

Abstract
The emerging trends in the combinatorial chemistry and drug design have led to the development of drug candidates with greater lipophilicity, high molecular weight and poor water solubility. Majority of the failures in new drug development have been attributed to poor water solubility of the drug. Issues associated with poor solubility can lead to low bioavailability resulting in suboptimal drug delivery. About 40% of drugs with market approval and nearly 90% of molecules in the discovery pipeline are poorly water-soluble. With the advent of various insoluble drug delivery technologies, the challenge to formulate poorly water soluble drugs could be achieved. Numerous drugs associated with poor solubility and low bioavailabilities have been formulated into successful drug products. Several marketed drugs were reformulated to improve efficacy, safety and patient compliance. In order to gain marketing exclusivity and patent protection for such products, revitalization of poorly soluble drugs using insoluble drug delivery technologies have been successfully adopted by many pharmaceutical companies. This review covers the recent advances in the field of insoluble drug delivery and business prospects.
KEY WORDS: Bioavailability, Cocrystals, Solubility, Inclusion complexation, Nanoparticles, Self-emulsifying formulations, Proliposomes
Graphical abstract
The improved formulation of existing drugs is turning out to be lucrative business for pharmaceutical industry which is facing innovation deficit these days for new molecules. New dosage form, change of forms of drugs (ester/salt), prodrug/active metabolite of drug, different routes of administration are few changes that pharmaceutical companies are exploring for 505(b)(2) fillings. Significant number of insoluble drugs in the market provides profitable strategies for pharmaceutical companies to file NDA under 505(b)(2) with improved formulations providing faster dissolution and enhanced bioavailability. Hence this review summarizes various solubilization technologies. The recent advances, clinical benefits and business potentials of these technologies are discussed in detail.
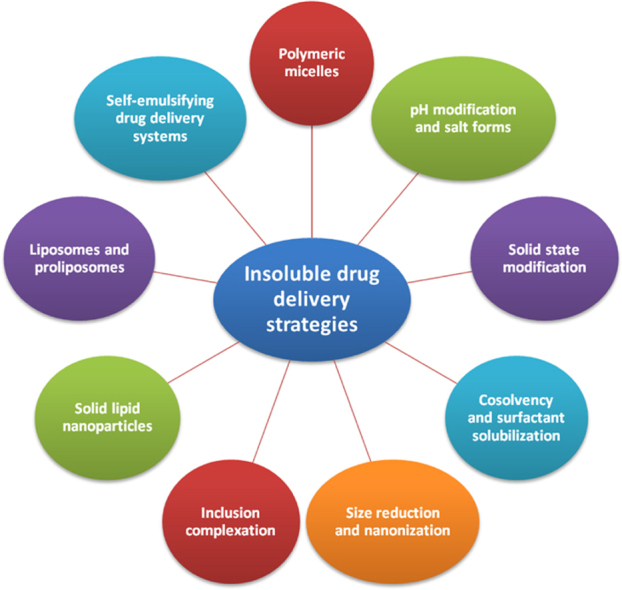
1. Introduction
The search for innovative medicines in disease management without compromising on safety and efficacy is a challenge. In spite of significant success in the discovery of new drugs, there are still unmet medical conditions which need effective therapy. Market potential, competition among companies, dry pipeline of developmental candidates of various companies have hastened the drug discovery and development process. As a result, a significant number of drugs getting approvals have poor biopharmaceutical properties. An estimated 40% of approved drugs and nearly 90% of the developmental pipeline drugs consist of poorly soluble molecules1. Several marketed drugs suffer from poor solubility, low permeability, rapid metabolism and elimination from the body along with poor safety and tolerability2.
Recent studies have revealed that discovery and development of new drugs alone are not sufficient to achieve therapeutic excellence and capture market economies3. Therefore, modified formulations of existing drugs are gaining more importance. The improved formulation of existing drugs is turning out to be lucrative business for pharmaceutical industry which is facing innovation deficit these days for new molecules4. New dosage form, change of forms of drugs (ester/salt), prodrug/active metabolite of drug, different routes of administration are few changes that pharmaceutical companies are exploring for 505(b)(2) fillings5. Significant number of insoluble drugs in the market provides profitable strategies for pharmaceutical companies to file NDA under 505(b)(2) with improved formulations providing faster dissolution and enhanced bioavailability. Hence this review summarizes various solubilization technologies. The recent advances, clinical benefits and business potentials of these technologies are discussed in detail. The potential benefits of insoluble drug delivery technologies are depicted in Fig. 1.
Figure 1.

Benefits of insoluble drug delivery strategies.
2. Insoluble drug delivery technologies
2.1. pH modification and salt forms
Nearly 70% of drugs are reported to be ionizable, of which a majority are weakly basic. A pH-dependent solubility is exhibited by ionizable drugs, wherein weakly acidic drugs are more soluble at pH>pKa (ionization constant) and weakly basic drugs are soluble at pH<pKa6. This pH dependent solubility was explored extensively to formulate insoluble drugs. On the other hand, salt formation of weakly acidic or basic drugs provided alternate strategies for formulation of drugs which have pH dependent solubility. Pharmaceutically acceptable counter ions in the salt can provide favorable pH conditions upon dissolution in water, and thus the pH of resulting solution would be close to maximum pH of drugs. Hence salt forms may sometimes avoid pH adjustments necessary for solubilization of drugs. In addition, salt formation has been reported to improve crystallinity, stability and pharmaceutical processibility of drugs6.
There are many insoluble drugs on the market which are formulated with pH modification technology. Ciprofloxacin is a classic drug which is weakly basic and practically insoluble in water at neutral pH. However it exhibits pH-dependent solubility with higher solubility at acidic condition. Most of the intravenous formulations contain lactic acid as pH modifier to improve solubility7. Intravenous ciprofloxacin infusions are essential for treating different kinds of severe bacterial infections. Telmisartan is another drug which exhibits pH-dependent solubility. The currently marketed oral formulation of telmisartan contain alkalis, such as sodium hydroxide and meglumine for pH modification7, 8, 9, 10. Telmisartan formulation marketed under brand name Micardis® is manufactured using a expensive spray-drying process, wherein drug and alkalis along with other excipients are dissolved in water and spray-dried to produce granules11. The spray-dried granules obtained were reported to have a pH-independent dissolution profile. However, generic versions of the telmisartan formulation are hard to come by, owing to the insoluble nature of the drug׳s free-acid-form and the critical steps involved in its manufacturing process that provided an additional market capitalization to the innovator12.
Repaglinide is an example of Zwitterion drug with poor water solubility of 37 µg/mL13. Currently repaglinide, marketed as Prandin® in USA, is formulated with meglumine as pH modifier. Various patents disclose the use of meglumine in the formulation and spray-drying as the process for preparing the granules14, 15, 16, 17. Tricky process and critical formulation sometimes prove to be hard to make generic copies. In case of both telmisartan and repaglinide, actual salt forms of drugs are not used in the formulation, instead the bases such as meglumine and sodium hydroxide were added to the formulation. This could be due to technical reasons, such as lack of crystallinity, poor stability and deliquescent nature of resulting salts. On other hand, including bases in the formulation could be due to commercial reasons, in order to build complexity in the process and product, such that it is hard to make generic versions. These are a few examples of how a clinically and commercially beneficial drug product could be launched in the market by altering the formulation strategies.
Aspirin is century old non-steroidal anti-inflammatory drug (NSAID), yet currently explored by various companies for commercial benefits. Soluble formulations of aspirin are currently available on the market. Aspro Clear, is soluble, effervescent tablet containing aspirin. The effervescence and favorable pH condition required for solubility of aspirin are facilitated by incorporating sodium bicarbonate and citric acid in the formulation. Aspro Clear reported to provide faster relief of pain than plain aspirin tablets18. This is another example, how insoluble drug formulation technology can be explored for commercial and clinical benefits.
Insoluble drugs are mostly formulated using the salt forms of weakly acid and basic drugs. Various salt forms of drugs have been the area of interest for pharmaceutical companies for commercial and clinical benefits. In the following section, few examples of such inventions are discussed. Identification of bisulfate salt form of atazenavir is an interesting example of how salt screening could help a molecule to progress from being dropped at preclinical development to clinical studies and finally to marketing approval. Atazenavir as free base is practically insoluble in water (<1 µg/mL) and had poor oral bioavailability in preclinical animal models19. Lack of sufficient absorption was reported to be a hurdle in the development of this molecule. In an effort to identify viable option to improve the bioavailability, series of salts were screened and finally atazenavir bisulfate was selected for further development19, 20. Atazenavir bisulfate exhibited distinct advantage over other salts such as methane sulfonate and hydrochloride in terms of solubility and solid state stability. Hydrochloride and methane sulfonate salts of atazenavir, when dissolved in water beyond saturation solubility of salts, there was solid state transformation leading to dissociation of salt to free base at pH>pHmax. Analysis of excess of solid in the suspension revealed that material was indeed free base. Under similar experimental conditions bisulfate was found to be stable and did not convert to free base, and rather excess of solid was found to be in hydrated sulfate salt20. Therefore, the absolute bioavailability of bisulfate was multifold-higher than the free base. This invention not only lead to superior protease inhibitor on the market but provided additional patent protection and marketing exclusivity to inventor company.
One of largest-selling anticancer drugs imatinib is marketed as a salt form, imatinib mesylate. The drug exhibits poor solubility and hence mesylate salt was used for its development, which is soluble in water at pH<5.521, 22. Among the two polymorphic forms (α and β), generated by imatinib salt, the β form is more stable with acceptable pharmaceutical properties. However, additional marketing rights were assigned to the innovator due to their patent protection of the β form23. Many old drugs have been reformulated as salt forms for commercial purposes. One such example is fenofibrate, which was approved in 1993 and was included in generic competition from the year 200024. Since then, Abbott laboratories24 continued filing NDA׳s altering the dose in order to gain market exclusivity. Interestingly, the active form of all fenofibrate formulations was found to be fenofibric acid, an active metabolite of fenofibrate which was responsible for the therapeutic activity. This fact was well explored by Abbot and developed cholin-fenofibrate, a soluble and light-stable salt of fenofibric acid25. This salt form was developed into a delayed release capsule formulation and was approved by the FDA. This delayed release formulation was proved to be one of blockbuster product in the recent time. In the times of innovation drought, such inventions are becoming huge commercial success thus improving overall investment drive in pharmaceutical research and development.
The use of aspirin for clinical management of migraine was tested recently. The soluble aspirin-d,l-lysine salt was formulated for intravenous injection (IV). The clinical studies revealed that intravenous administration of aspirin was effective in relieving migraine attack. Although sumatriptan was slightly more effective than aspirin IV in headache relief, aspirin was well tolerated26, 27. Hence the new salt form of aspirin demonstrated safe, effective and affordable alternative therapy for the treatment of migraine. Similarly aspirin-calcium was utilized in the formulation of soluble tablet (Solorpin®). The formulation showed faster onset of action compared to the tablet with plain aspirin28. Improved clinical benefits, as well as commercial profits, were accomplished with these salt forms.
Clopidogrel is an anti-platelet agent that works through irreversible binding of its active metabolite to the P2Y12 subtype of adenosine diphosphate (ADP) chemoreceptors on platelets cell membrane. Initially, it was available as Plavix®, consisting of the salt form, clopidogrel bisulfate. However, other salt forms like clopidogrel besilate and clopidogrel hydrochloride were approved in Europe. In this case salt forms are explored by generic maker for market exclusivity29. Recently FDA approved Advil®, a sodium salt of ibuprofen. This product is superior in terms of its rapid onset of action as compared to Advil Liqui-Gel® capsules containing ibuprofen30. Apart from enabling faster pain-relief to patients, this new salt form of ibuprofen provided market exclusivity of at least 3–5 years for the manufacturer.
Rosuvastatin (sparingly soluble) is available in the market as its calcium salt. Recently generic-maker Watson pharmaceuticals, Inc., gained approval for its NDA containing rosuvastatin zinc under section 505(b)(2)31, thus getting marketing exclusivity more than typical ANDA. However, the approval is subject to a court decision due to a legal petition filed by AstraZeneca. Pharmaceutical companies are continuously exploring the salt forms of drugs for better clinical performance. Sometimes it seems like reformulation is an alternative path for the pharmaceutical companies to exploit marketing exclusivity and captivity32. Further advancements in this technology will be more interesting, since there would be many more NDA׳s drugs to be approved with new salt forms in the future.
2.2. Co-solvency and surfactant solubilization
Formulation of insoluble drugs using co-solvents is also one of the oldest and widely used technique, especially for liquid formulation intended for oral and intravenous administration. Reduction of the dielectric constant is possible by the addition of co-solvents, which facilitates increased solubilization of non-polar drug molecules. In order to maximize the solubility and prevent precipitation upon dilution, co-solvents are used in conjunction with surfactants and pH modifiers33, 34.
Taxol, an intravenous injection of paclitaxel, is the most debated formulation using this approach. This was developed using 49% of dehydrated alcohol and 527 mg of cremophore EL35, which must be diluted before infusion. Additionally, pretreatment of patients with antihistamines is essential owing to a hypersensitivity reaction due to higher content of cremophore EL in the formulation. Later, several formulations of paclitaxel excluding cremophore EL were attempted and couple of them gained FDA approval after making a smooth means of access through clinical testing. Formulations devoid of cremophore EL included Abraxane (albumin microspheres containing Paclitaxel) and Genexol (PEG-PLA polymeric micelles with Paclitaxel)36, 37.
Similarly, docetaxel is another widely used anticancer drug and the original formulations of taxotere contains ethanol and Tween 80 to solubilize the drug (0.54 g polysorbate 80 and 0.395 g dehydrated alcohol)38. However, hypersensitivity reactions using this product were reported due to the surfactants in the formulation. Sandoz, Inc., Hospira Inc. and Apotex Inc. each has docetaxel containing a new drug product approved under section 505(b)(2)39, 40, 41. Most of the new formulations have PEG 300 as additional cosolvent and Tween 80 content significantly less than taxotere. These new formulations were claimed to be safer and stable than taxotere. Insoluble drug delivery technology utilizing the co-solvent-surfactant approach had indeed proved vital in providing an effective treatment option for cancer patients. Further improvement in the formulation of taxol׳s resulted in more patient compliance and new intellectual properties for pharmaceutical companies. A list of pharmaceutical formulations containing the highest amounts of co-solvents and surfactants are provided in Table 142. Though co-solvency and surfactant solubilization techniques are widely used for enhancing the solubility of hydrophobic drugs, they have some disadvantages: tolerability of formulations with high levels of synthetic surfactants may be poor in cases where long term chronic administration is intended; uncontrolled precipitation may occur upon dilution with aqueous media or physiological fluids. Precipitates may be amorphous or crystalline and can vary in size; precipitation of drug from a co-solvent mixture may result in embolism and local adverse effects at the injection site; concomitant solubilization of other ingredients such as preservatives may lead to consequent alteration in stability and effectiveness of the drug product.
Table 1.
List of parenteral drug formulations containing co-solvents and surfactants.
| Solvent | Percentage in marketed formulation (%) | Percentage administered (%) | Route of administration | Example |
|---|---|---|---|---|
| Cremophor EL | 11–65 | ≤10 | IV infusion | Paclitaxel |
| Cremophor RH 60 | 20 | ≤0.08 | IV infusion | Tacrolimus |
| Dimethylacetamide (DMA) | 6 | ≤3 | IV infusion | Teniposide |
| Ethanol | 5–80 | ≤6 | SC | Dihydroergotamine |
| Glycerin | 15–32 | ≤15 | IM, SC, IV | Dihydroergotamine |
| N-methyl-2-pyrrolidone | 100 | 100 | Subgingival | Doxycyclin |
| PEG 300 | ≤60 | ≤50 | IM, IV bolus | Methocarbamil |
| PEG 400 | 18–67 | ≤18 | IM | Lorazepam |
| Polysorbate 80 | 0.075–100 | ≤4 | IM | Chlordiazepoxide |
| Propylene glycol | 10–80 | ≤80 | IM | Lorazepam |
| Solutol HS-15 | 50 | 50 | IV | Propanidid |
IM: intramuscular; IV: intravenous; PEG: polyethylene glycol; SC: subcutaneous.
2.3. Amorphous forms, solid dispersions and cocrystals
Stable crystal forms of drugs pose problem in solubilization due to high lattice energy. Thus, disordered amorphous forms offer distinct advantage over crystal forms with regards to solubility. Hence, changing the solid state characteristics of active pharmaceutical ingredient (API) renders the molecule more water soluble. But, excess of enthalpy, entropy and free energies of amorphous forms makes them prone to crystallization, leading to the formation of stable crystals43. However, the advent of new techniques to improve stability of amorphous forms improved chances of their use in pharmaceutical formulations44. Complicated process of making amorphous drug systems and various factors affecting the stability of those forms resulted in reduced generic competition for already approved amorphous products. Cefuroxime axetil practically was insoluble in water and introduced as Ceftin® by GSK in amorphous form and was protected by a couple of patents, which barred the entry of generic players for a reasonable period45, 46. Another drug product, the amorphous zafirlukast is available commercially as Accolate®. The amorphous form is subject to various patents which precluded early generic entry47, 48. Amorphous forms of other drugs like nelfinavir mesylate, quinapril hydrochloride and rosuvastatin calcium are also commercially available in the market.
Solid dispersion technology was extensively explored in recent decades for the delivery of insoluble drugs. Physically, solid dispersions are eutectic mixtures or solid solutions in which drugs exist either in an amorphous form dispersed in the carrier or as a molecular dispersion in the carrier49, 50, 51. Solid dispersions favor enhanced dissolution of drugs due to the formation of a high-energy amorphous form or increased solubility leading to supersaturation. The increased solubility can be attributed to the dispersion of drugs at the molecular level and/or solubilization effects of the polymer. The drug remains in a metastable form for considerable time in the supersaturated state and polymeric carrier in turn can stabilize the metastable state by preventing nucleation51. Advances in melt-extrusion and spray-drying have accelerated industrial applications of solid dispersions for the delivery of insoluble drugs.
Sporanox® is a classic example of a drug (itraconazole) formulated using solid dispersion technology. At neutral pH itraconazole has a negligible solubility of 1 ng/mL52. For preparing solid dispersions of itraconazole, spray-layering technology was used in which an organic solution of drug and hydroxylpropyl methylcellulose (HPMC) was sprayed over sugar beads to form a thin film consisting of molecularly dispersed drug and polymer. This amorphous formulation significantly enhanced bioavailability compared to crystalline itraconazole. Apart from spray layering, itraconazole solid dispersions were also prepared using hot-melt extrusion with varying polymers such as HPMC, Eudragit and polyvinyl pyrrolidone (PVP) mixture. In vitro studies revealed a faster dissolution of solid dispersions containing Eudragit in comparison to HPMC and sporanox52. In contrast, clinical studies revealed a similarity between solid dispersions containing HPMC and sporanox, which can be attributed to the solubilization and stabilization effects of HPMC in physiological conditions (in vivo).
A list of currently marketed solid-dispersion products is shown in Table 251. All the listed products have generated clinically beneficial results by producing adequate drug levels in the body at desired therapeutic concentration, leading to improved bioavailability. Apart from potential clinical benefits, these products have generated considerable intellectual property and commercial success to the manufacturer.
Table 2.
List of marketed products in United States utilizing solid dispersion technology.
| Drug | Brand name | Carrier | Manufacturer | Year of FDA approval |
|---|---|---|---|---|
| Itraconazole | Sporanox® | HPMC | Janssen Pharmaceuticals, Inc., USA | 1992 |
| Tacrolimus | Prograf® | HPMC | AstellasPharma, US Inc. | 1994 |
| Lopinavir/Ritonavir | Kaletra® | PVP/VA | Abbot Labarotaries, USA | 2005 |
| Nabilone | Casamet® | PVP | Meda Pharmaceuticals Inc., USA | 2006 |
| Nimodipine | Nimotop® | PEG | Bayer (Pty) Ltd., USA | 2006 |
| Fenofibrate | Fenoglide® | PEG/Poloxamer | Santarus, Inc. | 2007 |
| Etravirine | Intelence® | HPMC | Janssen Therapeutics, USA | 2008 |
Pharmaceutical cocrystal technology has received greater attention in the last decade owing to its successful delivery of insoluble drugs. Stoichiometric solids of drug and conformer (second component), which exist as crystals at ambient temperature are referred to as cocrystals. Non-covalent forces like acid–amide, acid–acid, and amide–amide interactions, usually of hydrogen bonding nature, hold the drug and conformer together in the cocrystal. The enhanced solubility of drug in cocrystal is achieved by lower lattice energy and higher solvent affinity53. Any of the generally regarded as safe (GRAS)-listed excipients, organic acids (such as fumaric acid, malic acid, glutaric acid, succinic acid, oxalic acid), nutraceuticals (such as pterostilbene, quercetin, p-coumaric acid and saccharine) can act as a conformer. Co-crystal technology has been explored for solubility enhancement of drugs like itraconazole, carbamazepine, gabapentinin, modafinil, piroxicam, caffeine, etc.53. The cocrystal technology have been used to create intellectual property and large number of patents have been filed54. However there is no approved product with drug cocrystals, with enormous potential for delivery of insoluble drugs till today, but the future of cocrystals is promising.
2.4. Polymeric micelles
Water insoluble drugs often have greater affinity for hydrophobic solvents because of hydrophobic–hydrophobic interactions and also have affinity for hydrophobic region of micelles. Hence encapsulation of those drugs in micelles enables their formulation in aqueous vehicle. Initially the hydrophilic surfactants were used to solubilize the drug for oral and intravenous administration. However, limited solubilization, higher critical micellear concentrations (CMC) and potential adverse events after intravenous administration have limited their application. Polymeric micelles on other hand, offer greater advantage in terms of solubilization capacity, lower CMC and greater tolerability. Polymeric micelles are formed using diblcok polymers such as PEG-PLA or triblock polymers PLA-PEG-PLA. The PEG is usually the hydrophilic component in the polymer for micelles and hydrophobic chain can be of poly lactic acid, poly aspartic acid, polycaprolactic acid, etc.37, 55. Due to low CMC, the polymeric micelles remain stable at low polymer concentration after dilution with body fluids. The nano-sized nature of polymeric micelles provides opportunity for tumor-targeting via enhanced permeation and retention effect (EPR). The hydrophilic PEG surface makes micelles less susceptible for reticulo-endothelial scavenging, and thus drugs have longer circulation time. Polymeric micelles can also be tailored for pH-responsive release of drugs at specific tissues and for active-targeting using targeting ligands37.
The Genexol-PM is polymeric micelles comprising of PEG-(d,l-lactide) polymer with paclitaxel encapsulated in the micelles. This is the first polymeric micelle formulation approved by FDA56 and is reported to be superior in terms of safety and tolerability compared to other marketed formulations (ethanol/Cremophore EL). The pluronics-based polymeric micelles containing doxorubicin (SP1049C) are currently in phase III clinical trial and have been granted orphan status by FDA. Another paclitaxel polymeric micelles (NK105) and cisplatin micelles are in phase II clinical trials37. The polymeric micelles for delivery of insoluble drugs, especially parenteral formulations can offer intellectual property for companies and better treatment options for the patients in need of those drugs37. Table 3 presents the representative list of drug-loaded polymeric micelles products and their progress57, 58.
Table 3.
Representative list of drug-loaded polymeric micelles-based products.
| Product | Incorporated drug | Status | Company |
|---|---|---|---|
| Genexol PM | Paclitaxel | Marketed | Samyang |
| Estrasorb | Estrogen | Marketed | Novavax |
| Medicelle | DACH-platin-PEG-polyglutamic acid | phase I/II | NanoCarrier |
| Flucide | Anti-influenza | phase I/II | Nano Viricides |
| Basulin | Insulin | phase II/III | Flamel Technologies |
| DO/NDR/02 | Paclitaxel | phase I/II | Dabur Research Foundation |
| NK-911 | Doxorubicin | phase II | Nippon Kayaku Co. |
| NK-105 | Paclitaxel | phase II/III | Nippon Kayaku Co. |
| NK-012 | SN-38 | phase II | Nippon Kayaku Co. |
| NC-6004 | Cisplatin | phase III | Nanocarrier Co. |
| NC-4016 | Oxaliplatin | phase I/II | Nanocarrier Co. |
| SP-1049C | Doxorubicin | phase II/III | SupratekPharma Inc. |
| NC-6300 | Epirubicin | phase I/II | Nanocarrier Co. |
2.5. Inclusion complexation
Cyclodextrins (CD) are the versatile excipients studied extensively for pharmaceutical applications59. These are cyclic oligosaccharides consisting of glucopyranose units that are united via 1,4-linkage. Three major types of CDs include α, β and γ, varying with 6, 7 and 8 glucopyranose units, respectively. CDs have a truncated-cone structure with a hydrophobic interior and a hydrophilic exterior due to the cyclic orientation of pyranose units. Central cavity of cyclodextrin is hydrophobic due to skeletal carbon atoms and ethereal oxygen. Polarity of cavity is estimated to be somewhere close to aqueous ethanolic solution59. The hydrophobic nature of cavity enables entrapment of hydrophobic molecules of suitable size inside the cavity and hydrophilic surface of CD makes complex soluble in water. Apart from solubilization, cyclodextrins are also used for drug stabilization, drug protection from light, thermal and oxidative stress, taste masking of drugs, and reduced dermal, ocular or gastrointestinal irritation.
The relative size of CD to the guest molecule, the presence of key functional groups on the guest molecule, and thermodynamic interactions between CD, guest molecule and solvent are the key factors that enable the formation of an inclusion complex. In addition to natural CDs, insoluble drugs are formulated using synthetic CDs like hydroxy propyl-β-cyclodextrins, hydroxy propyl-γ-cyclodextrins and sulfobutyl cyclodextrin (Captisol®), since the latter have higher solubility and safety profiles when compared to the former59, 60.
The use of cyclodextrins in the formulation has enabled many product containing insoluble drugs to reach the market and eventually helped to treat many life-threatening disease conditions. Recently cyclodextrins are explored to reformulate existing drugs for better clinical applications and also for revenue generation via NDAs under section 505(b)(2). There were several cyclodextrin containing drug products in Japan, Germany and other European countries; however, Janssen Pharmaceuticals, Inc. was the first company to get US FDA approval for its antifungal drug product (sporanox oral and IV solution) containing itraconazole with 40% of hydroxy propyl-β-cyclodextin in the year 199958. This was proved to be huge commercial successes for Janssen Pharmaceuticals, Inc. and their efforts in finding modified CD were paid off. The introduction of spornox oral solution lead to effective treatment of fungal throat infections and intravenous formulation for severe systemic fungal infections. The intravenous formulations of ziprazidone mesylate and voriconazole formulated with sulphobutyl cyclodextrin are available in many countries including USA. The reformulation of existing drugs using cyclodextrin has been explored to get approval of new drug applications with greater marketing exclusivity and patent protection. Drugs such as aripriprazole, mitomycine, diclofenac sodium, chlodizepoxide, meloxicam, alfaxalone, cisapride, indomethacine, insulin (nasal spray) and omeprazole have been reformulated using cyclodextrins for both commercial and health benefits61.
2.6. Size reduction and nanonization
Over the past two decades, nanoparticle technology has become a well-established and proven formulation approach for poorly-soluble drugs. Reducing a drug׳s particle size to sub-micron range is referred to as ‘nanonization’. In the field of pharmaceuticals, the term ‘nanoparticle’ is applied to structures less than 1 μm in size. Higher intracellular uptake of nanoparticles due to their sub-micron-size range offers a distinct advantage over microparticles. Nanoparticles offer a potential opportunity to overcome the challenges associated with the formulation of insoluble drugs. Drug nanoparticles can be produced by various technologies, which can be broadly categorized into ‘bottom up’ and the ‘top down’ technologies.
In bottom-up technologies controlled precipitation of the solubilized drug is achieved by adding a suitable non-solvent. Hydrosol developed by Sandoz (presently Novartis) is an example for nanoformulation prepared using the precipitation technique62, 63, 64. In this process, the drug is dissolved in a solvent and this solution is subsequently added to a non-solvent solution. This results in high super saturation, rapid nucleation and the formation of many small nuclei65. Upon solvent removal, the dispersion can be filtered and lyophilized to obtain amorphous nanocrystals having a high solubility and dissolution rate.
High-pressure homogenization and milling methods are the alternate technologies that are frequently used for producing drug nanoparticles. However, a combination approach, with a pre-processing step followed by size reduction is also in application. Supercritical fluid technology is another approach for nanosizing, but it is industrially less successful when compared to the aforementioned technologies.
In early 19th century, heterogeneous catalysts were first among the reported techniques for nanosizing66, 67. Preclinical studies of danazol nanosuspension with a median diameter of 169 nm showed enhanced oral bioavailability 82.3±10.1%, when compared to conventional ‘as-is’ drug suspension 5.1±1.9%68, 69. Fine particles of atovaquone in the range of 100–300 nm have been successfully produced using the homogenization (microfluidization) technique. Following oral administration, the nanoparticle formulation enhanced the drug concentration in plasma from 15% to 40% in comparison to micronized Wellvone®, at equivalent doses. These results reflect the potency of the nanonization technique in terms of reducing drug load from 22.5 mg/kg (Wellvone®) to 7.5 mg/ kg, and increasing the activity 2.5-fold70.
In August 2000, the first product incorporating the NanoCrystal® technology was approved by the US FDA. Wyeth׳s Rapamune® (sirolimus, an immunosuppressant) developed using similar technology captured the market after its approval. Rapamune® was marketed as an oral solution and stored at refrigerated condition. The oral solution was given with orange juice prior to dosing. The development of a NanoCrystal® dispersion of sirolimus provided a drug product with enhanced bioavailability and improved stability.
In April 2003 an antiemetic drug, Emend® (aprepitant, MK 869) was approved and introduced into the market. Emend is capsule dosage form containing 80 or 125 mg of aprepitant formulated as drug nanoparticles. Following oral administration, the nanosuspension was able to overcome the significant food effect observed with the microsuspension formulation. Abraxane® (a reformulation of paclitaxel) is a nanoparticle-based product and was approved by FDA in 2006 for intravenous administration. It is a novel formulation consisting of lyophilized particles with 10% (w/w) paclitaxel and 90% (w/w) albumin71. The particle size of the nanosuspension is about 130 nm. The maximum tolerated dose observed from this study was higher than the commercial Taxol® formulation. Further studies confirmed that the nanoparticle formulation eliminated the need for premedication (since the toxic excipient Cremophor EL was not used in the formulation). Studies from intravenous and pulmonary applications of nanoparticles reported good tolerability and provided an alternative solution to insoluble drug therapeutics72, 73. A list of marketed products using drug nanoparticles is summarized in Table 474.
Table 4.
Overview of nanoparticle technology based marketed products.
| Trade name | Drug | Indication | Drug delivery company | Innovator company |
|---|---|---|---|---|
| Rapamune® | Rapamycin, sirolimus | Immunosuppressant | ElanNanosystems | Wyeth |
| Emend® | Aprepitant | Anti-emetic | ElanNanosystems | Merck & Co. |
| Tricor® | Fenofibrate | Hypercholesterolemia | Abbott Laboratories | Abbott laboratories |
| Megace ES® | Megestrol | Anti-anorexic | ElanNanosystems | Par Pharmaceuticals |
| Triglide® | Fenofibrate | Hypercholesterolemia | IDD-P Skyepharma | ScielePharma Inc. King |
| Avinza® | Morphine sulfate | Phychostimulant drug | ElanNanosystems | Pharmaceuticals |
| Focalin | Dexmethyl-phenidate HCl | Attention deficit hyperactivity disorder (ADHD) | ElanNanosystems | Novartis |
| Ritalin | Methyl phenidate HCl | CNS stimulant | ElanNanosystems | Novartis |
| Zanaflex Capsules | Tizanidine HCl | Muscle relaxant | ElanNanosystems | Acorda |
Nanoparticle technology serves as a screening aid during preclinical efficacy and safety studies of new chemical entities (NCEs). Fabrication of existing drugs with maximal drug exposure, less toxicity, expanded intellectual property by drug life cycle management and minimized competition during the drug׳s life time can be achieved through nanoparticle-based drug delivery systems. In fact, viable formulations for poorly soluble drugs with maximum drug exposure can be developed potentially by nanoparticle technology, which has opened the stage gates for reviving currently marketed products with suboptimal drug delivery, leading to better clinical and commercial benefits. Some of the key nanotechnology-based approaches for improving the oral bioavailability of poorly water-soluble drugs (according to Saffie-Siebert and co-workers75) are highlighted in Table 569, 76, 77, 78, 79, 80, 81.
Table 5.
Key nanotechnology-based approaches for the enhancement of drug solubility and oral bioavailability.
| Company | Nanotechnology-based formulation approach | Description and reference |
|---|---|---|
| American Biosciences (Blauvelt, USA) | Nanoparticle albumin-bound technology. e.g. paclitaxel-albumin nanoparticles | Paclitaxel albumin nanoparticles76 |
| Baxter Pharmaceuticals (Deerfield, USA) | Nanoedge technology: particle size reduction was achieved by homogenization, micro-precipitation, lipid emulsion and other dispersed systems. | Nano-lipid emulsion77 |
| BioSante Pharmaceuticals (Lincolnshire, USA) | Calcium phosphate based nanoparticles were produced for improved oral bioavailability of hormones/proteins and vaccine adjuvants | Calcium phosphate nanoparticles78 |
| ElanPharma International (Dublin, Ireland) | Nanoparticles (<1 µm) were produced by Wet milling technique using surfactants and stabilizers. The technology was applied successfully in developing apprepitant and reformulation of Sirolimus. | Nanocrystal drug particle79 |
| Eurand Pharmaceuticals (Vandalia, USA) | Nanocrystal or amorphous drug is produced by breakdown of crystal lattice and stabilized by using biocompatible carriers (swellablemicroparticles or cyclodextrins) | Cyclodextrin nanoparticle80 |
| iMEDDInc (Burlingame, USA) | Implantable drug delivery system using silicon membrane with nano-pores (10–100 nm) | Stretchable silicon nanomembrane81 |
| pSivida Ltd. (Watertown, USA) | The solubility and bioavailability of hydrophobic drugs was achieved by incorporating drug particles within the nano-width pores of biocompatible silicon membranes or fibers. | Silicon nanoparticles |
| PharmaSol GmbH (Berlin, Germany) | High pressure homogenization was used to produce nanostructured lipid particles dispersions with solid contents that provide high-loading capacity for hydrophilic drugs | Drug encapsulated in lipid nanoparticles69 |
| SkyePharmaPlc, (Piccadily, London, UK) | Nanoparticulate systems of water insoluble drugs were produced by applying high shear or impaction and stabilization was achieved by using phospholipids. | A polymer stabilizing nano- reactor with the encapsulated drug core69 |
2.7. Solid lipid nanoparticles
Solid lipid nanoparticles (SLN) are promising drug carriers with potential applications in the delivery of poorly soluble drugs82, 83. The lipid excipients used in the SLN formulations are biocompatible and biodegradable and most of them are physiological components that are generally regarded as safe (GRAS). Site-specific drug delivery, particularly for poorly soluble proteins and peptide drugs could be achieved by exploring SLN technology84. A significant increase in bioavailability was achieved when a poorly soluble compound, ofloxacin was formulated as SLN85. The enhancement in drug׳s bioavailability is attributed to the increase in surface area of the particles, improved dissolution rate and enhanced concentration of ofloxacin in gastrointestinal tract (GIT) fluids86, 87. The drug in lipid nanoparticles may adhere to the intestinal wall and thereby increases the drug residence time in the GIT, resulting in improved bioavailability88.
Pandita et al.89 developed an SLN formulation for a poorly soluble compound, paclitaxel. Improved oral bioavailability as compared to the control group was observed with the in vivo studies of SLN formulation. Studies also revealed an improved dissolution rate of poorly soluble drugs such as camptothecin, vinpocetine and fenofibrate by their successful incorporation into SLNs90, 91. Controlled release of drugs in the GIT with improved bioavailability by decreasing the variability in absorption can be achieved by these carrier systems. Apart from these, avoidance of organic solutions, increased drug stability in GIT and feasibility to scale-up are a few of the potential therapeutic benefits of solid lipid nanoparticles. However, a product with SLN is yet to hit the market. A list of examples of drugs developed using SLN technology and their biopharmaceutical applications are summarized Table 6.
Table 6.
List of examples of drugs developed using solid lipid nanoparticle technology.
| Drug | Lipid used | Biopharmaceutical application |
|---|---|---|
| 5-Fluoro uracil | Dynasan 114 and Dynasan 118 | Prolonged release in simulated colonic media |
| Apomorphine | Glycerylmonostearate, polyethylene glycol monostearate | Enhanced bioavailability in rats |
| Calcitonin | Trimyristin | Improvement of the efficacy of proteins |
| Clozapine | Trimyristin, Tristearin and Tripalmitin | Improvement of bioavailability |
| Cyclosporin A | Glycerylmonostearate and glycerylpalmitostearate. | Controlled release |
| Gonadotropin release hormone | Monostearin | Prolonged release |
| Ibuprofen | Stearic acid, Triluarin and Tripalmitin | Stable formulation with low toxicity |
| Idarubicin | Emulsifying wax | Delivery of oral proteins |
| Insulin | Stearin acid, octadecyl alcohol, cetylpalmitate, glycerylpalmitostearate, glyceryltripalmitate, glycerylbehenate and glycerylmonostearate. | Potential for oral delivery of proteins. |
| Lopinavir | Campritol 888 ATO | Bioavailability enhanced |
| Nimusulide | Glycerylbehanate, palmitostearate, glyceryltristearate | Sustained release of drug |
| Progesterone | Monostearin, stearic acid and oleic acid | Potential for oral drug delivery |
| Repaglinide | Glycerylmonostearate and tristearin | Reduced toxicity |
| Tetracycline | Gycerylmonostearate and stearic acid | Sustained release |
2.8. Liposomes and proliposomes
Liposomes are spherical closed vesicles of phospholipid bilayers with an entrapped aqueous phase, and may consist of one or more bilayers. Liposomes were first prepared by A.D. Bangham in the early 1960s and demonstrated that a wide variety of molecules can be encapsulated within aqueous spaces of liposomes or inserted into their membranes. Liposomes have been regarded as new drug delivery systems capable of transporting drug molecules to specific target site with enhanced efficacy and safety92. A potential advantage of liposomes is the encapsulation of hydrophobic as well as hydrophilic drugs, either in the phospholipid bilayer, at the bilayer interface or in the entrapped aqueous volume. Recent developments in liposome technology are generating more effective strategies for improving the vesicle stability after systemic administration93, 94.
Liposomal drug delivery offer significant therapeutic benefits to poorly soluble compounds. One such example is the formulation of cyclosporine and paclitaxel in which surfactants and organic co-solvents are used for systemic administration in humans. These solubilizers may cause toxicity at the administered doses. In comparison, liposomes are relatively non-toxic, non-immunogenic, biocompatible and biodegradable molecules, which can encapsulate a wide range of water-insoluble (lipophilic) compounds. Paclitaxel liposomes were able to deliver the drug systemically and increase the therapeutic index of paclitaxel in human ovarian tumor models95, 96. Currently, liposomes are being used as excipients for preparing better-tolerated clinical formulations of several lipophilic, sparingly water soluble drugs such as amphotericin B97. Developing liposome drug delivery improved solubility of lipophilic and amphiphilic drugs such as porphyrins, minoxidil, peptides and anthracyclines, respectively. Furthermore, in some cases anticancer agent such as acyclovir can be encapsulated in liposome interior at concentrations above their aqueous solubility98. A representative list of liposomal based drug delivery products is summarized in Table 7.
Table 7.
Representative list of liposomal based drug products.
| Product | Drug | Company | Indication target |
|---|---|---|---|
| Atragen™ | Tretinoin | Aronex Pharmaceuticals Inc. | Acute myeloid leukemia |
| Amphotec | Amphotericin B | Sequus Pharmaceutical Inc. | Fungal infections leishmaniasis |
| Ambisome™ | Amphotericin B | NeXstar Pharmaceutical Inc. Co. | Serious fungal infections |
| Amphocil™ | Amphotericin B | Sequus Pharmaceutical Inc. | Serious fungal infections |
| Abelcet™ | Amphotericin B | The Liposome Company, Inc. | Serious fungal infections |
| ALEC™ | Dry protein free powder of DPPC-PG | Britannia Pharmaceuticals Ltd. | Expanding lung diseases in infants |
| Avian retrovirus vaccine | Killed avian retrovirus | Vineland Laboratories, USA | Chicken pox |
| DaunoXome™ | Daunorubicin citrate | NeXstar Pharmaceutical Inc., Co. | Kaposi sarcoma in AIDS |
| DepoDur | Morphine | Pacira Pharmaceuticals Inc. | Post-surgical pain reliever |
| DaunoXome | Daunorubicin citrate | Galen Ltd. | Kaposi sarcoma in AIDS |
| Depocyt | Cytarabin | Pacira Pharmaceuticals Inc. | Treatment of lymphomatous meningitis |
| Doxil | Doxorubicin | SequusPharmaceutical Inc. | Kaposi sarcoma in AIDS |
| Estrasorb | estradiol | Novavax | Menopausal therapy |
| Evacet™ | Doxorubicin | The liposome company, USA | Metastatic breast cancer |
| EpaxalBerna® Vaccine | Inactivated hepatitis-A Virions | Swiss serum & vaccine institute, Switzerland. | Hepatitis A |
| Fungizone | Amphotericin B | Bristol-Myers Squibb, Netherland | Serious fungal infections |
| MiKasome® | Amikacin | NeXstar Pharmaceutical Inc., Co. | Bacterial infection |
| Nyotran™ | Nystatin | Aronex pharmaceuticals Inc. | Systemic fungal infections |
| Topex-Br | Terbutalinesulphate | Ozone Pharmaceuticals Ltd. | Asthma |
| Ventus | Prostoglandin-E1 | The liposome company, Inc. | Systemic inflammatory disease |
| VincaXome | Vincristine | NeXstar Pharmaceutical Inc., Co. | Solid tumors |
Proliposomes are dry, free flowing powders which can form multilamellar vesicles (MLVs) upon hydration with water. Proliposomes have been extensively studied as a potential carrier for oral delivery of drugs with poor bioavailability99. It provides a novel solution to product stability problems associated with the storage of aqueous liposomal dispersions, wherein it produces a dry product that can be stored for long duration and hydrated immediately before use100. Liposomes are either formed in vivo upon contact with the physiological fluids or prepared in vitro before administration using a hydrating solvent. The liposomes formed upon hydration are similar to conventional liposomes with uniform vesicle size101, 102.
Indomethacin proliposomes for oral administration were reported by Katare et al.103, in which the efficacy of the oral formulation was studied by measuring ulcerogenic index and anti-inflammatory activity using carrageenan-induced paw edema test in rats. The liposomal formulation showed enhanced performance in vivo with reference to their cytoprotective and anti-inflammatory properties.
Greater efficacy and less toxicity were reported by encapsulating vinpocetine in proliposomes. The study showed that the oral bioavailability of proliposomes was enhanced in New Zealand rabbits and thereby provided a new delivery platform to enhance the absorption of poorly soluble drugs in the GIT104. Therapeutic benefits of proliposomes include enhanced bioavailability, protection of drugs from degradation in the GIT, reduced toxicity and taste masking. The proliposomes can also provide target drug delivery and controlled drug release.
2.9. Microemulsions and self-emulsifying drug delivery systems
Micro-emulsions are thermodynamically stable, isotropic mixtures of oil, water, surfactant and a co-surfactant. In comparison to conventional emulsions, micro-emulsions produce a clear emulsion on mild agitation. The advantage of micro-emulsions over conventional emulsion and solution formulations is that the former produces a stable heterogeneous system. Micro-emulsion technology is widely used to address the challenges associated with poorly soluble compounds. Insoluble drugs can be administered through parenteral route by formulating into micro-emulsions. The micro-emulsions for parenteral delivery comprise lipid droplets (10%–20%), osmotic agent and an emulsifier. Apart from these, an antimicrobial agent is incorporated if the emulsion is packed in a multi-dose container. Propofol injection is a classic example of parenteral microemulsion formulation. Initially, Cremophore EL was used for formulating propofol, and then ethanol was included by changing the formulation. Finally, it was introduced into market by formulating into a microemulsion with soybean oil105, having a higher tolerable limit and safety profile. The representative list of marketed parenteral microemulsions is summarized in Table 8.
Table 8.
Representative list of marketed parenteral microemulsion products.
| Drug | Product name | Company | Therapeutic area |
|---|---|---|---|
| Cyclosporine A | Restasis | Allergan | Immunomodulation |
| Diazepam | Diazemuls | Braun Melsungen | Sedation |
| DexamethazonePalmitate | Limethason | Green Cross | Carticosteroid |
| Etomidate | Etomidat | Dumex (Denmark) | Anesthesia |
| Flurbiprofen | Lipfen | Green Cross | Analgesia |
| Prostaglandin-E1 | Liple | Green Cross | Vasodilator |
| Propofol | Propofol | Baxter Anesthesia | Anesthesia |
| Diprivan | AstraZeneca | Anesthesia | |
| Perflurodecalin+Perflurotripropylamine | Fluosol-DA | Green Cross | Analgesia |
| Vitamins A, D, E and K | Vitalipid | Kabi | Nutrition |
Self-emulsifying drug delivery systems have gained importance owing to their ability to enhance solubility and bioavailability of insoluble drugs106. Upon dilution by the aqueous environment in the GIT, these systems undergo rapid self-emulsification producing nano-sized globules of high surface area resulting in enhanced rate and extent of absorption with consistent plasma time profiles. An example of drug product developed using self-micro-emulsifying drug delivery system (SMEDDS) is Neoral®, an oral cyclosporine formulation which forms micro-emulsion in aqueous environment. The drug product showed improved bioavailability from 174%–239% as compared to cyclosporine-A, Sandimmune®107.
There are many examples and studies involving self-emulsifying systems for improving the in vitro and in vivo performances of poorly soluble drug candidates108. A significant enhancement in the bioavailability was observed with vinpocetin and atorvastatin in self-emulsifying systems as compared to their conventional tablet formulation, indicating the criticality of surfactant concentration in formulation for yielding the smaller particles with concomitant enhancement in drug permeation and absorption109, 110. The marketed oral products which yield an emulsion or micro-emulsion in the gastrointestinal tract are summarized in Table 9.
Table 9.
Marketed oral products which yield an emulsion or microemulsion in the gastrointestinal tract.
| Drug | Product name | Company | Therapeutic area |
|---|---|---|---|
| Cyclosporine | Sandimmune oral | Novartis | Immunosuppressant |
| Cyclosporine | Neoral | Novartis | Immunosuppressant |
| Calcitrol | Rocaltrol | Roche | Calcium regulator |
| Clofazimine | Lamprene | Geigy | Leprosy |
| Doxercalciferol | Hectoral | Bone care | Calcium regulator |
| Dronabionol | Marinol | Roxane | Anoxeria |
| Dutasteride | Avodart | GSK | Benign Prostatic Hyperplasia (BPH) |
| Isotretionoin | Accutane | Roche | Acne |
| Ritonavir | Norvir | Abbott | AIDS |
| Ritonavir/lopinavir | Kaletra | Abbott | AIDS |
| Paricalcitol | Zemplar | Abbott | Calcium regulator |
| Progesterone | Prometrium | Solvay | Endometrial hyperplasia |
| Saquinavir | Fortovase | Roche | AIDS |
| Sirolimus | Rapumune | Wyeth-ayerst | Immunosuppressant |
| Tritionoin | Vesanoid | Roche | Acne |
| Tipranavir | Aptivus | Boehringer Ingelheim | AIDS |
| Valproic acid | Depakene | Abbott | Epilepsy |
3. Conclusions
A great opportunity as well as potential challenge is foreseen from the large number of insoluble drugs that are approved by FDA, as well as those in the developmental pipeline. Exploring recent advances of insoluble drug delivery technologies will help in better therapeutic applications with improved patient compliance. On the other hand, the insoluble drug delivery technologies are being effectively utilized predominantly for commercial benefits through NDA route by developing improved formulations. Therefore, further advancement in the insoluble drug delivery technologies and their exploration for new drug applications will be much more promising in coming years.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Loftsson T, Brewster ME. Pharmaceutical applications of cyclodextrins: basic science and product development. J Pharm Pharmacol. 2010;62:1607–1621. doi: 10.1111/j.2042-7158.2010.01030.x. [DOI] [PubMed] [Google Scholar]
- 2.Hodgson J. ADMET—turning chemicals into drugs. Nat Biotechnol. 2001;19:722–726. doi: 10.1038/90761. [DOI] [PubMed] [Google Scholar]
- 3.The 505(b)(2) blog. 2012 approvals. Available from: 〈http://blog.camargopharma.com/index.php/scorecard/2012-approvals〉.
- 4.Drews J, Ryser S. Innovation deficit in the pharmaceutical industry. Drug Inf J. 1996;30:97–108. [Google Scholar]
- 5.US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Guidance for industry, applications covered by section 505(b)(2). Rockville, MD 20857: 1999.
- 6.Serajuddin AT. Salt formation to improve drug solubility. Adv Drug Deivl Rev. 2007;59:603–616. doi: 10.1016/j.addr.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 7.〈http://www.rxlist.com/cipro-iv-drug.htm〉
- 8.〈http://www.rxlist.com/micardis-drug.htm〉
- 9.Hauel N, Narr B, Ries U, Van Meel JC, Wienen W, Entzeroth M, inventors; Karl Thomae GmbH, assignee. Benzimidazoles useful as angiotensin-11 antagonists. United States Patent US 5591762. Januray 71997.
- 10.Heinrich S, Schneider M, inventors; Schneider Heinrich, Schneider Margarete, assignee. Polymorphs of telmisartan. United States Patent US 6358986. 2002 July 18
- 11.Giovanni G. Antihypertensive medicaments containing lacidipine and telmisartan. WIPO Patent No. WO/2000/027397A. 2000 May 18
- 12.Nakatani M, Ohki T, Takeshi S, Toyoshima K, inventors; Boehringer Ingelheim International GmbH, assignee. Solid pharmaceutical formulations comprising Telmisartan. European Patent EP 1545467. 2007 December 9
- 13.Mandić Z, Gabelica V. Ionization, lipophilicity and solubility properties of repaglinide. J Pharm Biomed Anal. 2006;41:866–871. doi: 10.1016/j.jpba.2006.01.056. [DOI] [PubMed] [Google Scholar]
- 14.Wolfgang G, Hurnaus R, Griss G, Sauter R, Reiffen M, Rupprecht E, inventors; Boehringer Ingelheim KG, assignee. Phenylacetic acid benzylamides. United States Patent US RE 37035. 2001 January 30
- 15.Wolfgang G, Hurnaus R, Griss G, Sauter R, Reiffen M, Ruprecht E, inventors; Karl Thomae GmbH, assignee. Phenylacetic acid benzylamides. United States Patent US 5312924. 1994 May 17
- 16.Wolfgang G, Hurnaus R, Griss G, Sauter R, Reiffen M, Ruprecht E, inventors; Karl Thomae GmbH, assignee. Phenylacetic acid benzylamides. United States Patent US 6143769. 2000 December 7
- 17.Müller PG, inventors; Novo, Nordisk A/s, assignee. NIDDM regimen. United States Patent US 6677358. 2004 January 13
- 18.〈http://www.onlinepharmacynz.com/product/485/Aspro_Clear_Regular_Strength_300mg.html〉
- 19.Kazmierski WM. Wiley; New Jersey: 2011. Antiviral drugs: from basic discovery through clinical trials. [Google Scholar]
- 20.Singh J, Pudipeddi M, Lindrud MD, inventors; Bristol-Myers Squibb Company, assignee. Bisulfate salt of HIV protease inhibitor. United States Patent US 6087383. 2000 July 11
- 21.Béni S, Szakács Z, Csernák O, Barcza L, Noszál B. Cyclodextrin/imatinib complexation: binding mode and charge dependent stabilities. Eur J Pharm Sci. 2007;30:167–174. doi: 10.1016/j.ejps.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 22.〈http://www.rxlist.com/gleevec-drug.htm〉
- 23.Zimmermann J, Sutter B, Burger HM. Crystal modification of a N-phenyl-2-pyrimidineamine derivative, processes for its manufacture and its use. WIPO Patent WO/1999/003854. 1999.
- 24.Downing NS, Ross JS, Jackevicius CA, Krumholz HM. Avoidance of generic competition by Abbott laboratories׳ fenofibrate franchise. Arch Intern Med. 2012;172:724–730. doi: 10.1001/archinternmed.2012.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.〈http://www.rxlist.com/trilipix-drug.htm〉
- 26.Weatherall MW, Telzerow AJ, Cittadini E, Kaube H, Goadsby PJ. Intravenous aspirin (lysine acetylsalicylate) in the inpatient management of headache. Neurology. 2010;75:1098–1103. doi: 10.1212/WNL.0b013e3181f39a11. [DOI] [PubMed] [Google Scholar]
- 27.Diener H, Asasumamig Study Group Efficacy and safety of intravenous acetylsalicylic acid lysinate compared to subcutaneous sumatriptan and parenteral placebo in the acute treatment of migraine. A double‐blind, double‐dummy, randomized, multicenter, parallel group study. Cephalalgia. 1999;19:581–588. doi: 10.1046/j.1468-2982.1999.019006581.x. [DOI] [PubMed] [Google Scholar]
- 28.Seymour RA, Williams FM, Luyk NM, Boyle MA, Whitfield PM, Nicholson E. Comparative efficacy of soluble aspirin and aspirin tablets in postoperative dental pain. Eur J Clin Pharmacol. 1986;30:495–498. doi: 10.1007/BF00607968. [DOI] [PubMed] [Google Scholar]
- 29.European Medicines Agency. Committee for medicinal products for human use, plenary meeting monthly report, EMEA/CHMP/389326/2009.
- 30.〈http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2012/201803s000ltr.pdf〉
- 31.〈http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2011/202172s000ltr.pdf〉
- 32.Patel A, Jones SA, Ferro A, Patel N. Pharmaceutical salts: a formulation trick or a clinical conundrum? Br J Cardiol. 2009;16:281–286. [Google Scholar]
- 33.Lee YC, Zocharski PD, Samas B. An intravenous formulation decision tree for discovery compound formulation development. Int J Pharm. 2003;253:111–119. doi: 10.1016/s0378-5173(02)00704-4. [DOI] [PubMed] [Google Scholar]
- 34.Kawakami K, Oda N, Miyoshi K, Funaki T, Ida Y. Solubilization behavior of a poorly soluble drug under combined use of surfactants and cosolvents. Eur J Pharm Sci. 2006;28:7–14. doi: 10.1016/j.ejps.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 35.〈http://www.rxlist.com/taxol-drug.htm〉
- 36.Hennenfent K, Govindan R. Novel formulations of taxanes: a review. Old wine in a new bottle? Ann Oncol. 2006;17:735–749. doi: 10.1093/annonc/mdj100. [DOI] [PubMed] [Google Scholar]
- 37.Oerlemans C, Bult W, Bos M, Storm G, Nijsen JFW, Hennink WE. Polymeric micelles in anticancer therapy: targeting, imaging and triggered release. Pharm Res. 2010;27:2569–2589. doi: 10.1007/s11095-010-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.〈http://www.rxlist.com/taxotere-drug.htm〉
- 39.〈http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/022234Orig1s000Approv.pdf〉
- 40.〈http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/201525Orig1s000ClinPharmR.pdf〉
- 41.〈http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2012/022312s000ltr.pdf〉
- 42.Powell MF, Nguyen T, Baloian L. Compendium of excipients for parenteral formulations. PDA J Pharm Sci Technol. 1998;52:238–311. [PubMed] [Google Scholar]
- 43.Yu L. Amorphous pharmaceutical solids: preparation, characterization and stabilization. Adv Drug Deliv Rev. 2001;48:27–42. doi: 10.1016/s0169-409x(01)00098-9. [DOI] [PubMed] [Google Scholar]
- 44.Laitinen R, Löbmann K, Strachan CJ, Grohganz H, Rades T. Emerging trends in the stabilization of amorphous drugs. Int J Pharm. 2013;453:65–79. doi: 10.1016/j.ijpharm.2012.04.066. [DOI] [PubMed] [Google Scholar]
- 45.Crisp HA, Clayton JC, Elliott LG, Wilson EM, inventor; Glaxo Group Limited, assignee. Preparation of a highly pure, substantially amorphous form of cefuroxime axetil. United States Patent US 4820833. April 111989.
- 46.Crisp HA, Clayton JC, inventors; Glaxo Group Limited, assignee. Amorphous form of cefuroxime ester. United States Patent US 4562181. 1985 December 31
- 47.Holohan JJ, Edwards IJ, inventors; Imperial Chemical Industries PLC, assignee. Pharmaceutical agents. United States Patent US 5319097. 1994 July 6
- 48.Corvari SJ, Creekmore JR, inventors; AstraZeneca UK Limited, assignee. Pharmaceutical compositions comprising Zafirlukast. European Patent EP 1041966. 2003 July 2
- 49.Sinha S, Ali M, Baboota S, Ahuja A, Kumar A, Ali J. Solid dispersion as an approach for bioavailability enhancement of poorly water-soluble drug ritonavir. AAPS PharmSciTech. 2010;11:518–527. doi: 10.1208/s12249-010-9404-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50:47–60. doi: 10.1016/s0939-6411(00)00076-x. [DOI] [PubMed] [Google Scholar]
- 51.Durate I, Temtem M, Gil M, Gaspar F. Overcoming poor bioavailability through amorphous solid dispersions. Ind Pharm. 2011;30:4–6. [Google Scholar]
- 52.Miller DA. Improved oral absorption of poorly water-soluble drugs by advanced solid dispersion systems [dissertation]. Austin: University of Texas; 2007
- 53.Thakuria R, Delori A, Jones W, Lipert MP, Roy L, Rodríguez-Hornedo N. Pharmaceutical cocrystals and poorly soluble drugs. Int J Pharm. 2013;453:101–125. doi: 10.1016/j.ijpharm.2012.10.043. [DOI] [PubMed] [Google Scholar]
- 54.Almarsson Ö, Peterson ML, Zaworotko M. The A to Z of pharmaceutical cocrystals: a decade of fast-moving new science and patents. Pharm Pat Anal. 2012;1:313–327. doi: 10.4155/ppa.12.29. [DOI] [PubMed] [Google Scholar]
- 55.Rios-Doria J, Carie A, Costich T, Burke B, Skaff H, Panicucci R. A versatile polymer micelle drug delivery system for encapsulation and in vivo stabilization of hydrophobic anticancer drugs. J Drug Deliv. 2012;2012:951741. doi: 10.1155/2012/951741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riggio C, Pagni E, Raffa V, Cuschieri A. Nano-oncology: clinical application for cancer therapy and future perspectives. J Nanomater. 2011;2011:164506. [Google Scholar]
- 57.Cabral H, Kataoka K. Progress of drug-loaded polymeric micelles into clinical studies. J Control Release. 2014;190:465–476. doi: 10.1016/j.jconrel.2014.06.042. [DOI] [PubMed] [Google Scholar]
- 58.Souto EB. Springer Science & Business Media; Berlin Heidelberg: 2012. Patenting nanomedicines: legal aspects, intellectual property and grant opportunities. [Google Scholar]
- 59.Loftsson T, Brewster ME. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J Pharm Sci. 1996;85:1017–1025. doi: 10.1021/js950534b. [DOI] [PubMed] [Google Scholar]
- 60.Nekkanti V, Karatgi P, Paruchuri S, Pillai R. Drug product development and pharmacological evaluation of a sparingly soluble novel camptothecin analog for peroral administration. Drug Deliv. 2011;18:294–303. doi: 10.3109/10717544.2010.544689. [DOI] [PubMed] [Google Scholar]
- 61.Loftsson T, Duchêne D. Cyclodextrins and their pharmaceutical applications. Int J Pharm. 2007;329:1–11. doi: 10.1016/j.ijpharm.2006.10.044. [DOI] [PubMed] [Google Scholar]
- 62.Gassmann P, List M, Schweitzer A, Sucker H. Hydrosols: alternatives for the parenteral application of poorly water soluble drugs. Eur J Pharm Biopharm. 1994;40:64–72. [Google Scholar]
- 63.List M, Sucker H. Pharmaceutical colloidal hydrosols for injection. GB Patent 2200048. 1998.
- 64.Sucker H, Gassmann P. Improvements in pharmaceutical compositions. GB Patent 2269536A. 1994.
- 65.Kipp JE, Wong JCT, Doty MJ, Rebbeck CL. Microprecipitation method for preparing submicron suspensions. Google Patents; 2003 January 2.
- 66.Rogers TL, Johnston KP, Williams RO. Solution-based particle formation of pharmaceutical powders by supercritical or compressed fluid CO2 and cryogenic spray-freezing technologies. Drug Dev Ind Pharm. 2001;27:1003–1015. doi: 10.1081/ddc-100108363. [DOI] [PubMed] [Google Scholar]
- 67.Kauffman GB. The origins of heterogeneous catalysis by platinum: Johann Wolfgang Döbereiner׳s contributions. Enantiomer. 1999;4:609–619. [PubMed] [Google Scholar]
- 68.Robertson AJB. The development of ideas on heterogeneous catalysis. Platin Met Rev. 1983;27:31–39. [Google Scholar]
- 69.Kaparissides C, Alexandridou S, Kotti K, Chaitidou S. Recent advances in novel drug delivery systems. J Nanotechnol. 2006;2:1–11. [Google Scholar]
- 70.Müller RH, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy: rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001;47:3–19. doi: 10.1016/s0169-409x(00)00118-6. [DOI] [PubMed] [Google Scholar]
- 71.Wong J, Brugger A, Khare A, Chaubal M, Papadopoulos P, Rabinow B. Suspensions for intravenous (IV) injection: a review of development, preclinical and clinical aspects. Adv Drug Deliv Rev. 2008;60:939–954. doi: 10.1016/j.addr.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 72.Mouton JW, Van Peer A, De Beule K, Van Vliet A, Donnelly J, Soons PA. Pharmacokinetics of itraconazole and hydroxyitraconazole in healthy subjects after single and multiple doses of a novel formulation. Antimicrob Agents Chemother. 2006;50:4096–4102. doi: 10.1128/AAC.00630-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kraft WK, Steiger B, Beussink D, Quiring JN, Fitzgerald N, Greenberg HE. The pharmacokinetics of nebulized nanocrystal budesonide suspension in healthy volunteers. J Clin Pharmacol. 2004;44:67–72. doi: 10.1177/0091270003261490. [DOI] [PubMed] [Google Scholar]
- 74.Nekkanti V, Vabalaboina V, Pillai R. Drugnanoparticles - an overview. In: Hashim AA, editor. The delivery of nanoparticles. Rijeka: InTech; 2012. Available from: http://www.intechopen.com/books/the-delivery-of-nanoparticles/drug-nanoparticles-an-overview.
- 75.Saffie-Siebert R, Ogden J, Parry-Billings M. Nanotechnology approaches to solving the problems of poorly water-soluble drugs. Drug Discov World. 2005;2005:71–76. [Google Scholar]
- 76.〈http://www.pharmafocusasia.com/research_development/sonication_assisted_nanoencapsulation.htm〉
- 77.Miller OJ, Bernath K, Agresti JJ, Amitai G, Kelly BT, Mastrobattista E. Directed evolution by in vitro compartmentalization. Nat Methods. 2006;3:561–570. doi: 10.1038/nmeth897. [DOI] [PubMed] [Google Scholar]
- 78.〈http://aiche.confex.com/aiche/2005/techprogram/P21745.htm〉
- 79.〈http://www.physorg.com/news3152.html〉
- 80.〈http://liambean.hubpages.com/hub/How-nano-technology-may-be-applied-to-medicine〉
- 81.〈http://phys.org/news154538439.html〉
- 82.Müller RH, Radtke M, Wissing SA. Nanostructured lipid matrices for improved microencapsulation of drug. Int J Pharm. 2002;242:121–128. doi: 10.1016/s0378-5173(02)00180-1. [DOI] [PubMed] [Google Scholar]
- 83.Gasco MR, inventors; Gasco, Maria R, assignee. Method for producing solid lipid microspheres having a narrow size distribution. United States Patent US 5250236. 1993 October 5
- 84.Trotta M, Carlotti ME, Gallarate M, Zara GP, Muntoni E, Battaglia L. Insulin-loaded SLN prepared with the emulsion dilution technique:in vivo tracking of nanoparticles after oral administration to rats. J Dispers Sci Technol. 2011;32:1041–1045. [Google Scholar]
- 85.Xie SY, Zhu LY, Dong Z, Wang XF, Wang Y, Li XH. Preparation, characterization and pharmacokinetics of enrofloxacin-loaded solid lipid nanoparticles: influences of fatty acids. Colloids Surf B:Biointerfaces. 2011;83:382–387. doi: 10.1016/j.colsurfb.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 86.Chakraborty S, Shukla D, Mishra B, Singh S. Lipid—an emerging platform for oral delivery of drugs with poor bioavailability. Eur J Pharm Biopharm. 2009;73:1–15. doi: 10.1016/j.ejpb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 87.Luo YF, Chen DW, Ren LX, Zhao XL, Qin J. Solid lipid nanoparticles for enhancing vinpocetine׳s oral bioavailability. J Control Release. 2006;114:53–59. doi: 10.1016/j.jconrel.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 88.Vasir JK, Tambwekar K, Garg S. Bioadhesive microspheres as a controlled drug delivery system. Int J Pharm. 2003;255:13–32. doi: 10.1016/s0378-5173(03)00087-5. [DOI] [PubMed] [Google Scholar]
- 89.Pandita D, Ahuja A, Lather V, Benjamin B, Dutta T, Velpandian T. Development of lipid-based nanoparticles for enhancing the oral bioavailability of paclitaxel. AAPS PharmSciTech. 2011;12:712–722. doi: 10.1208/s12249-011-9636-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Harms M, Müller-Goymann CC. Solid lipid nanoparticles for drug delivery. J Drug Deliv Sci Technol. 2011;21:89–99. [Google Scholar]
- 91.Wei W, Shi SJ, Liu J, Sun X, Ren K, Zhao D. Lipid nanoparticles loaded with 10-hydroxycamptothecin-phospholipid complex developed for the treatment of hepatoma in clinical application. J Drug Target. 2010;18:557–566. doi: 10.3109/10611861003599461. [DOI] [PubMed] [Google Scholar]
- 92.Bangham AD, Horne RW. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J Mol Biol. 1964;8:660–668. doi: 10.1016/s0022-2836(64)80115-7. [DOI] [PubMed] [Google Scholar]
- 93.Lasic DD, Papahadjopoulos D. Liposomes revisited. Science. 1995;267:1275–1276. doi: 10.1126/science.7871422. [DOI] [PubMed] [Google Scholar]
- 94.Kalepu S, Sunilkumar KT, Betha S, Mohanvarma M. Liposomal drug delivery system—a comprehensive review. Int J Drug Dev Res. 2013;5:62–75. [Google Scholar]
- 95.Sharma A, Mayhew E, Bolcsak L, Cavanaugh C, Harmon P, Janoff A. Activity of paclitaxel liposome formulations against human ovarian tumor xenografts. Int J Cancer. 1997;71:103–107. doi: 10.1002/(sici)1097-0215(19970328)71:1<103::aid-ijc17>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 96.Sharma A, Bernacki RJ, Straubinger RM, Ojima I. Antitumor efficacy of taxane liposomes on a human ovarian tumor xenograft in nude athymic mice. J Pharm Sci. 1995;84:1400–1404. doi: 10.1002/jps.2600841204. [DOI] [PubMed] [Google Scholar]
- 97.Proffitt RT, Adler-Moore J, Chiang SM, Inventors; NeXstar Pharmaceuticals, Inc., Assignee. Amphotericin B liposome preparation. United States Patent US 5965156. 1999 October 12
- 98.Lasic DD, Frederik PM, Stuart MCA, Barenholz Y, McIntosh TJ. Gelation of liposome interior: a novel method for drug encapsulation. FEBS Lett. 1992;312:255–258. doi: 10.1016/0014-5793(92)80947-f. [DOI] [PubMed] [Google Scholar]
- 99.Nekkanti V, Venkatesan N, Betageri GV. Proliposomes for oral delivery: progress and challenges. Curr Pharm Biotechnol. 2015;16:1–10. doi: 10.2174/1389201016666150118134256. [DOI] [PubMed] [Google Scholar]
- 100.Janga KY, Jukanti R, Velpula A, Sunkavalli S, Bandari S, Kandadi P. Bioavailability enhancement of zaleplon via proliposomes: role of surface charge. Eur J Pharm Biopharm. 2012;80:347–357. doi: 10.1016/j.ejpb.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 101.Betageri GV, inventors; Western University of Health Sciences, assignee. Proliposomal drug delivery system. United States Patent US 6849269. 2005 April 24
- 102.Betageri GV, inventors; Western Center for Drug Development College of Pharmacy Western University of Health Sciences, assignee. Enteric-coated proliposomal formulations for poorly water soluble drugs. United States Patent US 6759058. 2004 July 6
- 103.Katare OP, Vyas SP, Dixit VK. Proliposomes of indomethacin for oral administration. J Microencapsul. 1991;8:1–7. doi: 10.3109/02652049109021852. [DOI] [PubMed] [Google Scholar]
- 104.Xu HT, He L, Nie SF, Guan J, Zhang XN, Yang XG. Optimized preparation of vinpocetine proliposomes by a novel method and in vivo evaluation of its pharmacokinetics in New Zealand rabbits. J Control Release. 2009;140:61–68. doi: 10.1016/j.jconrel.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 105.Baker MT, Naguib M. Propofol: the challenges of formulation. Anesthesiology. 2005;103:860–876. doi: 10.1097/00000542-200510000-00026. [DOI] [PubMed] [Google Scholar]
- 106.Kalepu S, Manthina M, Padavala V. Oral lipid-based drug delivery systems—an overview. Acta Pharm Sin B. 2013;3:361–372. [Google Scholar]
- 107.Bhalani VT, Patel SB, inventors; Sidmak Laboratories, Inc., assignee. Pharmaceutical composition for cyclosporines. United States Patent US 5858401. 1999 January 12
- 108.Müllertz A, Ogbonna A, Ren S, Rades T. New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J Pharm Pharmacol. 2010;62:1622–1636. doi: 10.1111/j.2042-7158.2010.01107.x. [DOI] [PubMed] [Google Scholar]
- 109.Cui SX, Nie SF, Li L, Wang CG, Pan WS, Sun JP. Preparation and evaluation of self-microemulsifying drug delivery system containing vinpocetine. Drug Dev Ind Pharm. 2009;35:603–611. doi: 10.1080/03639040802488089. [DOI] [PubMed] [Google Scholar]
- 110.Shen HR, Zhong MK. Preparation and evaluation of self-microemulsifying drug delivery systems (SMEDDS) containing atorvastatin. J Pharm Pharmacol. 2006;58:1183–1191. doi: 10.1211/jpp.58.9.0004. [DOI] [PubMed] [Google Scholar]