

Abstract
Cancer metastasis is the major cause of cancer morbidity and mortality, and accounts for about 90% of cancer deaths. Although cancer survival rate has been significantly improved over the years, the improvement is primarily due to early diagnosis and cancer growth inhibition. Limited progress has been made in the treatment of cancer metastasis due to various factors. Current treatments for cancer metastasis are mainly chemotherapy and radiotherapy, though the new generation anti-cancer drugs (predominantly neutralizing antibodies for growth factors and small molecule kinase inhibitors) do have the effects on cancer metastasis in addition to their effects on cancer growth. Cancer metastasis begins with detachment of metastatic cells from the primary tumor, travel of the cells to different sites through blood/lymphatic vessels, settlement and growth of the cells at a distal site. During the process, metastatic cells go through detachment, migration, invasion and adhesion. These four essential, metastatic steps are inter-related and affected by multi-biochemical events and parameters. Additionally, it is known that tumor microenvironment (such as extracellular matrix structure, growth factors, chemokines, matrix metalloproteinases) plays a significant role in cancer metastasis. The biochemical events and parameters involved in the metastatic process and tumor microenvironment have been targeted or can be potential targets for metastasis prevention and inhibition. This review provides an overview of these metastasis essential steps, related biochemical factors, and targets for intervention.
Abbreviations: BM, basement membrane; CAFs, cancer-associated fibroblasts; CAMs, cell adhesion molecules; CAT, collective amoeboid transition; CCL2, chemokine (C–C motif) ligand 2; CCR3, chemokine receptor 3; Col, collagen; COX2, cyclooxygenase 2; CSF-1, chemokine colonystimulating factor–1; CTGF, connective tissue growth factor; CXCR2, chemokine receptor type 2; DISC, death-inducing signaling complex; ECM, extracellular matrix; EGF, epidermal growth factor; EGFR, EGF receptor; EMT, epithelial–mesenchymal transition; FAK, focal adhesion kinase; FAs, focal adhesions; FGF, fibroblast growth factor; FN, fibronectin; HA, hyaluronan; HGF, hepatocyte growth factor; HIFs, hypoxia-inducible factors; IKK, IκB kinase; JAK, the Janus kinases; LN, laminin; MAPK, mitogen-activated protein kinase; MAT, mesenchymal to amoeboid transition; MET, mesenchymal–epithelial transition; MMPs, matrix metalloproteinases; PDGF, platelet-derived growth factor; PI3K, phosphatidylinositol 3-kinase; STATs, signal transducers and activators of transcription; TAMs, tumor-associated macrophages; TGF-β, transforming growth factor β; TME, tumor microenvironment; VCAMs, vascular cell adhesion molecules; VEGF, vascular endothelial growth factor; VN, vitronectin
KEY WORDS: Metastasis, Detachment, Migration, Invasion, Adhesion, Cancer
Graphical abstract
Cancer metastasis is the major cause of cancer morbidity and mortality, and accounts for about 90% of cancer deaths. Limited progress has been made in the treatment of cancer metastasis due to various factors. This review is aimed to provide an overview of the metastasis process and targets for intervention with a focus on cancer cell detachment, migration, invasion and adhesion. It is hoped that this review can serve as a lead for readers who are interested in cancer metastasis and intervention.

1. Introduction
Cancer metastasis is a process in which cancer cells disseminate from the primary tumor, settle and grow at a site other than the primary tumor site. Most cancer deaths are caused by cancer metastasis not the primary tumor. Cancer metastasis is the primary cause of morbidity and mortality and responsible for about 90% of cancer deaths1. It is now accepted that tumor distribution and secondary site growth is not a matter of chance, but rather it is determined by the dependence of the ʻseeds’ (the cancer cells) on the ‘congenial soil’ (the target organ for metastasis) as proposed by the English surgeon Stephen Paget in 18892. Until recently, cancer research has primarily focused on the development of methods/agents that can detect tumor at the early stage, and on agents that inhibit tumor growth. Advances in early cancer detection and treatment have rendered that most solid tumors are now manageable or curable if they are diagnosed and treated before metastasis. However, once cancers spread beyond the initial primary site, they are usually highly incurable and fatal3. Due to a lack of understanding of the mechanisms that underlie the metastatic process, limited success has been made on prevention and inhibition of cancer metastasis.
Metastasis is a complicated event that involves multiple sequential and interrelated steps and multi-biochemical events with much to be elucidated. Metastasis is facilitated by four essential steps: detachment, migration, invasion and adhesion. Cancer cells first detach from the primary tumor, undergo migration, invasion, and travel to different sites through blood and lymphatic vessels, then settle (adhesion) and grow. Metastasis is regulated by various signaling pathways and is affected by the surrounding extracellular matrix (ECM). It is now known that metastasis genes are stress-response genes that physiologically contribute to inflammation, wound healing, and stress-induced angiogenesis4. This review is aimed to provide an overview of the metastasis process and targets for intervention with a focus on cancer cell detachment, migration, invasion and adhesion. It is not the intent of this review to provide an in-depth description of each parameters related to the four essential steps and relevant intervention targets since each topic itself can be a lengthy review. It is hoped that this review can serve as a lead for readers who are interested in cancer metastasis and intervention.
2. Cancer metastasis
Cancer metastasis is a process of dissemination of tumor cells from a primary tumor mass to a different site through blood vessels and lymphatic vessels (Fig. 1). It is a complex succession of a series of cell-biological events termed the “invasion–metastasis cascade”. The cascade involves the development of new blood vessels (angiogenesis), departure of metastatic cells from the primary tumor (detachment and migration), invasion through the basement membrane (BM) and ECM surrounding the tumor, invasion of the BM supporting the endothelium of local blood and lymphatic vessels, intravasation of the metastatic cells into the blood and/or lymphatic vessels, adhesion of the circulating metastatic cells to the endothelium of capillaries of the target organ site, invasion of the cells through the endothelial cell layer and the surrounding BM (extravasation), and finally the settling and growth of secondary tumors at the target organ site5, 6. Fig. 1 provides a brief overview of the process.
Figure 1.

Metastatic cascade. Metastatic cells detach from the primary tumor site, migrate and invade through the BM and ECM, enter the blood or lymphatic vessels (intravasation), travel in the blood/or lymphatic vessels, leave the blood or lymphatic vessels (extravasation), adhere and grow at a distal site.
Metastatic cell dissemination requires that cells first detach from the primary tumor7, 8, 9. Under normal circumstances, epithelial and endothelial cells will undergo apoptosis (programmed cell death) when detached, a phenomenon referred to as anoikis (induction of apoptosis caused by detachment from the ECM)10. During the process of anoikis, both death receptor pathways and mitochondrial pathway are activated10. This is a mechanism designed to protect multicellular organisms from cells establishing themselves outside their correct anatomical location. Normal epithelial cells require attachment to the ECM for survival. Metastatic cells must develop a mechanism to adapt and survive when detached from the ECM or in the absence of the ECM. In other words, they should develop a mechanism to resist anoikis7, 11. The resistance to anoikis together with some other property changes of the tumor cells (such as changes in cell-to-cell or cell-to-matrix adhesion, cell polarity, and cell invasive and migratory property) are collectively known as the epithelial–mesenchymal transition (EMT). EMT is a characteristic feature of most metastatic cells11. Specifically, epithelial cells are transformed from highly differentiated, polarized, and organized cells into undifferentiated, isolated, and mesenchymal-like cells with migratory and invasive properties12. Additionally, many tumor cells also show changes in their plasticity via morphological and phenotypical conversions during cancer progression. These changes, in addition to EMT, include collective amoeboid transition (CAT) and mesenchymal to amoeboid transition (MAT)11. EMT enables cells to increase migratory and invasive capabilities through formation of invasive protrusions (invadopodia) while CAT and MAT enables cells to increase migratory capability through formation of non-invasive protrusions (lamellipodia and filopodia). Protrusions are the extended parts formed at the leading edge of motile cells. Lamellipodia and filopodia are also present in normal epithelial cells while invadopodia are mostly observed with metastatic cells (more discussion of protrusions in Section 2.1)11. Interestingly, EMT in tumor cells is transient. Before a metastatic cell settles down and grows, it needs to reverse its mesenchymal to a more epithelial phenotype, a conversion known as mesenchymal–epithelial transition (MET). The contribution of MET to cancer progression is still unclear12.
It is known that not all tumor cells are metastatic, nor are all cells within metastatic tumors capable of metastasizing7. The four essential steps of the cancer metastatic process (detachment, migration, invasion and adhesion) are distinct from each other but also interrelated. For example, cell migration involves cell detachment, adhesion and invasion, while invasion involves migration and adhesion. An understanding of these four steps and their role in cancer metastasis helps understand the metastatic process and also identify targets for intervention.
2.1. Cancer cell adhesion, detachment, migration and invasion
2.1.1. Cell adhesion
Cell adhesion basically refers to cell attachment among cells (cell–cell adhesion) and with cells׳ environment, mostly the ECM (cell–matrix adhesion). Physiologically, cells are held within their defined boundary through tight cell–cell adhesion and cell–matrix adhesion. Cell adhesion helps establish tight connections both between cells and between cells and the matrix. Since cellular motility is an essential part of cancer metastasis, and adhesion and de-adhesion (detachment) are prerequisites for cellular motility3, cell adhesion is critical for cancer metastasis. Adhesion is also involved in the settling of metastatic cancer cells at a distal site. Further, cell adhesion is not just a way to link cells or link cells with the ECM, but it also serves as a mechanism to activate cell proliferation and survival pathways through integrins׳ interactions with downstream molecules that are essential for motile function and survival11.
Adhesion is primarily achieved by connecting intracellular cytoskeleton between cells (cell–cell adhesion) or connecting cellular cytoskeleton with ECM components such as collagen, fibronectin, fibrinogen, and laminin (cell–ECM adhesion) through a group of cell adhesion molecules (CAMs). CAMs are surface glycoproteins that are typically transmembrane receptors made up of three domains: intracellular domain, transmembrane domain, and extracellular domain. CAMs primarily include calcium-dependent CAMs (cadherins, integrins or selectins) and calcium-independent CAMs [the immunoglobulin superfamily (Ig-SF) and lymphocyte homing receptors (CD44)]13. Different types of CAMs are responsible for adhesion in different types of cells. For example, E-cadherins are responsible for epithelial cell–cell adhesion and R-cadherins are for retinal cell adhesion11, 13. CAMs are critical for cell adhesion. A brief description of the structures and functions of CAMs is presented below.
2.1.1.1. Integrins
Integrins are responsible for cell–ECM adhesion. They are members of a glycoprotein family that form heterodimeric receptors for ECM molecules such as fibronectin (FN), laminin (LN), collagen (Col), fibrinogen, and vitronectin (VN). They are composed of α and β subunits with non-covalent bonds connected to each other. Both α and β subunit contains a large extracellular domain, a transmembrane domain, and a short intracellular domain. There are at least 19α and 8β subunits that dimerize to yield at least 24 different integrin heterodimers with distinct ligand binding and signaling properties11.
Cell adhesion to ECM is essentially achieved through integrin-mediated linkage to extracellular ECM molecules and intracellular cytoskeleton. The large extracellular domain of integrins bind to ECM molecules while the intracellular domain is linked to cytoskeleton through intracellular focal adhesions (FAs) as demonstrated in Fig. 2. FAs are supramolecular complexes formed by more than 150 different proteins, including kinases, scaffold, and adaptor proteins, as well as actin linking proteins14. FAs also mediate intracellular signaling pathways and are dynamic structures which assemble, disperse, and recycle during cell migration11, 13. Binding of integrins to FAs and ECM molecules not only serve as a way for cell adhesion to ECM, but also relay the forces produced by the cytoskeleton onto ECM to generate the traction needed for cell adhesion and migration, and to transmit signals from the extracellular environment to the intracellular network, as well as signals from the intracellular network to extracellular environment. The transmission is mediated by integrin-activated signaling molecules, such as focal adhesion kinase (FAK), phosphatidylinositol 3-kinase (PI3K), and members of the extracellular signal-regulated kinase 1 and 2/mitogen activated protein (ERK1 and 2/MAP) kinase family to regulate cell proliferation, migration, and apoptosis of tumor and endothelial cells (Fig. 2)10. Therefore, integrins are not only involved in adhesion but also in migration and invasion. They are important for cell motility due to their ability to modulate physical interactions with ECMs and to regulate signaling pathways that control actin cytoskeleton dynamics and cell movement11. They also play critical roles in regulating other biological processes, such as apoptosis, proliferation, survival, and differentiation through integrin-mediated downstream signaling pathways13.
Figure 2.

Illustration of cell detachment, cell–cell adhesion and cell–matrix adhesion of epithelial cells by E-cadherins and integrins respectively. Cell detachment: cell detachment from ECM can occur through breakage of adhesion proteins at both intracellular site and extracellular site. Cytosolic cleavage can be achieved through both mechanic forces and enzymatic cleavage while extracellular cleavage is primarily achieved through cleavage by proteases such as MMP. Cell–matrix adhesion by integrins: cell–matrix adhesion is achieved through interaction of integrins with intracellular cytoskeleton and extracellular ECM components. The large extracellular domain of integrin binds to ECM components such as FN, LN, Col, fibrinogen, and VN. The intracellular domain is connected to cytoskeleton through focal adhesions. Cell–cell adhesion by cadherins: cell A and cell B are tightly linked by E-cadherins at adherent junction. The extracellular domain of the same type of cadherin (homodimers) (e.g., E-cadherin with E-cadherin) from the adjacent cells were tightly linked in a calcium-dependent manner. The intracellular domain of the cadherin is connected to cytoskeleton (α-actinin, vinculin, and actin cytoskeleton) through linker proteins (α-catenin, β-catenin and p120 catenin). Cell adhesion among other cells is achieved in a similar way except different CAMs are employed.
During cancer differentiation and metastasis processes, up-regulation of integrins has been linked to cancer invasiveness15, 16, 17. The subunits α3, α5, α6, αv, β1, and β3 are expressed in metastatic cells and can be considered as indicators for metastasis18. Integrins mediate the synthesis of cyclins and inositol lipids as well as activation of FAK and mitogen-activated protein kinase (MAPK)11, 19. Integrins also facilitate the metastatic process by proteolytically degrading the basement membrane through the activation of matrix metalloproteinases (MMPs)20, 21. MMPs are the primary proteases responsible for ECM degradation during cancer metastasis. In addition, integrins regulate tumor cell motility via Rho-A signaling cascade11. Further, integrins promote invasion by activation of PI3K and Src which is a proto-oncogene encoding a tyrosine kinase11. The various roles integrins play in cancer metastasis and proliferation make integrins an attractive target for cancer therapy.
2.1.1.2. Cadherins
Cadherins are a superfamily of transmembrane glycoproteins mediating homophilic (same type of cells) cell–cell adhesion11. More than 20 members of the cadherin molecules have been reported with cell type-specific expression manner such as E-cadherins in epithelial cells, N-cadherins in mesenchymal cells (such as stromal cells), VE-, P-, and R-cadherins in vascular endothelial, placental, and retinal tissues, respectively. Two cadherins of the same type from adjacent cells interact in a non-covalent manner to hold two cells tightly together11, 13.
Fig. 2 employs two epithelial cells (cells A and B) to demonstrate cell–cell adhesion with E-cadherins. These two cells are tightly linked by the extracellular domains of two E-cadherins with each from one of the two cells. The extracellular domain has five repeats and calcium binding sites. The calcium ions hold the two extracellular domains together forming the adherent junction between the cells. The intracellular domain of E-cadherin is linked to cytoskeleton (α-actinin, vinculin, and actin cytoskeleton) through linker proteins (catenin complex: α-catenin, β-catenin, γ-catenin, and p120 catenin) (Fig. 2). Formation of an intact E-cadherin–catenin complex not only stabilizes cell–cell adhesion, but also triggers downstream signal transduction, including Rho GTPases, PI3K, and MAPK, as well as other pathways22.
Modification and/or disruption of either E-cadherin or catenin reduce cellular adhesion23, and are early-caused incidents in cancer development. These include reduction or loss of E-cadherin by genetic and epigenetic incidents, shedding of E-cadherin, redistribution of E-cadherin to different sites in the cell, competition for binding sites from other proteins24, or phosphorylation of catenin. Down-regulation or decreased levels of E-cadherin is an essential event for EMT and has been found in metastatic cancer cells. Down-regulation or decreased levels of E-cadherin leads to loose cell–cell connection that allows tumor cells to disseminate and eventually metastasize. Loss of E-cadherin was also found to correlate with epithelial morphology loss and acquisition of metastatic potential by the carcinoma cells such as prostate, breast, and liver25, 26. Reconstitution of a functional E-cadherin adhesion complex suppresses the invasive properties of many different tumor cell types27, 28, 29. Interestingly, E-cadherin was found transiently vanished in migrating cells, but re-expressed with the start of cell differentiation in epithelial tissues11, 30.
In contrast to E-cadherin, N-cadherin, which is not expressed in normal mammary epithelial cells but expressed in stromal cells, e.g., fibroblasts, has been found to be increased in prostate cancer, breast cancer, and liver cancer. N-cadherin is one of the mesenchymal cadherins, and involved in adhesion of cells to stroma. Down-regulation of E-cadherin with the concomitant up-regulation of N-cadherin reduces cancer cell adhesion ability to epithelial cells, increases adhesion to stromal cells, and leads to subsequent invasion of tumor cells into stroma. N-cadherin promotes cell migration and metastasis regardless of the expression and function of E-cadherin31, 32. The critical roles of N-cadherin in tumor cell adhesion and migration make the protein an attractive target for cancer therapy11, 13.
2.1.1.3. Selectins
Selectins are vascular cell adhesion molecules (VCAMs) involved in adhesive interactions of leukocytes, platelets, and endothelial cells that mediate leukocyte trafficking and hemostasis11. Selectins are involved in processes of immune response, wound repair, inflammation, and hemostasis11. Reports showed that at least one selectin (P, L, or E) is capable of binding to any human carcinoma11, which demonstrates the potential of selectins to mediate contacts with tumor cells within vasculature. The absence of L-selectin, constitutively expressed on cell surfaces of almost all leukocyte subtypes, leads to significant attenuation of metastasis. Inhibition or downregulation of E-selectin expression results in attenuation of liver metastasis. On the other hand, its upregulation redirects metastasis to the liver11, 33.
2.1.1.4. The immunoglobulin superfamily (IgSF)
IgSF [Ig-cell adhesion molecules (Ig-CAM)] is a large group of cell surface proteins that are involved in the binding, recognition, and adhesion processes of cells, and are classified based on shared structural features with immunoglobulins. IgSF members mediate calcium-independent adhesion through their N-terminal Ig-like domains, which commonly bind other Ig-like domains of the same structure on an adjacent cell surface but may also interact with integrins and carbohydrates34. Its C-terminal intracellular domains often interact with cytoskeletal or adaptor proteins through which their extracellular interactions can lead to signaling within the cell, enabling these proteins to function in a range of normal biological processes and/or tumor genesis35. They play important roles in antigen recognition, leukocyte trafficking, and formation and maintenance of endothelial cell junctions11. A number of IgSF molecules have been identified as biomarkers for cancer progression. For example, melanoma CAM (MCAM) has been implicated in the progression of melanoma, breast, and prostate cancer36, 37, 38, neuronal cell adhesion molecule, member of the L1 protein family CAM (L1CAM), neural CAM (NCAM), platelet endothelial CAM (PECAM-1), aplysia CAM (ALCAM), and intercellular CAM-1 (ICAM-1) have been related with metastatic cancers including melanoma, glioma, breast, ovarian, endometrial, prostate, and colon35, 39, 40. Blockade of NCAM led to susceptibility to apoptosis in murine lung tumor cells41. Further, NCAM, MCAM, ALCAM, and L1CAM were found up-regulated in cells following the loss of E-cadherin expression and associated with an active, mobile state that retains enough cell–cell junctions to allow a group of cells to move as a unit in invading melanoma and colorectal carcinoma11, 35, 42, 43.
2.1.1.5. CD44 members
CD44 members have a single pass transmembrane glycoprotein involved in cell–cell, cell–matrix adhesion, and cell signaling. CD44 are lymphocyte-homing receptors and play an important role in lymphocyte homing, inflammation, cell signaling, adhesion, migration, aggregation and hyaluronan (HA) decomposition44, lymphocyte activation45, myelo- and lymphopoiesis45, angiogenesis46, and clearance cytokines11, 47. CD44 proteins also regulate growth, differentiation, survival, and migration, which are all involved in tumor development and metastasis. They are expressed in different tissues including lung, liver, central nervous system, and pancreas48, 49. CD44 family differs in extracellular domain by addition of variable states through alternative splicing50. The extracellular N-terminal of CD44 mediates binding to its primary physiologic ligand HA44 and to extracellular matrix proteins such as collagen, laminin51, fibronectin52, L-selectin, and osteopontin53. The intracellular C-terminal is attached to actin cytoskeleton54, ezrin55, and ankyrin56, which are vital not only in cell migration but also in signal transduction54, 57. All physiological functions of CD44 are related, in one way or another, to cell adhesion49, 58. The most important property of CD44 is its ability to bind HA, a vital factor for the metastatic process. Therefore, inhibition of HA binding to CD44 appears to interfere with events that are critical for tumor development like angiogenesis, apoptosis inhibition, and invasion11, 59.
2.1.2. Cell detachment
Cell detachment refers to a process by which cells detach mostly from the ECM. It is the first required step in the metastatic cascade60. Detachment is not a simple reversal process of cell adhesion and remains poorly understood61. Kirfel and colleagues61 describe a rear detachment during cell migration process in embryogenesis, tissue repair and regeneration as well as cancer and the inflammatory response. The paper provides some light on the cell detachment process. Cell detachment involves both mechanical forces and protease-mediated cleavage. As presented in Fig. 2, mechanical forces generated by actomyosin-driven contraction are believed to contribute to the dissociation of substrate adhesions at both the cytosolic site and the extracellular site. The cytosolic dissociation of cell–substrate adhesions can also be performed by the calpain cysteine proteases, by phosphorylation/dephosphorylation of cytosolic adapter proteins and by posttranslational modification of integrins or adapter proteins. Extracellular dissociation of cell–substrate adhesions can be achieved by proteolytic cleavage of matrix constituents mediated by matrix proteases or through shedding of matrix receptors such as integrins by specific sheddases leaving parts of the receptors on the substrate (Fig. 2)61.
Cell detachment from the ECM in normal epithelial cells and endothelial cells simultaneously triggers down-regulation of Bcl-xL (an anti-apoptotic component of the mitochondrial pathway) and up-regulation of Fas ligand (FasL) (an activator of the death receptor pathway) within a minute resulting in anoikis10. Metastatic cancer cells need to develop a mechanism to resist anoikis in order to survive. Although our knowledge on ECM-detachment and cell death has grown exponentially, how metastatic cancer cells adapt and adopt mechanism(s) that help evade anoikis is only beginning to be understood60. This includes the alteration of enzyme systems in the signaling pathways that regulate anoikis, such as small GTPases and effectors, receptor tyrosine kinases and other kinases, NF-κB, and EMT factors60. In addition to anoikis, it is also noted that there are multiple mechanisms (anoikis-independent) by which normal epithelial cells would die once detached from the ECM60. Metastatic cancer cells must overcome these anoikis-dependent and anoikis-independent barriers in order to survive once they lose the attachment to the ECM. To effectively eliminate metastatic cancer cells, it is suggested that both anoikis-dependent and anoikis-independent pathways should be targeted60. Fortunately, many of the signaling pathways are already the targets of current FDA-approved therapeutic drugs such as bevacizumab (Avastin) against VEGF, ramucirumab (Cyramza) against VEGF receptor, cetuximab (Erbitux) and panitumumab (Vectibix) against EGF receptor60. An excellent review on current understanding of the signaling pathways that regulate anoikis (anti-anoikis and pro-anoikis) and how the pathways are altered to evade anoikis by cancerous cells is provided by Buchheit and colleagues60. The review also provides an insight of anoikis-independent cell death.
2.1.3. Cell migration and invasion
The migratory and invasive abilities of cancer cells are two critical parameters of the metastatic cascade. Metastatic cells achieve penetration of the ECM through two different mechanisms: mesenchymal (fibroblastoid) cell migration and amoeboid cell migration. Mesenchymal cell migration depends on protease activities (protease-dependent) to degrade the ECM for cell passage. Inhibition of ECM degrading proteases, such as MMPs, is effective in inhibiting mesenchymal cell migration. It needs to be noted that mesenchymal cell migration is not only used by cancer cells; it is also seen with normal untransformed cells, such as fibroblasts, and endothelial and smooth muscle cells. Amoeboid cell migration is a protease-independent process where cells employ mechanical forces to open a path in the ECM instead of degrading them11. The hallmarks for amoeboid cell invasive migration are ECM loose attachment, complete cell polarity loss, and chemotaxis capability11. Invasion of single amoeboid cells was observed in breast cancer, lymphoma, small cell lung and prostate carcinomas as well as in melanoma and sarcoma11. Amoeboid cell invasion is described as the fastest migratory phenotype in comparison with mesenchymal cell migration11, 62.
The majority of cancers originate from epithelial tissues. These tumor cells need to remodel their tight cell–cell and cell–matrix adhesion to gain migratory capabilities, and thus to invade adjacent tissues62. The processes involve a dramatic reorganization of actin cytoskeleton and concomitant formation of F-actin-rich membrane protrusions (lamellipodia, filopodia, podosomes, and invadopodia) at the leading edge of motile cells. The protrusions are critical for migration and invasion through the use of mechanic forces and protease activities. Fig. 3 demonstrates the process of an epithelial cancer cell migrating and penetrating BM. The protrusions extend forward adhesion to their surroundings followed by trailing end contraction11.
Figure 3.
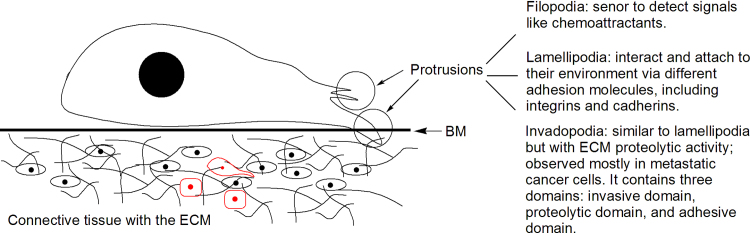
Invasive migration of metastatic cancer cells. A metastatic cell reorganizes its actin cytoskeleton and concomitantly forms F-actin-rich membrane protrusions (lamellipodia, filopodia, podosomes, and invadopodia) at the leading edge. The protrusions are critical for migration and invasion through the use of mechanic forces and protease activities. They serve as cells׳ sensory organ (filopodia) for signals like chemoattractants, or the main organelle for cell locomotion (lamellipodia), or for both cell locomotion and degradation of the ECM through the use of proteases (invadopodia).
The F-actin-rich protrusions include non-invasive protrusions (lamellipodia and filopodia) and invasive protrusions (invadopodia). Lamellipodia and filopodia are present in normal epithelial cells while invadopodia are mostly observed with metastatic cells11. Protrusions are named based on their shapes, i.e., filopodia (needle shape), pseudopodia (round), lobopodia (cylindrical), and lamellipodia (flat veils) and invadopodia (similar to lamellipodia but with ECM proteolytic activity). Functionally, protrusions serve as the cells׳ sensory organ (filopodia) for signals like chemoattractants and nutrients, or the main organelle for cell locomotion (lamellipodia), or for both cell locomotion and degradation of the ECM through the use of proteases (invadopodia)11. Lamellipodia also interact and attach to their environment via different adhesion molecules, including integrins and cadherins11. Filopodia and lamellipodia are highly interactive and interconvertible structures11.
Invadopodia appear when cell adhesion junctions and matrix need to be concomitantly degraded. Invadopodia share an actin filament-rich core and a multimeric protein complex surrounding the actin core, including integrins and integrin-associated proteins like vinculin, talin, and paxillin. They mediate ECM proteolysis through different MMP expression11. Formation of invadopodia involves initiation, assembly, and maturation63 and is initiated by growth factors such as EGF (epidermal growth factor), PDGF (platelet-derived growth factor), TGF-β (transforming growth factor β), and/or α6β1 integrin11. Growth factor receptor signaling activates PI3K leading to Src activation, which in turn phosphorylates multiple proteins including Tks5 (tyrosine kinase substrate). Assembly involves actin polymerization. Proteins involved in this process include cortactin (key regulator of actin polymerization), MENA (regulator of actin polymerization), Tks, etc. Maturation involves Src kinase, integrins, and proteases recruiting (including MMPs). Src kinase is a major regulator of invadopodia formation and function63, 64.
Structurally, invadopodia can be divided into three parts: proteolytic domain, invasive domain, and adhesive domain (Fig. 3). The proteolytic domain contains primarily proteases [MMPs, ADAM (a disintegrin and metalloproteinase, also known as sheddases) and serine proteases]. The invasive domain is localized inside invadopodial protrusions into the ECM and composed of actin and actin-associate proteins such as cortactin and MENA. The protein interactions within this domain lead to actin polymerization that provides mechanic forces to move the cell. The adhesive domain is localized at the edges and anchored to the ECM. It achieves adhesive function through integrin-mediated adhesion.
Invadopodia have been identified in numerous cancer cell lines, including malignant melanoma, breast cancer, glioma, and head and neck malignancies11. It needs to be noted that invadopodia also appear in some normal cells such as immune cells when they have to cross tissue boundaries11.
Metastatic cancer can invade and migrate either as single cells or as a collective group of cells11, 62. While cancer cells migrating as single cells can employ either the protease-dependent mesenchymal migration or protease-independent amoeboid-like migration, collective cell migration employs only mesenchymal cell migration.
Single cells that leave a primary tumor at its periphery involve the loss of epithelial polarity and the achievement of a mesenchymal morphology through EMT. The essential characteristics of EMT are the disruption of tight cell–cell contacts, and acquisition of a fibroblastoid spindle-shape morphology, with increased invasiveness and cell–stroma interactions, as well as slower rates of cell division; altogether, these can result in the release of single cells from a solid epithelial tumor62.
In contrast to single cell migration, cells that migrate collectively retain their cell–cell junctions through continuous expression of adhesion molecules. They migrate as sheets, strands, tubes or clusters and can remain connected to the primary tumor (coordinated invasion) or move as detached cell groups or clusters (cohort migration). Collective cell migration requires force generation for pulling cells from the front or pushing them from the rear. This energy is provided by substrate binding integrins in leading cells. Therefore the leading edge expresses β1 and β3 integrins to mediate adhesion complexes in order to connect to ECM components such as fibronectin11. ECM attachment activates cytoskeletal adaptor proteins such as cortactin, vinculin, paxilin, and talin. As with single invasive cells, collectively migrating cells form membrane protrusions and integrin-mediated focal adhesions that are connected to the actin cytoskeleton. To penetrate the ECM, the leading cells generate an invasion path by the use of β1 integrin-mediated focal adhesions and local expression of MT1-MMP (MMP14) at their leading edges to cleave the collagen fibers and to orient them in tube-like structures in which the following cell mass can migrate65, 66. Collectively migrating cells do not retract their cellular tails, but instead exert mechanical forces, such as moving by pulling on adjacent cells that are connected by adhesion junctions67. This clustered cohort-like cancer cell dissemination appears to be highly efficient in embolizing lymphatic or blood vessels and in cell survival in the circulation11, 62. Collective cell migration is mainly seen for basal cell carcinomas and squamous cell carcinomas of various origins62.
2.2. Tumor microenvironment and cell motility3, 11, 68
It is now well understood that tumor proliferation and metastasis are not an act-alone events of tumor cells, since tumor is required to interact with its microenvironment [tumor microenvironment (TME)]69, 70, 71, 72, 73. As presented earlier, tumor interactions with ECM components are essential in EMT and acquisition of tumor invasive abilities (invadopodia formation and function, proteases secretion and function, actin polymerization, etc.). TME is closely involved with all four essential steps (adhesion, detachment, migration, and invasion) of the metastatic process.
Structurally, TME includes tumor׳s surrounding and supportive stroma, different effectors of the immune system, blood platelets, fibroblasts, endothelial cells, proteases, cytokines, hormones and other humoral factors. Together, these components are involved in a complex crosstalk with tumor cells that affects growth, angiogenesis, and metastasis69, 74. The mechanisms of the interactions between tumor cells and the TME are complex and can fall into two main categories: contact-dependent mechanisms that involve cell–cell and cell–ECM adhesion molecules, and contact-independent mechanisms that involve soluble molecules such as growth factors, chemokines, and cytokines.
2.2.1. Chemokines
Chemokines are peptide signaling cytokines that act as a chemoattractants to guide the migration of cells. They are involved in a variety of physiological and pathological conditions including lymph node organogenesis, inflammation, infection, tissue repair, initiation, and progression of cancer11. Chemokine receptors are expressed in a variety of cancers11. Additionally, to shift the microenvironment to a metastasis-promoting state, cancer cells need to either transform the resident normal stroma cells to facilitate their growth/invasion or recruit other metastasis-promoting stromal cells to remodel the microenvironment. Some of chemokines are shown to have involvement in this process75.
2.2.2. Growth factors
In addition to cell growth stimulation and cell proliferation, many growth factors, such as VEGF (vascular endothelial growth factor), FGF (fibroblast growth factor), PDGF, CTGF (connective tissue growth factor), HGF (hepatocyte growth factor), and EGF, are fully involved in cell migration, angiogenesis, lymphangiogenesis, EMT, and regulation of cell adhesion, etc76. For example, activation of ErbBs ligands, a family member of EGF-related peptides, and overexpression of EGFR (EGF receptor) significantly increase tumor cell motility and intravasation77. In addition, HGF/Met signaling was found to be involved in a variety of cellular processes including cell motility. HGF and Met have also been indicated in the modulation of actin cytoskeleton and MMPs secretion11, 78. CTGF/CCN2 family member 2 (CCN2) has been reported to be associated with extracellular matrix remodeling, angiogenesis, chemotaxis, cell adhesion and migration, and expression of MMPs. Overexpression of CCN2/CTGF was associated with invasive potential of lung and breast cancers. Neutralizing CCN2/CTGF with an antibody attenuated metastasis of pancreatic cancer79. Inhibition of CCN2/CTGF expression could lead to inhibition of migration79. Growth factor signaling pathways have been effective targets for cancer growth inhibition and metastasis inhibition76.
2.2.3. Cancer-associated fibroblasts (CAFs)
CAFs are distinctive cell types recognized as constituting part of the carcinoma and increasingly implicated as functional participants in tumor formation and progression12. CAFs have been shown to enhance primary tumor growth and promote malignancy via HGF secretion80. Moreover, fibroblast HGF expression can be induced by tumor cells due to cytokine secretion such as IL-181. In a similar manner, CAFs have been found to express highly stromal cell-derived factor-1 (SDF-1), implicating the tumor growth-promoting role of fibroblasts, and to provide chemotactic signals for cancer cell invasion and migration82, 83. CAF signaling can be a target for cancer treatment11.
2.2.4. Tumor-associated macrophages (TAMs)
The macrophages within tumor are referred to as tumor-associated macrophages. Upon activation by cancer cells, the TAMs can release a vast diversity of growth factors, proteolytic enzymes, cytokines, and inflammatory mediators. TAMs promote cancer metastasis through several mechanisms that include tumor angiogenesis, tumor growth, and tumor cell migration and invasion. Highly motile TAMs can also control actin cytoskeleton remodeling pathways in metastasis11.
2.2.5. Proteases and MMPs
Proteases are enzymes that cleave protein peptide bonds. At least 500–600 proteases have been identified that cleave peptide bonds through different catalytic mechanisms11. Proteases employ serine, cysteine, and threonine residues or a water molecule as a nucleophile in the active site. They serve various different functions such as activation and inactivation of enzymes, activation of growth factors, gene expression, cell differentiation, cell cycle progression, cell proliferation, and cell death. The link between proteases and cancer was identified in 1946 by Fisher who proposed that tumor-associated proteolytic activity could be responsible for the degradation of the cell matrix and tumor cell invasion into the surrounding normal tissues. The major enzymes responsible for matrix degradation are matrix metalloproteases (MMPs). In addition to MMPs, serine proteases are also involved in matrix degradation11, 84.
MMPs are a family of zinc-dependent endopeptidases capable of cleaving the basement membrane and all ECM constituents. MMPs are produced by different cells including endothelial cells, leukocytes, macrophages, fibroblasts, and tumor cells11. They are synthesized as inactive enzymes and activated outside the cell by other MMPs or serine proteinases11. MMPs consist of at least 26 proteases and are subdivided into four groups: collagenases, gelatinases, stromelysins, and matrilysins11.
MMPs׳ role in metastasis is complex. In addition to cleavage of ECM, their targets also involve growth factor receptors, cytokines, chemokines, CAMs, apoptotic ligands, and angiogenic factors that contribute to all stages of tumor progression such as proliferation, adhesion, migration, angiogenesis, apoptosis, and evasion of the immune system11.
The serine proteases (SPs) are one of the largest preserved multigene proteases with well-described roles in the different processes including blood coagulation, wound healing, digestion, immune response, tumor growth, invasion, and metastasis85, 86. uPA (urokinase plasminogen activator) was first demonstrated by Duffy et al.87 and is a cell surface serine protease that is involved in ECM degradation, cancer invasion, and metastasis11. It has been demonstrated that the uPA system is able to induce human cancer cell proliferation by the proteolytic activation of factors such as HGF, TGF-β, and basic fibroblast growth factor (bFGF) or through uPA receptor interaction with integrins and following activation of the FAK and EGF tyrosine kinase receptors11.
3. Targets for intervention
Because metastatic cancer shares a number of common biochemical parameters and steps, targeting these parameters and steps has unique advantages in controlling metastasis as compared to targeting the parameters that control cancer growth; the latter is the base of most current chemotherapeutic agents. Various steps related to EMT, anoikis, cell motility, and tumor microenvironment have been targeted. The biochemical parameters and steps involved in motility are especially metastatic unique. The intervention methods employed include the use of miRNA, monoclonal antibodies, and small molecules4.
3.1. Targeting EMT and anoikis
Blocking EMT and/or reversing anoikis-resistance are rational approaches to inhibit cancer proliferation and metastasis10. Sakamoto and Kyprianou10 provide an excellent review for the current understanding of molecular signaling networks involved in anoikis and the development of agents that reverse anoikis-resistance. The review also provides the current status of various classes of agents developed. These agents include quinazoline-based anoikis inducers as well as inhibitors of PPARγ, TrkB, and SRC (Fig. 4)10.
Figure 4.

Agents that reverse anoikis-resistance.
3.1.1. The quinazoline-based anoikis inducers
The quinazoline-based anoikis inducers started with the finding that two quinazoline α1-adrenoceptor antagonists [doxazosin and terazosin (Fig. 4)] exhibit significant anti-tumor activity through induction of receptor-mediated apoptosis involving death-inducing signaling complex (DISC) formation/caspase-8 activation and inhibition of Akt activation88, 89, 90. Structural modification of these two lead compounds led to more potent analogs DZ-3 and DZ-50 (Fig. 4) that showed effective induction of anoikis and inhibition of cell migration and adhesion of prostate cancer cells10.
3.1.2. PPARγ inhibitor
Peroxisome proliferator-activated receptor gamma (PPAR-γ) belongs to the nuclear hormone receptor family. PPARγ has recently become a putative therapeutic cancer target in a variety of epithelial cell tumors91. Inhibition of PPAR-γ by T0070907 (Fig. 4) caused cell death by reducing adhesion and inducing anoikis91.
3.1.3. TrkB inhibitor
The Neurotrophic tyrosine kinase receptor (TrkB) has been found to be a potent anoikis suppressor. TrkB overexpression in nonmalignant cells promotes the formation of lung and heart metastases. Consistently, overexpression of TrkB is frequently found in many aggressive gastric and prostate carcinomas11, 92, 93, 94 Trk inhibitor CEP-751 (Fig. 4) was demonstrated to exhibit antitumor activity95. Later, a soluble lysinyl-β-alanyl ester of CEP-751 named CEP-2563 dihydrochloride (Fig. 4) was also reported and underwent clinical trials96.
3.1.4. SRC inhibitor
The Src kinases are currently being investigated as valuable therapeutic targets for cancer treatment. Several SRC inhibitors are now in clinical development, including dasatinib, bosutinib, and saracatinib. Among these SRC inhibitors, dasatinib (Fig. 4) is the most clinically studied SRC inhibitor97. In orthotopic nude mouse models, dasatinib treatment effectively inhibits both tumor growth and development of lymph node metastases in both androgen-sensitive and androgen-resistant prostate cancer98. Dasatinib suppresses cell adhesion, migration, and invasion of prostate cancer cells by blocking the kinase activities of the SFKs, Lyn, and Src98.
3.1.5. Metastatic-related endogenous miRNAs
Metastatic-related endogenous miRNAs have been found to play a significant role in various steps of the metastatic cascade. Depending on their roles, metastatic-related miRNAs are referred to as pro-metastatic or anti-metastatic. Potentially this can be a basis to develop therapeutic intervention for the prevention and cure of metastatic cancer through either inhibiting pro-metastatic or by over-expressing anti-metastatic miRNAs. Efforts have been made to develop anti-miRNA oligonucleotides with different chemical modifications to affect EMT and anoikis12. Profumo and Gandellini provide a good overview of miRNA-based therapy12.
3.2. Targeting cell motility
Cell motility plays an essential role in cancer cell detachment, migration, invasion, and adhesion. Interference of cell motility becomes an appealing approach in developing agents for the treatment of metastatic cancer3. The approaches primarily include inference of interactions of CAMs with their targets and related signaling pathways, and interference of the formation and function of invadopodia.
3.2.1. Interference of CAMs׳ interaction with their targets and related signaling pathways
3.2.1.1. N-cadherin inhibitors
The critical roles of N-cadherin in tumor cell motility make N-cadherin an attractive target for the inhibition of cancer metastasis. The first synthetic N-cadherin antagonist, a linear decapeptide (N-Ac-LRAHAVDVNGNH2), was described in 1990 by Blaschuk and co-workers99. Since then, several types of N-cadherin antagonists have been reported. They include synthetic linear peptides, synthetic cyclic peptides, and non-peptidyl peptidomimetics designed based on the cell adhesion recognition (CAR) sequence His-Ala-Val100. Another type of antagonist is a synthetic linear peptide that harbors a Trp residue in the second position from the N-terminus (similar to N-cadherin)13. One of these peptides is His-Ser-Trp-Thr-Leu-Tyr-Thr-Pro-Ser-Gly-Gln-Ser-Lys-NH2. The peptide inhibits endothelial cell tube formation in vitro indicating that it has anti-angiogenic properties13. Additionally, two monoclonal antibodies against the extracellular domain of N-cadherin were reported to inhibit the invasiveness and proliferation of N-cadherin expressing PC3 human prostate carcinoma cells in vitro13. Among all these inhibitors, the most studied cyclic peptide is N-Ac-Cys-His-Ala-Val-NH2 (designated ADH-1) (Fig. 5). ADH1 (Exherin) was the first N-cadherin antagonist that entered clinical trials. ADH1 selectively and competitively binds to N-cadherin and blocks its function. ADH1 has been tested in a phase II clinical trial as a monotherapy and in various phase I combination trials with cytotoxic drugs such as docetaxel, carboplatin, capecitabine, and melphalan13.
Figure 5.

Structures of CAM inhibitors.
3.2.1.2. Integrin antagonists
Integrins play key roles in cell motility because of their ability to mediate physical interactions with the ECM, cytoskeleton, and signaling pathways that control actin cytoskeleton dynamics and cell movement11. In addition, integrins also play roles in EMT and anoikis through signaling pathways. The α5β1, αvβ3 and αvβ5 integrins are widely expressed in different cancers and recognize the tripeptide Arg-Gly-Asp (RGD) motif present in several ECM proteins101. Integrin antagonists have been found effective in controlling cell proliferation and metastasis11, 13, 102.
The first small molecule integrin antagonist developed was cilengitide (EMD 121974) [Cyclo-l-Arg-Gly-l-Asp-d-Phe-N (Me) l-Val]103 which is a cyclic peptide belonging to the RGD-peptide family. Cilengitide binds to the integrin β chain and prevents the interaction of integrins with their endogenous ECM ligands. Cilengitide is effective in treating lung cancer, prostate cancer, melanoma, glioblastoma, leukemia, brain and CNS tumors, breast cancer, and squamous cell cancer and has undergone Phase I and II clinical trials11.
1a-RGD (Fig. 5) is an RGD-like integrin antagonist containing a bicyclic pseudopentapeptide that binds αvβ3, αvβ5 and α5β1 integrins with in vitro preferential affinity towards αvβ3. 1a-RGD was demonstrated to decrease cell migration and attachment, disassemble the actin cytoskeleton, reduce FAK phosphorylation, decrease the expression of target integrins at transcriptional level and induce anoikis in human U251 and U373 glioblastoma cell lines that express αvβ3 and αvβ5 and α5β1 integrins101.
ATN-161, an acetylated pentapeptide (Ac-Pro-His-Ser-Cys-Asn-NH2), was designed based on a sequence of fibronectin (Pro-His-Ser-Arg-Asn) through replacement of Arg with Cys. ATN-161 interferes binding of α5β1 integrin with this region of fibronectin to inhibit cancer growth and metastasis104, 105, 106 and has undergone phase I trials11, 13.
In addition to these small molecule antagonists, monoclonal antibodies against integrins have also been reported. They include CNTO95, etaracizumab (MEDI-522), and volociximab. These antibodies have undergone various phases of clinical trials11, 13.
3.2.1.3. Selectin inhibitors
Selectins have been implicated in mediating contacts with tumor cells within vasculature. Inhibition or downregulation of E-selectin expression results in attenuation of liver metastasis. In contrast, its upregulation redirects metastasis to the liver33. Heparin is shown to inhibit P-selectin-mediated interactions of platelets with cancer cell ligands in mice metastatic states107. Thus, compounds having both heparanase and selectin inhibition properties are promising for cancer therapy. Borsig et al.108 reported the novel semisynthetic sulfated trimannose CC-linked dimers (STMCs) (Fig. 5) with inhibitory activity for heparanase and selectin. This STMC hexa-saccharide is an in vivo effective inhibitor of P-selectin with antimetastasis activities in animal models11, 108.
3.2.1.4. CD44 antagonists
The CD44 transmembrane glycoprotein family, a hyaluronan receptor, mediates cellular responses to the microenvironment through binding of hyaluronic acid (HA) and other proteins of the ECM. These interactions start intracellular signaling cascades that foster tumor growth, survival and spread109. CD 44 is associated with aggressive tumor growth, proliferation, and metastasis, and has been a target for metastasis treatment11, 59. In normal physiology, this receptor has a crucial role in cell adhesion, inflammation, and repair processes110. Systemic use of antibodies against CD44v epitope decreased pancreatic adenocarcinoma metastasis11. Anti-CD44 antibody-targeting radiolabels or anticancer chemotherapeutics have been adopted in some patients11. Immunotoxin, a humanized antibody complexed with a cytotoxic drug mertansine against CD44v6, has entered into Phase 1 clinical trials and was reported to improve the conditions in 30 incurable squamous cell carcinoma patients11. Liu et al.11 showed that miRNA-34a was a negative CD44+ prostate cancer cell regulator. It enforced expression of miRNA-34a in CD44+ prostate cancer cells and inhibited clonogenic expansion, tumor regeneration, and metastasis.
3.2.2. Interference with the formation and function of invadopodia
Unlike other actin-based protrusions such as lamellipodia and filopodia that are present in normal cells, invadopodia are uniquely present in invasive cancer cells and considered as the transformed version of podosomes which are present in highly invasive normal cells such as macrophages, osteoclasts and dendritic cells64. The main function of invadopodia in tumors is to promote matrix degradation and tumor invasion. Invadopodia play critical roles during three steps of the metastatic process: invasion into the surrounding stroma, intravasation into the vasculature and extravasation64. Invadopodia has emerged as an appealing target for metastasis prevention and inhibition111.
Intervention of invadopodia can be achieved by impacting the formation, structure and function of invadopodia. For example, since formation of invadopodia involves growth factors, growth factor inhibitors affect invadopodia formation. Suppressing invadopodia formation, structure, and function by inhibiting Src, Twist1 or Tks5 has been convincingly shown to inhibit tumor metastasis in various tumor models64. MMP inhibition also inhibits invadopodia formation112. Blocking MMP activity by inhibitors, antibodies, or siRNA impairs invadopodia function and matrix degradation63, 112.
Since invadopodia are only involved in metastasis not cell proliferation, it is suggested that invadopadia inhibitors should be used in combination with an inhibitor of cell proliferation to prevent metastasis as well as to inhibit cancer growth.
3.3. Targeting tumor microenvironment (TME)
As described earlier, TME plays essential roles in the metastasis cascade. Extensive research efforts have been made to interfere with communication of tumors with TME to achieve the inhibition of cancer growth and metastasis. Various TME components and signaling pathways that impact cancer metastasis have been targeted. They include inhibition of proteases, interference with inflammatory processes, inhibition of integrin signaling, interference with hypoxia processes, and remodeling of ECM11, 73, 113.
3.3.1. Inhibition of growth factor signaling
As presented earlier, growth factor signaling is not only involved in cancer proliferation, but also essential in EMT and acquisition of a tumor invasive abilities. Various inhibitors against VEGF, FGF, PDGF, and EGFR signaling have been developed. These inhibitors include small molecules (Fig. 6) and monoclonal antibodies such as bevacizumab against VEGF, ramucirumab against VEGF receptor, and cetuximab and panitumumab against EGF receptor73.
Figure 6.

Chemical structures of small molecules that inhibit growth factor signaling pathways.
3.3.2. Inhibition of proteases
A hallmark of tumor cell invasion is upregulation of proteolytic enzymes, especially MMPs3. Inhibition of MMPs was considered to be a very promising approach and was studied in a variety of clinical trials as therapy for various types of cancers. Unfortunately, those trials were largely unsuccessful3. The disappointing outcomes are probably caused by various factors that include the development of drug resistance by the tumor cells, lack of sufficient specificity of the inhibitors, and changes in the cancer cell migration and invasion mechanism from proteolysis-dependent migration to proteolysis-independent migration (amoeboid cell migration through mesenchymal-amoeboid transition)11, 112. It has been suggested that the combination of a MMP inhibitor with other chemotherapeutic agents would probably yield a better therapeutic outcome. Presented in Fig. 7 are representative small molecule MMP inhibitors, which are grouped chemically as hydroxamates, thiol-based analogs, pyrimidine-2,4,6-triones and others4, 11. Additionally, monoclonal antibodies against MMPs have also been reported4, 11.
Figure 7.

Structures of representative MMP inhibitors.
uPA is a serine proteases involved in ECM degradation, cancer growth and metastasis. A number of small molecule uPA inhibitors have been reported. They include A6 [a capped, eight l-amino acid peptide (Ac-Lys-Pro-Ser-Ser-Pro-Pro-Glu-Glu-NH2) derived from the biologically active connecting peptide domain of the serine protease], Suramin, WX-UK1, and WX-671 (Fig. 8)11.
Figure 8.

Structures of representative uPA inhibitors.
3.3.3. Interference with inflammation11, 73, 114, 115
It is well accepted that inflammation is closely associated with cancer growth and metastasis116. Inflammation can impact cancer by providing bioactive molecules from cells infiltrating the tumor microenvironment. These bioactive molecules include cytokines, growth factors, chemokines, cell survival signals to avoid apoptosis, proangiogenic factors, and ECM modifying enzymes116. Interference with the function of these bioactive molecules has been demonstrated to be effective in the inhibition of`cancer growth and/or metastasis. Fig. 9 presents representative small molecules that interfere with the inflammation process. In addition, neutralizing antibodies against CCL2 [chemokine, (C–C motif) ligand 2], CSF-1 (chemokine, colony stimulating factor-1), IL-6 (Interleukin-6, cytokine), IL-6 receptor, TNF, TGF-β have also been reported73. IL-6 plays a key role in promoting proliferation and inhibition of apoptosis by binding to its receptor (IL-6Rα) and co-receptor gp130 (glycoprotein 130). The binding activates the JAK/STAT signaling pathway of the Janus kinases (JAK) and signal transducers and activators of transcription (STATs) STAT1 and STAT372. STATs belong to a family of transcription factors closely associated with the tumorigenic processes. Several studies have highlighted the effect of the IL-6/JAK/STAT signaling pathway on cancer initiation and progression72, 73, 116. Current attempts to target the IL-6/JAK-STAT3 pathway include the clinical use of IL-6 and IL-6 receptor blocking antibodies, specific STAT3 inhibitors and JAK inhibitors72.
Figure 9.

Chemical structures of representative compounds that interfere with the inflammation process. CCL2: chemokine (C–C motif) ligand 2; CSF-1: chemokine colonystimulating factor-1; JAK: Janus kinases; IKK: The IκB kinase (IKK) complex; TGF-β: transforming Growth Factor β; CXCR2: chemokine receptor type 2; CCR3: chemokine receptor; COX2: cyclooxygenase 2.
The IκB kinase (IKK) complex is the key enzyme in activation of the NF-κB pathway116. NF-κB is a transcription factor that controls the expression of a number of important genes for mediating immune and inflammatory responses. IKK inhibitor PS-1145 (Fig. 9) has been found effective in treating lymphoma73.
3.3.4. Inhibition of integrin signaling4, 11, 116
Inhibition of integrin signaling has been presented in the earlier section and will not be discussed further..
3.3.5. Interfering with hypoxia process
Hypoxia is a characteristic microenvironment in the majority of solid tumors117. Tumor hypoxia is a result of rapid tumor cell proliferation that exceeds the development of the tumor׳s blood supply. Hypoxia-inducible factors (HIFs) are transcription factors that respond to a decrease in oxygen in the cellular environment. Activation of HIF-l (hypoxia-inducible factor 1) in cancer can increase the transcription of many genes involved in glucose metabolism, apoptosis resistance, invasion, metastasis and angiogenesis. HIF-l has been considered to be an attractive target for the development of novel cancer therapeutics. Fig. 10 presents some selected inhibitors of HIF-1118.
Figure 10.

Chemical structures of representative compounds that interfere with HIF-1 function118.
3.3.6. Remodeling of ECM
Remodeling of the ECM has also been explored for metastasis inhibition113. Hyaluronan [hyaluronic acid (HA) or hyaluronate] is one of the chief components of the ECM and plays a critical role in tumor cell adhesion and migration. Hyaluronidases are a family of enzymes that degrade hyaluronan. There is compelling evidence that the administration of exogenous hyaluronidase can impose significant anticancer activity in HA-overexpressing tumors113. Recombinant human hyaluronidase, halozyme (Hylenex™) also known as rHuPH20, is an FDA-approved enzyme that can reversibly degrade HA, lower the viscosity of hyaluronan, increase tissue permeability, and hence enhance the absorption and dispersion of chemotherapeutic agents. Its PEGylated form (PEGPH20), which exhibits longer survival time in vivo, has recently been introduced into clinical trials113.
Heparan sulfate proteoglycans (HSPGs) are an integral and dynamic part of normal tissue architecture at the cell surface and within the ECM. Sulfatases and heparanase are key enzymes for the degradation of HSPGs. Recently, these enzymes have been reported to be required in tumor initiation and progress. Inhibitors of heparanase, mostly heparan sulfate mimetics, have been found to be effective as anti-metastatic agents and some of them have undergone various stages of clinical trials119. While heparanase inhibitors exhibit promising potential for cancer treatment, the effect on cancer growth and metastasis of sulfatase inhibitors is less certain119.
3.3.7. Cancer-associated fibroblasts (CAFs)
CAFs are distinctive cell types recognized as constituting part of the carcinoma and increasingly implicated as functional participants in tumor formation and progression12. CAFs have shown to enhance primary tumor growth and promote malignancy via HGF secretion11. Moreover, fibroblast HGF expression can be induced by tumor cells due to cytokine secretion such as IL-111. In a similar manner, CAFs have been found to highly express SDF-1, implicating the tumor growth-promoting role of fibroblasts, and provide chemotactic signals for cancer cell invasion and migration11. CAF signaling can be a target for cancer treatment11. Inhibitors of CAFs include those that inhibit growth factor signaling such as TGF-β, PDGFR, VEGF, VEGFR, HGF/MET, and IGF-1R11. They also include monoclonal antibody (Avastin/bevacizumab), antisense oligo (AP12009/trabedersen), fusion protein (Aflibercept/VEGF-TRAP) and small molecule inhibitor such as LY2157299 (Fig. 11), a receptor kinase inhibitor targeting cancer-associated fibroblasts11.
Figure 11.
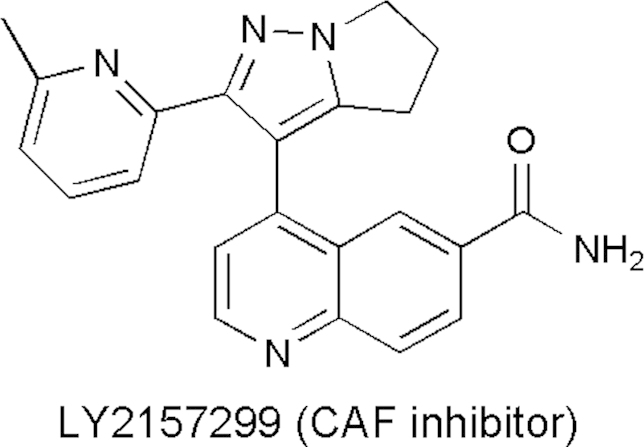
Chemical structure of LY2157299.
4. Summary
Despite extensive research efforts on cancer treatment, overall survival has not been improved significantly in metastasis cancer patients. This is primarily due to the reason that the predominant cancer treatment focuses on inhibition of cancer growth, with little emphasis on metastasis. Limited success has been made in terms of treating cancer metastasis, though the current new generation anti-cancer drugs (predominantly neutralizing antibodies for growth factors and small molecule kinase inhibitors) do have effects on cancer metastasis in addition to their effects on cancer growth4.
The rapid advance in our understanding of cancer metastasis at molecular and cellular levers as well as signaling pathways in the past 30 years provides numerous potential targets for the intervention of cancer metastasis. This is especially true in terms of intervention with biochemical processes and signaling pathways involved in cell detachment, migration, invasion, adhesion, and cancer cell communication with tumor microenvironment. In view of the complexity of cancer metastatic cascade such as anoikis-dependent vs. anoikis-independent pathways, mesenchymal cell migration (protease-dependent) vs. amoeboid cell migration (protease-independent), a combination that inhibits multiple elements in the cancer metastasis cascade might be needed to produce effective metastasis inhibition. Further, simultaneous inhibition of both cancer growth and metastasis will likely be required to produce clinically effective therapeutic outcomes.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev Oncog. 2013;18:43–73. doi: 10.1615/critrevoncog.v18.i1-2.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Damsky WE, Theodosakis N, Bosenberg M. Melanoma metastasis: new concepts and evolving paradigms. Oncogene. 2014;33:2413–2422. doi: 10.1038/onc.2013.194. [DOI] [PubMed] [Google Scholar]
- 3.Wells A, Grahovac J, Wheeler S, Ma B, Lauffenburger D. Targeting tumor cell motility as a strategy against invasion and metastasis. Trends Pharmacol Sci. 2013;34:283–289. doi: 10.1016/j.tips.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber GF. Why does cancer therapy lack effective anti-metastasis drugs? Cancer Lett. 2013;328:207–211. doi: 10.1016/j.canlet.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 5.Hu YY, Zheng MH, Zhang R, Liang YM, Han H. Notch signaling pathway and cancer metastasis. Adv Exp Med Biol. 2012;727:186–198. doi: 10.1007/978-1-4614-0899-4_14. [DOI] [PubMed] [Google Scholar]
- 6.Daenen LGM, Roodhart JML, van Amersfoort M, Dehnad M, Roessingh W, Ulfman LH. Chemotherapy enhances metastasis formation via VEGFR-1-expressing endothelial cells. Cancer Res. 2011;71:6976–6985. doi: 10.1158/0008-5472.CAN-11-0627. [DOI] [PubMed] [Google Scholar]
- 7.Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369:1742–1757. doi: 10.1016/S0140-6736(07)60781-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haraguchi M. The role of the transcriptional regulator snail in cell detachment, reattachment and migration. Cell Adhes Migr. 2009;3:259–263. doi: 10.4161/cam.3.3.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zanotti S, Gibertini S, Bragato C, Mantegazza R, Morandi L, Mora M. Fibroblasts from the muscles of Duchenne muscular dystrophy patients are resistant to cell detachment apoptosis. Exp Cell Res. 2011;317:2536–2547. doi: 10.1016/j.yexcr.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 10.Sakamoto S, Kyprianou N. Targeting anoikis resistance in prostate cancer metastasis. Mol Asp Med. 2010;31:205–214. doi: 10.1016/j.mam.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alizadeh AM, Shiri S, Farsinejad S. Metastasis review: from bench to bedside. Tumour Biol. 2014;35:8483–8523. doi: 10.1007/s13277-014-2421-z. [DOI] [PubMed] [Google Scholar]
- 12.Profumo V, Gandellini P. MicroRNAs: cobblestones on the road to cancer metastasis. Crit Rev Oncog. 2013;18:341–355. doi: 10.1615/critrevoncog.2013007182. [DOI] [PubMed] [Google Scholar]
- 13.Li DM, Feng YM. Signaling mechanism of cell adhesion molecules in breast cancer metastasis: potential therapeutic targets. Breast Cancer Res Treat. 2011;128:7–21. doi: 10.1007/s10549-011-1499-x. [DOI] [PubMed] [Google Scholar]
- 14.Bravo-Cordero JJ, Hodgson L, Condeelis JS. Spatial regulation of tumor cell protrusions by RhoC. Cell Adhes Migr. 2014;8:263–267. doi: 10.4161/cam.28405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White DE, Muller WJ. Multifaceted roles of integrins in breast cancer metastasis. J Mammary Gland Biol Neoplasia. 2007;12:135–142. doi: 10.1007/s10911-007-9045-5. [DOI] [PubMed] [Google Scholar]
- 16.Jin H, Varner J. Integrins: roles in cancer development and as treatment targets. Br J Cancer. 2004;90:561–565. doi: 10.1038/sj.bjc.6601576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janik ME, Lityńska A, Vereecken P. Cell migration-the role of integrin glycosylation. Biochim Biophys Acta. 2010;1800:545–555. doi: 10.1016/j.bbagen.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 18.Pawelek JM, Chakraborty AK. Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nat Rev Cancer. 2008;8:377–386. doi: 10.1038/nrc2371. [DOI] [PubMed] [Google Scholar]
- 19.Schlaepfer DD, Jones KC, Hunter T. Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: summation of both c-Src- and focal adhesion kinase-initiated tyrosine phosphorylation events. Mol Cell Biol. 1998;18:2571–2585. doi: 10.1128/mcb.18.5.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks PC, Strömblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin αvβ3. Cell. 1996;85:683–693. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
- 21.Deryugina EI, Bourdon MA, Luo GX, Reisfeld RA, Strongin A. Matrix metalloproteinase-2 activation modulates glioma cell migration. J Cell Sci. 1997;110:2473–2482. doi: 10.1242/jcs.110.19.2473. [DOI] [PubMed] [Google Scholar]
- 22.Rivard N. Phosphatidylinositol 3-kinase: a key regulator in adherens junction formation and function. Front Biosci (Landmark Ed) 2009;14:510–522. doi: 10.2741/3259. [DOI] [PubMed] [Google Scholar]
- 23.Hazan RB, Qiao R, Keren R, Badano I, Suyama K. Cadherin switch in tumor progression. Ann NY Acad Sci. 2004;1014:155–163. doi: 10.1196/annals.1294.016. [DOI] [PubMed] [Google Scholar]
- 24.Jiang WG. E-cadherin and its associated protein catenins, cancer invasion and metastasis. Br J Surg. 1996;83:437–446. doi: 10.1002/bjs.1800830404. [DOI] [PubMed] [Google Scholar]
- 25.Pećina-Šlaus N. Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. 2003;3:17. doi: 10.1186/1475-2867-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riethmacher D, Brinkmann V, Birchmeier C. A targeted mutation in the mouse E-cadherin gene results in defective preimplantation development. Proc Natl Acad Sci U S A. 1995;92:855–859. doi: 10.1073/pnas.92.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo J, Lubaroff DM, Hendrix MJ. Suppression of prostate cancer invasive potential and matrix metalloproteinase activity by E-cadherin transfection. Cancer Res. 1999;59:3552–3556. [PubMed] [Google Scholar]
- 28.Hsu MY, Meier FE, Nesbit M, Hsu JY, van Belle P, Elder DE. E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am J Pathol. 2000;156:1515–1525. doi: 10.1016/S0002-9440(10)65023-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bogenrieder T, Herlyn M. Axis of evil: molecular mechanisms of cancer metastasis. Oncogene. 2003;22:6524–6536. doi: 10.1038/sj.onc.1206757. [DOI] [PubMed] [Google Scholar]
- 30.Larue L, Antos C, Butz S, Huber O, Delmas V, Dominis M. A role for cadherins in tissue formation. Development. 1996;122:3185–3194. doi: 10.1242/dev.122.10.3185. [DOI] [PubMed] [Google Scholar]
- 31.Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. 2000;148:779–790. doi: 10.1083/jcb.148.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nieman MT, Prudoff RS, Johnson KR, Wheelock MJ. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J Cell Biol. 1999;147:631–644. doi: 10.1083/jcb.147.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bendas G, Borsig L. Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol. 2012;2012:676731. doi: 10.1155/2012/676731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barclay AN. Membrane proteins with immunoglobulin-like domains—a master superfamily of interaction molecules. Semin Immunol. 2003;15:215–223. doi: 10.1016/s1044-5323(03)00047-2. [DOI] [PubMed] [Google Scholar]
- 35.Wai Wong C, Dye DE, Coombe DR. The role of immunoglobulin superfamily cell adhesion molecules in cancer metastasis. Int J Cell Biol. 2012;2012:340296. doi: 10.1155/2012/340296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson JP, Bar-Eli M, Jansen B, Markhof E. Melanoma progression-associated glycoprotein MUC18/MCAM mediates homotypic cell adhesion through interaction with a heterophilic ligand. Int J Cancer. 1997;73:769–774. doi: 10.1002/(sici)1097-0215(19971127)73:5<769::aid-ijc26>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 37.Wu GJ, Wu MWH, Wang CS, Liu Y. Enforced expression of METCAM/MUC18 increases tumorigenesis of human prostate cancer LNCaP cells in nude mice. J Urol. 2011;185:1504–1512. doi: 10.1016/j.juro.2010.11.052. [DOI] [PubMed] [Google Scholar]
- 38.Zeng GF, Cai SX, Wu GJ. Up-regulation of METCAM/MUC18 promotes motility, invasion, and tumorigenesis of human breast cancer cells. BMC Cancer. 2011;11:113. doi: 10.1186/1471-2407-11-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roland CL, Harken AH, Sarr MG, Barnett CC., Jr. ICAM-1 expression determines malignant potential of cancer. Surgery. 2007;141:705–707. doi: 10.1016/j.surg.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 40.Siesser PF, Maness PF. L1 cell adhesion molecules as regulators of tumor cell invasiveness. Cell Adhes Migr. 2009;3:275–277. doi: 10.4161/cam.3.3.8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jezierska A, Matysiak W, Motyl T. ALCAM/CD166 protects breast cancer cells against apoptosis and autophagy. Med Sci Monit. 2006;12:BR263–BR273. [PubMed] [Google Scholar]
- 42.Gavert N, Ben-Shmuel A, Raveh S, Ben-Ze׳ev A. L1-CAM in cancerous tissues. Expert Opin Biol Ther. 2008;8:1749–1757. doi: 10.1517/14712598.8.11.1749. [DOI] [PubMed] [Google Scholar]
- 43.Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- 44.Underhill C. CD44: the hyaluronan receptor. J Cell Sci. 1992;103:293–298. doi: 10.1242/jcs.103.2.293. [DOI] [PubMed] [Google Scholar]
- 45.Shimizu Y, van Seventer GA, Siraganian R, Wahl L, Shaw S. Dual role of the CD44 molecule in T cell adhesion and activation. J Immunol. 1989;143:2457–2463. [PubMed] [Google Scholar]
- 46.Trochon V, Mabilat C, Bertrand P, Legrand Y, Smadja-Joffe F, Soria C. Evidence of involvement of CD44 in endothelial cell proliferation, migration and angiogenesis in vitro. Int J Cancer. 1996;66:664–668. doi: 10.1002/(SICI)1097-0215(19960529)66:5<664::AID-IJC14>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 47.Webb DS, Shimizu Y, van Seventer GA, Shaw S, Gerrard TL. LFA-3, CD44, and CD45: physiologic triggers of human monocyte TNF and IL-1 release. Science. 1990;249:1295–1297. doi: 10.1126/science.1697984. [DOI] [PubMed] [Google Scholar]
- 48.Fox SB, Fawcett J, Jackson DG, Collins I, Gatter KC, Harris AL. Normal human tissues, in addition to some tumors, express multiple different CD44 isoforms. Cancer Res. 1994;54:4539–4546. [PubMed] [Google Scholar]
- 49.Richter U, Wicklein D, Geleff S, Schumacher U. The interaction between CD44 on tumour cells and hyaluronan under physiologic flow conditions: implications for metastasis formation. Histochem Cell Biol. 2012;137:687–695. doi: 10.1007/s00418-012-0916-5. [DOI] [PubMed] [Google Scholar]
- 50.Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4:33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 51.Ishii S, Ford R, Thomas P, Nachman A, Steele G, Jr., Jessup JM. CD44 participates in the adhesion of human colorectal carcinoma cells to laminin and type IV collagen. Surg Oncol. 1993;2:255–264. doi: 10.1016/0960-7404(93)90015-q. [DOI] [PubMed] [Google Scholar]
- 52.Jalkanen S, Jalkanen M. Lymphocyte CD44 binds the COOH-terminal heparin-binding domain of fibronectin. J Cell Biol. 1992;116:817–825. doi: 10.1083/jcb.116.3.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Naor D, Sionov RV, Ish-Shalom D. CD44: structure, function, and association with the malignant process. Adv Cancer Res. 1997;71:241–319. doi: 10.1016/s0065-230x(08)60101-3. [DOI] [PubMed] [Google Scholar]
- 54.Marhaba R, Zöller M. CD44 in cancer progression: adhesion, migration and growth regulation. J Mol Histol. 2004;35:211–231. doi: 10.1023/b:hijo.0000032354.94213.69. [DOI] [PubMed] [Google Scholar]
- 55.Legg JW, Lewis CA, Parsons M, Ng T, Isacke CM. A novel PKC-regulated mechanism controls CD44-ezrin association and directional cell motility. Nat Cell Biol. 2002;4:399–407. doi: 10.1038/ncb797. [DOI] [PubMed] [Google Scholar]
- 56.Lokeshwar VB, Bourguignon LY. Post-translational protein modification and expression of ankyrin-binding site(s) in GP85 (Pgp-1/CD44) and its biosynthetic precursors during T-lymphoma membrane biosynthesis. J Biol Chem. 1991;266:17983–17989. [PubMed] [Google Scholar]
- 57.Iczkowski KA. Cell adhesion molecule CD44: its functional roles in prostate cancer. Am J Transl Res. 2010;3:1–7. [PMC free article] [PubMed] [Google Scholar]
- 58.Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B. CD44 is the principal cell surface receptor for hyaluronate. Cell. 1990;61:1303–1313. doi: 10.1016/0092-8674(90)90694-a. [DOI] [PubMed] [Google Scholar]
- 59.Orian-Rousseau V. CD44, a therapeutic target for metastasising tumours. Eur J Cancer. 2010;46:1271–1277. doi: 10.1016/j.ejca.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 60.Buchheit CL, Weigel KJ, Schafer ZT. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer. 2014;14:632–641. doi: 10.1038/nrc3789. [DOI] [PubMed] [Google Scholar]
- 61.Kirfel G, Rigort A, Borm B, Herzog V. Cell migration: mechanisms of rear detachment and the formation of migration tracks. Eur J Cell Biol. 2004;83:717–724. doi: 10.1078/0171-9335-00421. [DOI] [PubMed] [Google Scholar]
- 62.Spano D, Heck C, De Antonellis P, Christofori G, Zollo M. Molecular networks that regulate cancer metastasis. Semin Cancer Biol. 2012;22:234–249. doi: 10.1016/j.semcancer.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 63.Jacob A, Prekeris R. The regulation of MMP targeting to invadopodia during cancer metastasis. Front Cell Dev Biol. 2015;3:4. doi: 10.3389/fcell.2015.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Paz H, Pathak N, Yang J. Invading one step at a time: the role of invadopodia in tumor metastasis. Oncogene. 2014;33:4193–4202. doi: 10.1038/onc.2013.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wolf K, Friedl P. Mapping proteolytic cancer cell-extracellular matrix interfaces. Clin Exp Metastasis. 2009;26:289–298. doi: 10.1007/s10585-008-9190-2. [DOI] [PubMed] [Google Scholar]
- 66.Friedl P, Wolf K. Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Res. 2008;68:7247–7249. doi: 10.1158/0008-5472.CAN-08-0784. [DOI] [PubMed] [Google Scholar]
- 67.Yilmaz M, Christofori G. Mechanisms of motility in metastasizing cells. Mol Cancer Res. 2010;8:629–642. doi: 10.1158/1541-7786.MCR-10-0139. [DOI] [PubMed] [Google Scholar]
- 68.McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol. 2014;16:717–727. doi: 10.1038/ncb3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goubran HA, Kotb RR, Stakiw J, Emara ME, Burnouf T. Regulation of tumor growth and metastasis: the role of tumor microenvironment. Cancer Growth Metastasis. 2014;7:9–18. doi: 10.4137/CGM.S11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Naora H. Heterotypic cellular interactions in the ovarian tumor microenvironment: biological significance and therapeutic implications. Front Oncol. 2014;4:18. doi: 10.3389/fonc.2014.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wood SL, Pernemalm M, Crosbie PA, Whetton AD. The role of the tumor-microenvironment in lung cancer-metastasis and its relationship to potential therapeutic targets. Cancer Treat Rev. 2014;40:558–566. doi: 10.1016/j.ctrv.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 72.Bournazou E, Bromberg J. Targeting the tumor microenvironment: JAK-STAT3 signaling. JAKSTAT. 2013;2:e23828. doi: 10.4161/jkst.23828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fang H, Declerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res. 2013;73:4965–4977. doi: 10.1158/0008-5472.CAN-13-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chou J, Shahi P, Werb Z. microRNA-mediated regulation of the tumor microenvironment. Cell Cycle. 2013;12:3262–3271. doi: 10.4161/cc.26087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Y, Yang PY, Wang XF. Microenvironmental regulation of cancer metastasis by miRNAs. Trends Cell Biol. 2014;24:153–160. doi: 10.1016/j.tcb.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang WG, Ablin RJ. Cancer metastasis, challenges, progress and the opportunities. Front Biosci (Elite Ed) 2011;3:391–394. doi: 10.2741/e254. [DOI] [PubMed] [Google Scholar]
- 77.Xue CS, Wyckoff J, Liang FB, Sidani M, Violini S, Tsai KL. Epidermal growth factor receptor overexpression results in increased tumor cell motility in vivo coordinately with enhanced intravasation and metastasis. Cancer Res. 2006;66:192–197. doi: 10.1158/0008-5472.CAN-05-1242. [DOI] [PubMed] [Google Scholar]
- 78.Ma PC, Maulik G, Christensen J, Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003;22:309–325. doi: 10.1023/a:1023768811842. [DOI] [PubMed] [Google Scholar]
- 79.Aguiar DP, de Farias GC, de Sousa EB, de Mattos Coelho-Aguiar J, Lobo JC, Casado PL. New strategy to control cell migration and metastasis regulated by CCN2/CTGF. Cancer Cell Int. 2014;14:61. doi: 10.1186/1475-2867-14-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Micke P, Ostman A. Tumour-stroma interaction: cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer. 2004;45 Suppl 2:S163–S175. doi: 10.1016/j.lungcan.2004.07.977. [DOI] [PubMed] [Google Scholar]
- 81.Qian LW, Mizumoto K, Maehara N, Ohuchida K, Inadome N, Saimura M. Co-cultivation of pancreatic cancer cells with orthotopic tumor-derived fibroblasts: fibroblasts stimulate tumor cell invasion via HGF secretion whereas cancer cells exert a minor regulative effect on fibroblasts HGF production. Cancer Lett. 2003;190:105–112. doi: 10.1016/s0304-3835(02)00517-7. [DOI] [PubMed] [Google Scholar]
- 82.Luker KE, Luker GD. Functions of CXCL12 and CXCR4 in breast cancer. Cancer Lett. 2006;238:30–41. doi: 10.1016/j.canlet.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 83.Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 84.Shuman Moss LA, Jensen-Taubman S, Stetler-Stevenson WG. Matrix metalloproteinases: changing roles in tumor progression and metastasis. Am J Pathol. 2012;181:1895–1899. doi: 10.1016/j.ajpath.2012.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rawlings ND, Barrett AJ. Families of serine peptidases. Methods Enzymol. 1994;244:19–61. doi: 10.1016/0076-6879(94)44004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Netzel-Arnett S, Hooper JD, Szabo R, Madison EL, Quigley JP, Bugge TH. Membrane anchored serine proteases: a rapidly expanding group of cell surface proteolytic enzymes with potential roles in cancer. Cancer Metastasis Rev. 2003;22:237–258. doi: 10.1023/a:1023003616848. [DOI] [PubMed] [Google Scholar]
- 87.Duffy MJ, O׳Grady P, Devaney D, O׳Siorain L, Fennelly JJ, Lijnen HJ. Urokinase-plasminogen activator, a marker for aggressive breast carcinomas. Preliminary report. Cancer. 1988;62:531–533. doi: 10.1002/1097-0142(19880801)62:3<531::aid-cncr2820620315>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 88.Keledjian K, Garrison JB, Kyprianou N. Doxazosin inhibits human vascular endothelial cell adhesion, migration, and invasion. J Cell Biochem. 2005;94:374–388. doi: 10.1002/jcb.20240. [DOI] [PubMed] [Google Scholar]
- 89.Keledjian K, Kyprianou N. Anoikis induction by quinazoline based α1-adrenoceptor antagonists in prostate cancer cells: antagonistic effect of Bcl-2. J Urol. 2003;169:1150–1156. doi: 10.1097/01.ju.0000042453.12079.77. [DOI] [PubMed] [Google Scholar]
- 90.Rennebeck G, Martelli M, Kyprianou N. Anoikis and survival connections in the tumor microenvironment: is there a role in prostate cancer metastasis? Cancer Res. 2005;65:11230–11235. doi: 10.1158/0008-5472.CAN-05-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schaefer KL, Wada K, Takahashi H, Matsuhashi N, Ohnishi S, Wolfe MM. Peroxisome proliferator-activated receptor γ inhibition prevents adhesion to the extracellular matrix and induces anoikis in hepatocellular carcinoma cells. Cancer Res. 2005;65:2251–2259. doi: 10.1158/0008-5472.CAN-04-3037. [DOI] [PubMed] [Google Scholar]
- 92.Zhang Y, Fujiwara Y, Doki Y, Takiguchi S, Yasuda T, Miyata H. Overexpression of tyrosine kinase B protein as a predictor for distant metastases and prognosis in gastric carcinoma. Oncology. 2008;75:17–26. doi: 10.1159/000151615. [DOI] [PubMed] [Google Scholar]
- 93.Tanaka K, Mohri Y, Nishioka J, Kobayashi M, Ohi M, Miki C. Neurotrophic receptor, tropomyosin-related kinase B as an independent prognostic marker in gastric cancer patients. J Surg Oncol. 2009;99:307–310. doi: 10.1002/jso.21232. [DOI] [PubMed] [Google Scholar]
- 94.Desmet CJ, Peeper DS. The neurotrophic receptor TrkB: a drug target in anti-cancer therapy? Cell Mol Life Sci. 2006;63:755–759. doi: 10.1007/s00018-005-5490-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Camoratto AM, Jani JP, Angeles TS, Maroney AC, Sanders CY, Murakata C. CEP-751 inhibits TRK receptor tyrosine kinase activity in vitro exhibits anti-tumor activity. Int J Cancer. 1997;72:673–679. doi: 10.1002/(sici)1097-0215(19970807)72:4<673::aid-ijc20>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 96.Undevia SD, Vogelzang NJ, Mauer AM, Janisch L, Mani S, Ratain MJ. Phase I clinical trial of CEP-2563 dihydrochloride, a receptor tyrosine kinase inhibitor, in patients with refractory solid tumors. Investig New Drugs. 2004;22:449–458. doi: 10.1023/B:DRUG.0000036687.26604.8c. [DOI] [PubMed] [Google Scholar]
- 97.Araujo J, Logothetis C. Dasatinib: a potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat Rev. 2010;36:492–500. doi: 10.1016/j.ctrv.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Park SI, Zhang J, Phillips KA, Araujo JC, Najjar AM, Volgin AY. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008;68:3323–3333. doi: 10.1158/0008-5472.CAN-07-2997. [DOI] [PubMed] [Google Scholar]
- 99.Blaschuk OW, Sullivan R, David S, Pouliot Y. Identification of a cadherin cell adhesion recognition sequence. Dev Biol. 1990;139:227–229. doi: 10.1016/0012-1606(90)90290-y. [DOI] [PubMed] [Google Scholar]
- 100.Blaschuk OW. Discovery and development of N-cadherin antagonists. Cell Tissue Res. 2012;348:309–313. doi: 10.1007/s00441-011-1320-5. [DOI] [PubMed] [Google Scholar]
- 101.Russo MA, Paolillo M, Sanchez-Hernandez Y, Curti D, Ciusani E, Serra M. A small-molecule RGD-integrin antagonist inhibits cell adhesion, cell migration and induces anoikis in glioblastoma cells. Int J Oncol. 2013;42:83–92. doi: 10.3892/ijo.2012.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ganguly KK, Pal S, Moulik S, Chatterjee A. Integrins and metastasis. Cell Adhes Migr. 2013;7:251–261. doi: 10.4161/cam.23840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alva A, Slovin S, Daignault S, Carducci M, Dipaola R, Pienta K. Phase II study of cilengitide (EMD 121974, NSC 707544) in patients with non-metastatic castration resistant prostate cancer, NCI-6735. A study by the DOD/PCF prostate cancer clinical trials consortium. Investig New Drugs. 2012;30:749–757. doi: 10.1007/s10637-010-9573-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stoeltzing O, Liu WB, Reinmuth N, Fan F, Parry GC, Parikh AA. Inhibition of integrin α5β1 function with a small peptide (ATN-161) plus continuous 5-FU infusion reduces colorectal liver metastases and improves survival in mice. Int J Cancer. 2003;104:496–503. doi: 10.1002/ijc.10958. [DOI] [PubMed] [Google Scholar]
- 105.Khalili P, Arakelian A, Chen GP, Plunkett ML, Beck I, Parry GC. A non-RGD-based integrin binding peptide (ATN-161) blocks breast cancer growth and metastasis in vivo. Mol Cancer Ther. 2006;5:2271–2280. doi: 10.1158/1535-7163.MCT-06-0100. [DOI] [PubMed] [Google Scholar]
- 106.Livant DL, Brabec RK, Pienta KJ, Allen DL, Kurachi K, Markwart S. Anti-invasive, antitumorigenic, and antimetastatic activities of the PHSCN sequence in prostate carcinoma. Cancer Res. 2000;60:309–320. [PubMed] [Google Scholar]
- 107.Läubli H, Borsig L. Selectins promote tumor metastasis. Semin Cancer Biol. 2010;20:169–177. doi: 10.1016/j.semcancer.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 108.Borsig L, Vlodavsky I, Ishai-Michaeli R, Torri G, Vismara E. Sulfated hexasaccharides attenuate metastasis by inhibition of P-selectin and heparanase. Neoplasia. 2011;13:445–452. doi: 10.1593/neo.101734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.D׳Arena G, Calapai G, Deaglio S. Anti-CD44 mAb for the treatment of B-cell chronic lymphocytic leukemia and other hematological malignancies: evaluation of WO2013063498. Expert Opin Ther Pat. 2014;24:821–828. doi: 10.1517/13543776.2014.915942. [DOI] [PubMed] [Google Scholar]
- 110.Negi LM, Talegaonkar S, Jaggi M, Ahmad FJ, Iqbal Z, Khar RK. Role of CD44 in tumour progression and strategies for targeting. J Drug Target. 2012;20:561–573. doi: 10.3109/1061186X.2012.702767. [DOI] [PubMed] [Google Scholar]
- 111.Leong HS, Robertson AE, Stoletov K, Leith SJ, Chin CA, Chien AE. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 2014;8:1558–1570. doi: 10.1016/j.celrep.2014.07.050. [DOI] [PubMed] [Google Scholar]
- 112.Revach OY, Geiger B. The interplay between the proteolytic, invasive, and adhesive domains of invadopodia and their roles in cancer invasion. Cell Adhes Migr. 2014;8:215–225. doi: 10.4161/cam.27842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Omidi Y, Barar J. Targeting tumor microenvironment: crossing tumor interstitial fluid by multifunctional nanomedicines. Bioimpacts. 2014;4:55–67. doi: 10.5681/bi.2014.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gao F, Liang B, Reddy ST, Farias-Eisner R, Su XL. Role of inflammation-associated microenvironment in tumorigenesis and metastasis. Curr Cancer Drug Targets. 2014;14:30–45. doi: 10.2174/15680096113136660107. [DOI] [PubMed] [Google Scholar]
- 115.Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers. 2014;6:1670–1690. doi: 10.3390/cancers6031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185. doi: 10.1155/2014/149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dhani N, Fyles A, Hedley D, Milosevic M. The clinical significance of hypoxia in human cancers. Semin Nucl Med. 2015;45:110–121. doi: 10.1053/j.semnuclmed.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 118.Barar J, Omidi Y. Dysregulated pH in tumor microenvironment checkmates cancer therapy. Bioimpacts. 2013;3:149–162. doi: 10.5681/bi.2013.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hammond E, Khurana A, Shridhar V, Dredge K. The role of heparanase and sulfatases in the modification of heparan sulfate proteoglycans within the tumor microenvironment and opportunities for novel cancer therapeutics. Front Oncol. 2014;4:195. doi: 10.3389/fonc.2014.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]