidative stress. The antioxidant responsive element. Biochemistry. 2006;71:962–974. doi: 10.1134/s0006297906090033. [DOI] [PubMed] [Google Scholar]
- 7.Magesh S, Chen Y, Hu LQ. Small molecule modulators of Keap1–Nrf2–ARE pathway as potential preventive and therapeutic agents. Med Res Rev. 2012;32:687–726. doi: 10.1002/med.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JM, Johnson JA. An important role of Nrf2–ARE pathway in the cellular defense mechanism. J Biochem Mol Biol. 2004;37:139–143. doi: 10.5483/bmbrep.2004.37.2.139. [DOI] [PubMed] [Google Scholar]
- 9.Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H: quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem J. 2003;374:337–348. doi: 10.1042/BJ20030754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266:11632–11639. [PubMed] [Google Scholar]
- 11.Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW. When Nrf2 talks, who׳s listening? Antioxid Redox Signal. 2010;13:1649–1663. doi: 10.1089/ars.2010.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katsuoka F, Motohashi H, Ishii T, Aburatani H, Engel JD, Yamamoto M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol Cell Biol. 2005;25:8044–8051. doi: 10.1128/MCB.25.18.8044-8051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMahon M, Itoh K, Yamamoto M, Chanas SA, Henderson CJ, McLellan LI. The cap ׳n׳ collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001;61:3299–3307. [PubMed] [Google Scholar]
- 14.Li Y, Paonessa JD, Zhang Y. Mechanism of chemical activation of Nrf2. PLoS One. 2012;7:e35122. doi: 10.1371/journal.pone.0035122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the β-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Itoh K, Mimura J, Yamamoto M. Discovery of the Negative Regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal. 2010;13:1665–1678. doi: 10.1089/ars.2010.3222. [DOI] [PubMed] [Google Scholar]
- 17.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y. An Nrf2/Small maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 18.Jain AK, Bloom DA, Jaiswal AK. Nuclear import and export signals in control of Nrf2. J Biol Chem. 2005;280:29158–29168. doi: 10.1074/jbc.M502083200. [DOI] [PubMed] [Google Scholar]
- 19.Jung KA, Kwak MK. The Nrf2 system as a potential target for the development of indirect antioxidants. Molecules. 2010;15:7266–7291. doi: 10.3390/molecules15107266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katoh Y, Iida K, Kang MI, Kobayashi A, Mizukami M, Tong KI. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch Biochem Biophys. 2005;433:342–350. doi: 10.1016/j.abb.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells. 2001;6:857–868. doi: 10.1046/j.1365-2443.2001.00469.x. [DOI] [PubMed] [Google Scholar]
- 23.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J Biol Chem. 2004;279:31556–31567. doi: 10.1074/jbc.M403061200. [DOI] [PubMed] [Google Scholar]
- 24.Tkachev VO, Menshchikova EB, Zenkov NK. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry. 2011;76:407–422. doi: 10.1134/s0006297911040031. [DOI] [PubMed] [Google Scholar]
- 25.Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med. 2004;36:1208–1213. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- 26.Zipper LM, Mulcahy RT. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem. 2002;277:36544–36552. doi: 10.1074/jbc.M206530200. [DOI] [PubMed] [Google Scholar]
- 27.Chauhan N, Chaunsali L, Deshmukh P, Padmanabhan B. Analysis of dimerization of BTB-IVR domains of Keap1 and its interaction with Cul3, by molecular modeling. Bioinformation. 2013;9:450–455. doi: 10.6026/97320630009450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li XC, Zhang DN, Hannink M, Beamer LJ. Crystal structure of the kelch domain of human Keap1. J Biol Chem. 2004;279:54750–54758. doi: 10.1074/jbc.M410073200. [DOI] [PubMed] [Google Scholar]
- 32.Uruno A, Motohashi H. The Keap1–Nrf2 system as an in vivo sensor for electrophiles. Nitric Oxide. 2011;25:153–160. doi: 10.1016/j.niox.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 33.Stępkowski TM, Kruszewski MK. Molecular cross-talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radic Biol Med. 2011;50:1186–1195. doi: 10.1016/j.freeradbiomed.2011.01.033. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26:221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang DD, Lo SC, Sun Z, Habib GM, Lieberman MW, Hannink M. Ubiquitination of Keap1, a BTB-kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J Biol Chem. 2005;280:30091–30099. doi: 10.1074/jbc.M501279200. [DOI] [PubMed] [Google Scholar]
- 36.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a "tethering" mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem. 2006;281:24756–24768. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- 37.Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1–Nrf2 pathway. Arch Toxicol. 2011;85:241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1–Nrf2 pathway. Trends Pharmacol Sci. 2013;34:340–346. doi: 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 39.Eggler AL, Small E, Hannink M, Mesecar AD. Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem J. 2009;422:171–180. doi: 10.1042/BJ20090471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol. 2006;26:2887–2900. doi: 10.1128/MCB.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem Res Toxicol. 2008;21:705–710. doi: 10.1021/tx700302s. [DOI] [PubMed] [Google Scholar]
- 42.He XQ, Ma Q. NRF2 cysteine residues are critical for oxidant/electrophile-sensing, kelch-like ECH-associated protein-1-dependent ubiquitination–proteasomal degradation, and transcription activation. Mol Pharmacol. 2009;76:1265–1278. doi: 10.1124/mol.109.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28:2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun Z, Zhang S, Chan JY, Zhang DD. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol Cell Biol. 2007;27:6334–6349. doi: 10.1128/MCB.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kobayashi M, Li L, Iwamoto N, Nakajima-Takagi Y, Kaneko H, Nakayama Y. The antioxidant defense system Keap1–Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol Cell Biol. 2009;29:493–502. doi: 10.1128/MCB.01080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tong KI, Kobayashi A, Katsuoka F, Yamamoto M. Two-site substrate recognition model for the Keap1–Nrf2 system: a hinge and latch mechanism. Biol Chem. 2006;387:1311–1320. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- 47.Tong KI, Padmanabhan B, Kobayashi A, Shang CW, Hirotsu Y, Yokoyama S. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol Cell Biol. 2007;27:7511–7521. doi: 10.1128/MCB.00753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 49.Rajendran P, Nandakumar N, Rengarajan T, Palaniswami R, Gnanadhas EN, Lakshminarasaiah U. Antioxidants and human diseases. Clin Chim Acta. 2014;436:332–347. doi: 10.1016/j.cca.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 50.Lewis KN, Mele J, Hayes JD, Buffenstein R. Nrf2 a guardian of healthspan and gatekeeper of species longevity. Integr Comp Biol. 2010;50:829–843. doi: 10.1093/icb/icq034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kansanen E, Kuosmanen SM, Leinonen H, Levonen A.. The Keap1–Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 53.Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31:90–99. doi: 10.1093/carcin/bgp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiang MJ, Namani A, Wu SJ, Wang XL. Nrf2: bane or blessing in cancer? J Cancer Res Clin Oncol. 2014;140:1251–1259. doi: 10.1007/s00432-014-1627-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gañán-Gómez I, Wei Y, Yang H, Boyano-Adánez MC, García-Manero G. Oncogenic functions of the transcription factor Nrf2. Free Radic Biol Med. 2013;65:750–764. doi: 10.1016/j.freeradbiomed.2013.06.041. [DOI] [PubMed] [Google Scholar]
- 56.Ren DM, Villeneuve NF, Jiang T, Wu TD, Lau A, Toppin HA. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A. 2011;108:1433–1438. doi: 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chian S, Thapa R, Chi ZX, Wang XJ, Tang XW. Luteolin inhibits the Nrf2 signaling pathway and tumor growth in vivo. Biochem Biophys Res Commun. 2014;447:602–608. doi: 10.1016/j.bbrc.2014.04.039. [DOI] [PubMed] [Google Scholar]
- 58.De Vries HE, Witte M, Hondius D, Rozemuller AJM, Drukarch B, Hoozemans J. Nrf2-induced antioxidant protection: a promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic Biol Med. 2008;45:1375–1383. doi: 10.1016/j.freeradbiomed.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 59.Sandberg M, Patil J, D׳Angelo B, Weber SG, Mallard C. NRF2-regulation in brain health and disease: implication of cerebral inflammation. Neuropharmacology. 2014;79:298–306. doi: 10.1016/j.neuropharm.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang MJ, An CR, Gao YQ, Leak RK, Chen J, Zhang F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol. 2013;100:30–47. doi: 10.1016/j.pneurobio.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gan L, Johnson JA. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim Biophys Acta. 2014;1842:1208–1218. doi: 10.1016/j.bbadis.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 62.Yoh K, Hirayama A, Ishizaki K, Yamada A, Takeuchi M, Yamagishi S. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells. 2008;13:1159–1170. doi: 10.1111/j.1365-2443.2008.01234.x. [DOI] [PubMed] [Google Scholar]
- 63.Pi JB, Zhang Q, Fu JQ, Woods CG, Hou YY, Corkey BE. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol Appl Pharmacol. 2010;244:77–83. doi: 10.1016/j.taap.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bai Y, Cui WP, Xin Y, Miao X, Barati MT, Zhang C. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. J Mol Cell Cardiol. 2013;57:82–95. doi: 10.1016/j.yjmcc.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 65.Jiménez-Osorio AS, Picazo A, González-Reyes S, Barrera-Oviedo D, Rodríguez-Arellano ME, Pedraza-Chaverri J. Nrf2 and redox status in prediabetic and diabetic patients. Int J Mol Sci. 2014;15:20290–20305. doi: 10.3390/ijms151120290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Velmurugan GV, Sundaresan NR, Gupta MP, White C. Defective Nrf2-dependent redox signalling contributes to microvascular dysfunction in type 2 diabetes. Cardiovasc Res. 2013;100:143–150. doi: 10.1093/cvr/cvt125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Uruno A, Yagishita Y, Yamamoto M. The Keap1–Nrf2 system and diabetes mellitus. Arch Biochem Biophys. 2014;566:76–84. doi: 10.1016/j.abb.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 68.Bhakkiyalakshmi E, Sireesh D, Rajaguru P, Paulmurugan R, Ramkumar KM. The emerging role of redox-sensitive Nrf2–Keap1 pathway in diabetes. Pharmacol Res. 2015;91:104–114. doi: 10.1016/j.phrs.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 69.Jiang T, Huang ZP, Lin YF, Zhang ZG, Fang DY, Zhang DD. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes. 2010;59:850–860. doi: 10.2337/db09-1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Choi BH, Kang KS, Kwak MK. Effect of redox modulating NRF2 activators on chronic kidney disease. Molecules. 2014;19:12727–12759. doi: 10.3390/molecules190812727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. New Engl J Med. 2011;365:327–336. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- 72.Boutten A, Goven D, Artaud-Macari E, Boczkowski J, Bonay M. NRF2 targeting: a promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol Med. 2011;17:363–371. doi: 10.1016/j.molmed.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 73.Cho HY, Kleeberger SR. Nrf2 protects against airway disorders. Toxicol Appl Pharmacol. 2010;244:43–56. doi: 10.1016/j.taap.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 74.Reddy SP. The antioxidant response element and oxidative stress modifiers in airway diseases. Curr Mol Med. 2008;8:376–383. doi: 10.2174/156652408785160925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hübner RH, Schwartz JD, De Bishnu P, Ferris B, Omberg L, Mezey JG. Coordinate control of expression of Nrf2-modulated genes in the human small airway epithelium is highly responsive to cigarette smoking. Mol Med. 2009;15:203–219. doi: 10.2119/molmed.2008.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rangasamy T, Cho CY, Thimmulappa RK, Zhen LJ, Srisuma SS, Kensler TW. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Investig. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Biswal S, Thimmulappa RK, Harvey CJ. Experimental therapeutics of Nrf2 as a target for prevention of bacterial exacerbations in COPD. Proc Am Thorac Soc. 2012;9:47–51. doi: 10.1513/pats.201201-009MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li J, Ichikawa T, Janicki JS, Cui T. Targeting the Nrf2 pathway against cardiovascular disease. Expert Opin Ther Targets. 2009;13:785–794. doi: 10.1517/14728220903025762. [DOI] [PubMed] [Google Scholar]
- 79.Dreger H, Westphal K, Wilck N, Baumann G, Stangl V, Stangl K. Protection of vascular cells from oxidative stress by proteasome inhibition depends on Nrf2. Cardiovasc Res. 2010;85:395–403. doi: 10.1093/cvr/cvp279. [DOI] [PubMed] [Google Scholar]
- 80.Collins AR, Lyon CJ, Xia X, Liu JZ, Tangirala RK, Yin F. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ Res. 2009;104:e42–e54. doi: 10.1161/CIRCRESAHA.108.188771. [DOI] [PubMed] [Google Scholar]
- 81.Ruotsalainen A., Inkala M, Partanen ME, Lappalainen JP, Kansanen E, Mäkinen PI. The absence of macrophage Nrf2 promotes early atherogenesis. Cardiovasc Res. 2013;98:107–115. doi: 10.1093/cvr/cvt008. [DOI] [PubMed] [Google Scholar]
- 82.Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium–induced colitis. Cancer Res. 2006;66:11580–11584. doi: 10.1158/0008-5472.CAN-06-3562. [DOI] [PubMed] [Google Scholar]
- 83.Jena G, Trivedi PP, Sandala B. Oxidative stress in ulcerative colitis: an old concept but a new concern. Free Radic Res. 2012;46:1339–1345. doi: 10.3109/10715762.2012.717692. [DOI] [PubMed] [Google Scholar]
- 84.Stachel I, Geismann C, Aden K, Deisinger F, Rosenstiel P, Schreiber S. Modulation of nuclear factor E2–related factor 2 (Nrf2) activation by the stress response gene immediate early response-3 (IER3) in colonic epithelial cells: a novel mechanism of cellular adaption to inflammatory stress. J Biol Chem. 2014;289:1917–1929. doi: 10.1074/jbc.M113.490920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aleksunes LM, Manautou JE. Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol Pathol. 2007;35:459–473. doi: 10.1080/01926230701311344. [DOI] [PubMed] [Google Scholar]
- 86.Okawa H, Motohashi H, Kobayashi A, Aburatani H, Kensler TW, Yamamoto M. Hepatocyte-specific deletion of the Keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem Biophys Res Commun. 2006;339:79–88. doi: 10.1016/j.bbrc.2005.10.185. [DOI] [PubMed] [Google Scholar]
- 87.Qu Q, Liu J, Zhou HH, Klaassen CD. Nrf2 protects against furosemide-induced hepatotoxicity. Toxicology. 2014;324:35–42. doi: 10.1016/j.tox.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 88.García-Niño WR, Pedraza-Chaverrí J. Protective effect of curcumin against heavy metals-induced liver damage. Food Chem Toxicol. 2014;69:182–201. doi: 10.1016/j.fct.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 89.Klaassen CD, Reisman SA. Nrf2 the rescue: effects of the antioxidative/electrophilic response on the liver. Toxicol Appl Pharmacol. 2010;244:57–65. doi: 10.1016/j.taap.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kurzawski M, Dziedziejko V, Urasińska E, Post M, Wójcicki M, Miętkiewski J. Nuclear factor erythroid 2–like 2 (Nrf2) expression in end-stage liver disease. Environ Toxicol Pharmacol. 2012;34:87–95. doi: 10.1016/j.etap.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 91.Deramaudt TB, Dill C, Bonay M. Regulation of oxidative stress by Nrf2 in the pathophysiology of infectious diseases. Méd Mal Infect. 2013;43:100–107. doi: 10.1016/j.medmal.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 92.Schaedler S, Krause J, Himmelsbach K, Carvajal-Yepes M, Lieder F, Klingel K. Hepatitis B virus induces expression of antioxidant response element–regulated genes by activation of Nrf2. J Biol Chem. 2010;285:41074–41086. doi: 10.1074/jbc.M110.145862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Investig. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maicas N, Ferrándiz ML, Brines R, Ibáñez L, Cuadrado A, Koenders MI. Deficiency of Nrf2 accelerates the effector phase of arthritis and aggravates joint disease. Antioxid Redox Signal. 2011;15:889–901. doi: 10.1089/ars.2010.3835. [DOI] [PubMed] [Google Scholar]
- 95.Wruck CJ, Fragoulis A, Gurzynski A, Brandenburg LO, Kan YW, Chan KM. Role of oxidative stress in rheumatoid arthritis: insights from the Nrf2-knockout mice. Ann Rheum Dis. 2011;70:844–850. doi: 10.1136/ard.2010.132720. [DOI] [PubMed] [Google Scholar]
- 96.Córdova EJ, Velázquez-Cruz R, Centeno F, Baca V, Orozco L. The NRF2 gene variant, -653G/A, is associated with nephritis in childhood-onset systemic lupus erythematosus. Lupus. 2010;19:1237–1242. doi: 10.1177/0961203310367917. [DOI] [PubMed] [Google Scholar]
- 97.Braun S, Hanselmann C, Gassmann MG, Auf Dem Keller U, Born-Berclaz C, Chan KM. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol Cell Biol. 2002;22:5492–5505. doi: 10.1128/MCB.22.15.5492-5505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Beyer TA, Auf Dem Keller U, Braun S, Schäfer M, Werner S. Roles and mechanisms of action of the Nrf2 transcription factor in skin morphogenesis, wound repair and skin cancer. Cell Death Differ. 2007;14:1250–1254. doi: 10.1038/sj.cdd.4402133. [DOI] [PubMed] [Google Scholar]
- 99.Nagai N, Thimmulappa RK, Cano M, Fujihara M, Izumi-Nagai K, Kong XN. Nrf2 is a critical modulator of the innate immune response in a model of uveitis. Free Radic Biol Med. 2009;47:300–306. doi: 10.1016/j.freeradbiomed.2009.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Eggler AL, Gay KA, Mesecar AD. Molecular mechanisms of natural products in chemoprevention: induction of cytoprotective enzymes by Nrf2. Mol Nutr Food Res. 2008;52 Suppl 1:S84–S94. doi: 10.1002/mnfr.200700249. [DOI] [PubMed] [Google Scholar]
- 101.Hur W, Gray NS. Small molecule modulators of antioxidant response pathway. Curr Opin Chem Biol. 2011;15:162–173. doi: 10.1016/j.cbpa.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 102.Hu LQ, Magesh S, Chen L, Wang LL, Lewis TA, Chen Y. Discovery of a small-molecule inhibitor and cellular probe of Keap1–Nrf2 protein–protein interaction. Bioorg Med Chem Lett. 2013;23:3039–3043. doi: 10.1016/j.bmcl.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jiang ZY, Lu MC, Xu LL, Yang TT, Xi MY, Xu XL. Discovery of potent Keap1–Nrf2 protein–protein interaction inhibitor based on molecular binding determinants analysis. J Med Chem. 2014;57:2736–2745. doi: 10.1021/jm5000529. [DOI] [PubMed] [Google Scholar]
- 104.Chen Y, Inoyama D, Kong A., Beamer LJ, Hu L. Kinetic analyses of Keap1–Nrf2 interaction and determination of the minimal Nrf2 peptide sequence required for Keap1 binding using surface plasmon resonance. Chem Biol Drug Des. 2011;78:1014–1021. doi: 10.1111/j.1747-0285.2011.01240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huang XY. Fluorescence polarization competition assay: the range of resolvable inhibitor potency is limited by the affinity of the fluorescent ligand. J Biomol Screen. 2003;8:34–38. doi: 10.1177/1087057102239666. [DOI] [PubMed] [Google Scholar]
- 106.Inoyama D, Chen Y, Huang XY, Beamer LJ, Kong A., Hu L. Optimization of fluorescently labeled Nrf2 peptide probes and the development of a fluorescence polarization assay for the discovery of inhibitors of Keap1–Nrf2 interaction. J Biomol Screen. 2012;17:435–447. doi: 10.1177/1087057111430124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Smirnova NA, Haskew-Layton RE, Basso M, Hushpulian DM, Payappilly JB, Speer RE. Development of Neh2-luciferase reporter and its application for high throughput screening and real-time monitoring of Nrf2 activators. Chem Biol. 2011;18:752–765. doi: 10.1016/j.chembiol.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lo SC, Li XC, Henzl MT, Beamer LJ, Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006;25:3605–3617. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hancock R, Bertrand HC, Tsujita T, Naz S, El-Bakry A, Laoruchupong J. Peptide inhibitors of the Keap1–Nrf2 protein–protein interaction. Free Radic Biol Med. 2012;52:444–451. doi: 10.1016/j.freeradbiomed.2011.10.486. [DOI] [PubMed] [Google Scholar]
- 110.Hancock R, Schaap M, Pfister H, Wells G. Peptide inhibitors of the Keap1–Nrf2 protein–protein interaction with improved binding and cellular activity. Org Biomol Chem. 2013;11:3553–3557. doi: 10.1039/c3ob40249e. [DOI] [PubMed] [Google Scholar]
- 111.Steel R, Cowan J, Payerne E, O׳Connell MA, Searcey M. Anti-inflammatory effect of a cell-penetrating peptide targeting the Nrf2/Keap1 interaction. ACS Med Chem Lett. 2012;3:407–410. doi: 10.1021/ml300041g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sun HP, Jiang ZY, Zhang MY, Lu MC, Yang TT, Pan Y. Novel protein–protein interaction inhibitor of Nrf2-Keap1 discovered by structure-based virtual screening. Med Chem Commun. 2014;5:93–98. [Google Scholar]
- 113.Jnoff E, Albrecht C, Barker JJ, Barker O, Beaumont E, Bromidge S. Binding mode and structure-activity relationships around direct inhibitors of the Nrf2-Keap1 complex. ChemMedChem. 2014;9:699–705. doi: 10.1002/cmdc.201300525. [DOI] [PubMed] [Google Scholar]
- 114.Marcotte D, Zeng WK, Hus J., McKenzie A, Hession C, Jin P. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg Med Chem. 2013;21:4011–4019. doi: 10.1016/j.bmc.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 115.Zhuang CL, Narayanapillai S, Zhang WN, Sham YY, Xing CG. Rapid identification of Keap1–Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J Med Chem. 2014;57:1121–1126. doi: 10.1021/jm4017174. [DOI] [PubMed] [Google Scholar]
- 116.Sato M, Aoki T, Inoue H, Tanaka T, Kunishima N, inventors. Keap1 protein binding compound, crystal of complex between the same and Keap1 protein, and method for producing the same. Toray Ind Inc., assignee. Japanese Patent JP 2013-028575A. 2013 February 7.
- 117.Beamer LJ, Li X, Bottoms CA, Hannink M. Conserved solvent and side-chain interactions in the 1.35 angstrom structure of the Kelch domain of Keap1. Biol Crystallogr. 2005;61:1335–1342. doi: 10.1107/S0907444905022626. [DOI] [PubMed] [Google Scholar]